
Poly(silyl ether)s (silyl ether copolymers) via hydrosilylation of carbonyl compounds
Serter
Luleburgaz
,
Umit
Tunca
 * and
Hakan
Durmaz
*
* and
Hakan
Durmaz
*
Istanbul Technical University, Department of Chemistry, Maslak 34469, Istanbul, Turkey. E-mail: tuncau@itu.edu.tr; durmazh@itu.edu.tr
First published on 6th June 2023
Abstract
Hydrosilylation of carbonyl compounds gives the corresponding silyl ethers in the presence of various catalysts. Previously, transition metals and metal halides, such as Ni, NiCl2, ZnCl2, and H2PtCl6, were efficiently used for the hydrosilylation of carbonyls. The hydrosilylation strategy using dicarbonyls (diketones and dialdehydes) or hydroxy ketones with dihydrosilanes was then implemented in polymer production to afford the synthesis of poly(silyl ether)s (PSE)s. The first preparation of PSEs by the Weber group used aromatic α,ω-dicarbonyl, and dihydrosilane catalyzed by transition metal complexes, expensive/low abundance ruthenium, and rhodium, whereas nowadays, inexpensive/high abundance catalysts (Mn, Zn, and Cu) and metal-free catalyst (tris(pentafluorophenyl)borane (B(C6F5)3)) have attracted much interest in the synthetic approach toward PSEs. Furthermore, the metal-free catalysts utilized in the hydrosilylation of carbonyls have recently found practical applications in polymer chemistry. Moreover, the chiral PSEs developed by the Zhou group have promising applications in asymmetric catalysis and chiral separation as chiral stationary phases. Particularly, this review focuses on the synthesis of PSEs through various dicarbonyls (or hydroxyl carbonyls) and disilanes. We excluded papers regarding methods involving the polycondensation of diols with dichlorosilanes, diaminosilanes, or dialkoxysilanes and ring-opening polymerization (ROP) of cyclic carbosiloxane.
 Serter Luleburgaz | Serter Luleburgaz received his BSc degree in 2016 from the Chemistry Department of Istanbul Technical University (ITU), Turkey. He completed his MSc in 2017 and PhD in 2023 under the supervision of Prof. Durmaz. His PhD thesis focused on the reductive etherification reaction (RER) in macromolecular engineering and he has been involved in other projects including polymer synthesis, modification, and applications from various aspects. |
 Umit Tunca | Umit Tunca He has been a professor at the Chemistry Department of Istanbul Technical University, Turkey since 1998. He has published over 150 papers and three book chapters. His research interests are mainly living radical polymerizations and click reactions. His awards include Monbusho (Japan) (1987) and Alexander von Humboldt (Germany) (1990) research fellowships, and TUBITAK (The Scientific and Technological Research Council of Turkey) Young Investigator (1997). |
 Hakan Durmaz | Hakan Durmaz completed his PhD in 2010 in the Chemistry Department of Istanbul Technical University (ITU), Turkey. He continued his post-doctoral studies at the University of Michigan (USA) between 2012 and 2014. He became an associate professor in 2015 and a full professor in 2020 at ITU. He has received many research awards from various institutions in Turkey. He has published more than 100 peer-reviewed articles. His research interests include polymer synthesis, modification, and applications from various aspects. |
Introduction and background
Metal halides such as NiCl2, ZnC12, or H2PtCl6 are efficient catalysts for the hydrosilylation reaction of carbonyl compounds, although the mechanism has not been completely identified.1,2 Calas and Frainnet's group reported that a metal halide such as ZnCl2, or NiCl2 could be utilized as a catalyst in the hydrosilylation of benzaldehyde with Et3SiH, resulting in silyl ether disproportionating immediately to dibenzylether and hexaethyldisiloxane under vigorous reaction conditions (high temperatures and long reaction times) (Scheme 1).3 | ||
| Scheme 1 Silylation of aldehydes with Et3SiH using ZnCl2 (or Ni) as the catalyst.3 | ||
It should be noted that the disproportionation of the silyl ether product was frequently observed in the case of aldehydes under the reaction conditions given above.
Ojima et al. first employed a rhodium(I) complex ((Ph3P)3RhCl) as a catalyst in the hydrosilylation of a variety of carbonyl compounds (simple aldehydes and ketones, α-diketones, α,β-unsaturated carbonyl compounds, etc.) under mild reaction conditions (Scheme 2).4 The authors selected mono-, di-, and tri-hydrosilanes as organosilane sources for the hydrosilylation of carbonyl compounds. The same group also noted that no disproportionation was detected when the rhodium(I) complex was utilized in the hydrosilylation of compounds; for example, with benzaldehyde, only benzyloxysilane was obtained in high yields.
 | ||
| Scheme 2 A plausible mechanism proposed by the Ojima group for the hydrosilylation of carbonyl compounds catalyzed by (Ph3P)3RhCl. Reproduced from ref. 4 with permission from Elsevier, copyright 1975. | ||
Piers group for the first time presented the hydrosilylation of aromatic aldehydes, ketones, and esters at room temperature in the presence of 1–4 mol% of tris(pentafluorophenyl)borane (B(C6F5)3) and 1 equiv. of triphenyl silane (Ph3SiH). Scheme 3A illustrates the reaction mechanism for the B(C6F5)3-catalyzed hydrosilylation of ketones.5,6 They also highlighted that limiting the silane reagent to 1 equiv. was essential for clean reactions since further reduction of the silyl ether or silyl acetal products was observed when excess silane was present. More recently, the Chandrasekhar group reported that an efficient synthesis of symmetrical and unsymmetrical ethers could be achieved by reductive coupling of carbonyl compounds with alkoxysilanes.7 This reaction is performed using inert polymethylhydrosiloxane (PMHS) (2 equiv.) as the hydride source and B(C6F5)3 (1 mol%) as the catalytic activator of the PMHS. They proposed a plausible mechanism for this reductive etherification reaction (RER) platform in which coordination of the carbonyl compound with B(C6F5)3 is followed by the formation of an acetal intermediate, which abstracts hydride from PMHS and gives the corresponding ether (Scheme 3B).
 | ||
| Scheme 3 (A) B(C6F5)3-catalyzed hydrosilylation of carbonyl compounds using R3SiH. R1 and R2 were not indicated. Reproduced from ref. 5 with permission from American Chemical Society, copyright 1996 and ref. 6 with permission from the Royal Chemical Society, copyright 2015, (B) reductive etherification reaction (RER) of carbonyl compounds using PMHS catalyzed by B(C6F5)3. Reproduced from ref. 7 with permission from Elsevier, copyright 2004. | ||
The hydrosilylation of carbonyl groups in ketones and aldehydes follows the RER mechanism. In the RER protocol, carbonyl compounds in the presence of silane and a Brønsted or Lewis acid catalyst afford the synthesis of symmetric and unsymmetrical ethers.8–28 However, the silyl ether is produced rather than the corresponding ether in the hydrosilylation. Notably, both mechanisms seem to cover the competing reaction pathways.
More interestingly, triethylsilane (Et3SiH) with B(C6F5)3 also reduces ethers (dialkyl and aryl alkyl ethers) to the corresponding alkanes under ambient conditions.29 Various research groups have employed the silane–B(C6F5)3 combination in the reduction of carbonyl to alkanes (deoxygenation) as well.30,31 The use of an R3SiH Lewis acid catalyst in combination with B(C6F5)3 in both silyl ether and alkyl ether syntheses using carbonyl compounds seems intriguing and unusual.
Synthesis of poly(silyl ether)s (PSEs) via hydrosilylation of carbonyl compounds
PSEs, similar to poly(siloxane)s, are high-performance materials, which demonstrate high thermal stability, but they have low hydrolytic stability due to acid-catalyzed hydrolysis of their silyl ether (Si–O–C) backbone. PSEs as high-performance materials can be employed as elastomeric products, gas-permeable membranes, and biocompatible coatings for commodity applications.32,33 On the other hand, the labile silyl ether connections of PSEs enable environmental and biomedical advantages and applications.34PSEs have been prepared via several routes involving the polycondensation reaction and ring-opening polymerization (ROP) (Table 1). The polycondensation reaction occurs between dialkoxysilane and α,ω-diol. As an alternative, dichlorosilane cannot be used with α,ω-diol due to the formation of HCl, which leads to the hydrolysis of the silyl ether linkage.35 PSE has also been synthesized by acid-catalyzed ROP of 1-oxa-2-silacyclopentane, which has been achieved by platinum-catalyzed intramolecular hydrosilylation of dimethylsilyl allyl ether.36 Following the ROP methodology, PSE has been produced by the reaction of bis(glycidyl) ether with dichlorosilane. In this route, tetra n-butyl ammonium chloride catalyzes the reaction, and the chloride nucleophile attacking the oxirane ring results in ring-opening and the formation of chloromethyl and a secondary alkoxide group. The polymerization occurs when a secondary alkoxide sequentially reacts with the silyl halide to produce a silyl ether linkage and regenerate the tetra n-butyl ammonium chloride catalyst.37–39 Weber and coworkers carried out seminal work on the hydrosilylation of carbonyl compounds catalyzed by transition metal complexes, including ruthenium and rhodium, to result in the synthesis of PSEs.40 In this protocol, 1,4-diacetyldiphenyl ether, 1,4-diacetylbenzene, or terephthalaldehyde with 1,3-tetramethyldisiloxane were copolymerized using dihydridocarbonyltris(triphenylphosphine)ruthenium (Ph3P)2RuH2CO catalyst in toluene at 100 °C for 12 h (Scheme 4). The molecular weight (Mw) and dispersity (Đ) of the polymers were determined by GPC and found in the range of 93 to 48 kDa and 3.52–2.74, respectively. The authors carried out the methanolysis reaction of PSEs and observed significant differences in stability toward methanolysis. The PSE structure having the primary C linked to the O atom is unstable upon precipitation in methanol at room temperature. However, PSEs with a secondary C bonded to the O atom display more resistance to methanolysis. The authors indicated that the PSEs obtained in this work might similarly offer controlled degradation upon hydrolysis. The authors also noted that the PSEs are thermally stable up to almost 300 °C.
| Methods for the synthesis of PSEs | Compounds | Structure of PSE | Ref. |
|---|---|---|---|
| Polycondensation | Dichlorosilane, diaminosilanes or dialkoxysilane and diol |

|
35 |
| Ring-opening polymerization | 1-Oxa-2-silacyclopentane |

|
36 |
| Hydrosilylation polymerization | Bis(glycidyl) ether and dihalosilane |

|
38 |
| Aldehyde (or ketone) and silane |
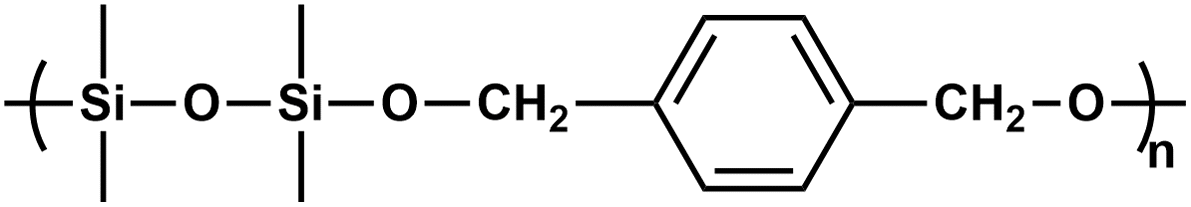
|
40 | |
| Hydrosilylation and dehydrogenative cross-coupling polymerization | Hydroxy-aldehyde or -ketone and silane |

|
52 |
| Dehydrogenative cross-coupling polymerization | Diol and silane |

|
56 |
 | ||
| Scheme 4 Synthesis of PSE using terephthalaldehyde and 1,3-tetramethyldisiloxane catalyzed by ruthenium. Reproduced from ref. 40 with permission from the American Chemical Society, copyright 1998. | ||
As an extension of this work, the Weber group reported the Ru-catalyzed hydrosilylation polymerization of o-quinones with α,ω-dihydridooligodimethylsiloxane (ODMS) to yield high-molecular-weight PSEs (Mn varies in the range of 8.6–41 kDa) (Scheme 5).41 The polymerization was carried out in bulk using activated (Ph3P)2RuH2CO as a catalyst under an argon atmosphere at 125 °C for 18 h. As expected, the Tgs of the polymers decreased in parallel to the increasing number of repeating units (n) of ODMS. When the n of the ODMS is 6, the Tg of the PSE was found to be −100 °C, which is fairly close to the Tg of polydimethylsiloxane (−125 °C).
 | ||
| Scheme 5 PSE synthesis from phenanthrene-9,10-dione and 1,9-dihydridodecamethylpentasiloxane. Reproduced from ref. 41 with permission from the American Chemical Society, copyright 2001. | ||
In the following work, aliphatic ω-dimethylsilyloxy ketones or aliphatic a,ω-diketones with ODMS were polymerized in bulk at 125 °C for 18 h using the abovementioned Ru catalyst to yield unsymmetrical and symmetrical PSEs, respectively (Scheme 6).42
 | ||
| Scheme 6 Preparation of unsymmetrical PSE using 5-dimethylsilyloxy-2-pentanone using Ru catalyst at 125 °C for 18 h. Reproduced from ref. 42 with permission from the American Chemical Society, copyright 2002. | ||
The Mws determined by GPC display PSEs with broad molecular weights ranging from 4.5 to 85.5 kDa. The Tgs for PSEs display similar behavior to that described previously for PSEs with aromatic precursors. A similar behavior was observed if the C atom of the Si–O–C linkage is secondary; PSEs having this structure are considerably more resistant to hydrolysis and methanolysis than if it is primary.
A similar strategy was also applied to the synthesis of PSEs starting from dimethylsilyloxyaryl ketones or aldehydes using an activated Ru catalyst under the abovementioned reaction conditions (Scheme 7).43
 | ||
| Scheme 7 PSE synthesis from hydrosilylation of 4-dimethylsilyloxyacetophenone. Reproduced from ref. 43 with permission from Elsevier, copyright 2000. | ||
It should be noted here that the hydrosilylation polymerization gave high-molecular-weight PSEs (up to Mw = 168 kDa) with high isolated yields (in the range of 80–85%). The Weber group noticed that the Tgs of PSEs decrease as the number of dimethylsiloxane units between the aromatic moieties increases.
The Oro group reported the rhodium-catalyzed synthesis of PSEs.44 They first showed that rhodium(I)-N-heterocyclic carbenes (Rh(I)-NHCs) with labile NHCs are very efficient and selective catalysts for alkyne hydrosilylation45 and hydrogen transfer hydrogenation.46 They used a Rh(COD)(2-methoxyethyl-NHC-(CH2)3Si(OiPr3)3)Br homogenous catalyst in the hydrosilylation of terephthalaldehyde with 1,1,3,3,5,5-hexamethyltrisiloxane (hexMTS) in dioxane at 110 °C for 4 days (Scheme 8) (Table 1). They obtained the final PSE as a yellow oil (Mw = 5.2 kDa and Đ = 1.7) in quantitative yield. As a second step, authors prepared a heterogeneous catalyst immobilizing the Rh(COD)(2-methoxyethyl-NHC-(CH2)3Si(OiPr3)3)Br on a mobile crystalline material-41 (MCM-41) and employed it in similar reaction conditions to those given above. The PSE was obtained with bimodal distribution containing two portions: 22% of total mass with Mw = 94 kDa (Đ = 1.6) and 78% of total mass with Mw = 7.4 kDa (Đ = 1.5).
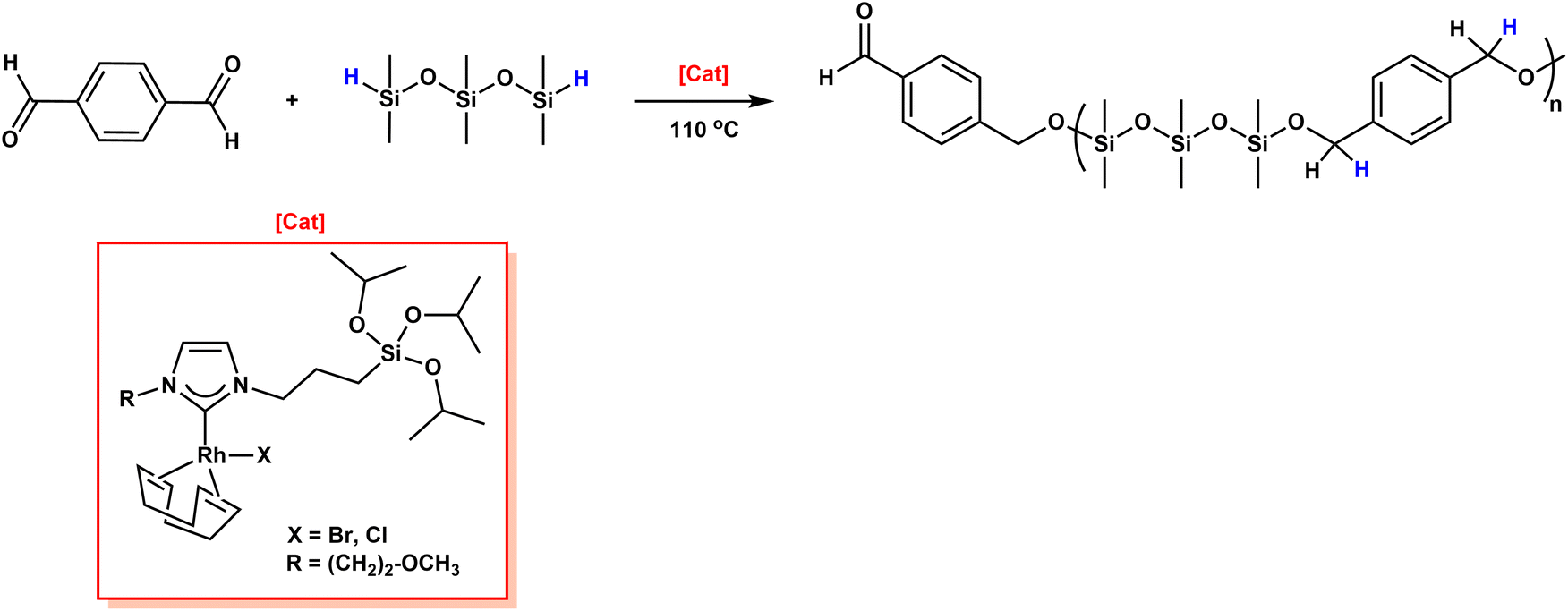 | ||
| Scheme 8 Hydrosilylation of terephthalaldehyde with hexMTS using Rh(COD)(2-methoxyethyl-NHC-(CH2)3Si(OiPr3)3)Br homogenous catalyst. Reproduced from ref. 44 with permission from John Wiley and Sons, copyright 2013. | ||
In the following work, the Oro group changed 2-methoxy ethyl to 2,6-diisopropylphenyl in the Rh-NHC complex, then immobilized it in mesoporous materials, such as MCM-41 and KIT-6, and finally utilized the immobilized catalysts in the preparation of PSE by catalytic hydrosilylation under the same reaction conditions as those mentioned above.47 These heterogeneous catalyst systems gave PSEs with high molecular weights (Mw = 2.61 × 106 and Mw = 4.43 × 105 g mol−1) for those immobilized on MCM-41 and KIT-6, respectively.
Recently, the Cui group utilized the zwitterionic heteroscopionate zinc hydrido complex (LZnH) as a cheaper and less toxic catalyst compared to traditional rhodium and ruthenium catalysts.48 The authors employed the LZnH catalyst in the hydrosilylation of terephthalaldehyde, isophthalaldehyde, and phthalaldehyde using diphenylsilane (DPS) to afford PSEs (Scheme 9). The polymerizations were carried out at 40 °C in THF under N2 in the range of 24 to 72 h. PSEs with high molecular weights (Mn = 20.3–17.6 kDa; Đ = 2.35–2.60) were achieved when terephthalaldehyde and isophthalaldehyde were used as monomers with DPS in the polymerization. Nevertheless, phthalaldehyde afforded only a seven-membered cyclic silyl ether.
 | ||
| Scheme 9 The plausible mechanism for the polymerization of aromatic dialdehydes and DPS in the presence of LZnH. Reproduced from ref. 48 with permission from John Wiley and Sons, copyright 2017. | ||
Du and coworkers presented a manganese–salen (MnN(salen-3,5-tert-Bu2)) catalyst as a cheap and nontoxic metal for hydrosilylation of dicarbonyl compounds as well as dehydrogenative cross-coupling of diols to yield PSEs (Scheme 10).49 The authors also employed compounds containing both carbonyl and hydroxyl functionalities via a combination of hydrosilylation and dehydrogenative cross-coupling mechanisms for PSE production. The polymerization reactions of dicarbonyls with DPS were carried out using the manganese catalyst at the reflux temperature of high boiling solvents for longer reaction times (12–48 h) to produce PSEs with rather low molecular weights ranging from Mn = 1.8 to 2.4 kDa (Đ = 1.73–1.88) in low yields. However, analogous structures of diols compared to dicarbonyls gave moderately high-molecular-weight PSEs (Mn = 9–15 kDa, Đ = 1.41–2.38) with high yields. This may be ascribed to the stronger nucleophilicity of hydroxyl than that of carbonyl. When p-hydroxybenzaldehyde was selected as a mixed monomer to react with DPS, a PSE with Mn = 3.8 kDa (Đ = 1.73) was achieved under similar reaction conditions. More recently, extending this system (hydrosilylation and dehydrogenative cross-coupling polymerization) to the furan-based monomers, 5-hydroxymethylfurfural (HMF), 2,5-bis-(hydroxymethyl)furan, 2,5-diformylfuran, and 5,5′-[oxybis-(methylene)]di(2-furaldehyde), derived from biorenewable sources, Du and coworkers obtained a series of PSEs with high molecular weight (up to Mn = 25 kDa) under similar reaction conditions to those given above.50
 | ||
| Scheme 10 A plausible mechanism for the synthesis of PSEs from dicarbonyl and DPS using a manganese–salen (MnN(salen-3,5-tert-Bu2)) catalyst. Reproduced from ref. 49 with permission from the American Chemical Society, copyright 2017. | ||
Cui and coworkers exploited a previously mentioned HMF monomer as a renewable source with DPS or methyl phenyl silane in the presence of a new pair of catalysts, B(C6F5)3 and the heteroscorpionate zinc hydride complex LZnH (L = (MePz)2CP(Ph)2NPh, MePz = 3,5-dimethylpyrazolyl), to result in the corresponding PSE (Scheme 11).51 When DPS was employed as a comonomer with a ratio of HMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DPS:LZnH = 100:100:1, in toluene at room temperature for 24 h, followed by 48 h at 40 °C, a PSE was obtained with Mw = 12.2 kDa and Đ = 1.23. On increasing the ratio of HMF:DPS:LZnH to 200:200:1, the Mw of the PSE increased to 19.8 kDa with Đ = 1.31. Similar results were observed in the case of the methylphenyl silane, suggesting that both polymerizations proceeded in a controlled fashion.
:DPS:LZnH = 100:100:1, in toluene at room temperature for 24 h, followed by 48 h at 40 °C, a PSE was obtained with Mw = 12.2 kDa and Đ = 1.23. On increasing the ratio of HMF:DPS:LZnH to 200:200:1, the Mw of the PSE increased to 19.8 kDa with Đ = 1.31. Similar results were observed in the case of the methylphenyl silane, suggesting that both polymerizations proceeded in a controlled fashion.
 | ||
| Scheme 11 LZnH and B(C6F5)3 pair as synergistic catalysts employed in the hydrosilylation of HMF. Reproduced from ref. 51 with permission from John Wiley and Sons, copyright 2019. | ||
The bio-based building block HMF (or vanillin) and disilanes (DPS, methylphenyl silane, or diethylsilane) were polymerized using Pt(II) complexes as catalysts in 2-methyl tetrahydrofuran (MeTHF) at room temperature for 24 h and 50 °C for 4 h to yield the corresponding PSEs.52 When HMF and DPS were polymerized, a catalytic loading of 500 ppm of [Pt]Me was sufficient to obtain a PSE with high molecular weight (Mw = 57.5 kDa and Đ = 3.5) and high yield (Scheme 12).
 | ||
| Scheme 12 Preparation of PSE containing HMF and DPS using [Pt]Me catalyst in MeTHF. Reproduced from ref. 52 with permission from John Wiley and Sons, copyright 2022. | ||
Recently, Li and Hawker utilized only B(C6F5)3 in a metal-free catalyst concept for the preparation of PSEs using aryl-substituted α-diketone and bis(silane) monomers under mild reaction conditions (Scheme 13).53
 | ||
| Scheme 13 Preparation of a PSE using benzil, 1,4-bis-(dimethylsilyl)benzene, and B(C6F5)3 in CHCl3 or toluene. Reproduced from ref. 53 with permission from American Chemical Society, copyright 2019. | ||
Notably, considering the monomers benzil and 1,4-bis(dimethylsilyl)benzene, 0.5 mol% of B(C6F5)3 catalyst loading and equimolar reaction conditions in CHCl3 and toluene produced PSEs with Mw = 47 kDa (Đ = 1.8) and Mw = 98 kDa (Đ = 1.8) in 80% yield, respectively. The authors confirmed that a cyclic dimer was detected in 15% yield in all polymerization systems. Moreover, methanolysis and hydrolysis of the selected PSEs were monitored as a function of time against changes in the Mn values by GPC. The PSEs containing silphenylene units demonstrated no change in Mn for up to 15 days in methanolysis. However, the Mn of the siloxane-based PSE decreased to half of the initial molecular weight after 15 days. Moreover, both PSEs with silphenylene and siloxane units were stable in the presence of trimethylamine over the same time. As expected, both polymers are prone to acidic hydrolysis while exhibiting a sharp decrease in Mn values to complete degradation within 12 h and 7 days for siloxane- and silphenylene-based PSEs, respectively.
Chiral PSEs have been prepared using a wide diversity of dicarbonyls and disilanes in the presence of CuH catalysts in various solvents and temperatures.54 The Zhou group employed 1,1,3,3-tetramethyldisiloxane and 4,4′-diacetylbiphenyl as model monomers and investigated the effects of solvents, copper precursors, and bisphosphine ligands, both on the reactivity and diastereoselectivity for optimization of the reaction conditions. As a result, model monomers were polymerized utilizing the copper(II) cyclohexanebutyrate/L1 catalyst in tert-butyl methyl ether at 30 °C for 24 h, and therefore, the resultant PSE was achieved with high yield, moderately high molecular weight (Mn = 11.8 kDa, Đ = 2.39) and excellent stereoselectivity (up to 99% ee and 96:4 dl/meso) (Scheme 14).
 | ||
| Scheme 14 Synthesis of chiral PSE utilizing monomers 1,1,3,3-tetramethyldisiloxane and 4,4′-diacetylbiphenyl in the presence of copper(II) cyclohexanebutyrate/L catalyst in tert-butyl methyl ether at 30 °C for 24 h. Reproduced from ref. 54 with permission from the American Chemical Society, copyright 2020. | ||
The authors claimed that the enantiopure PSEs with good thermal properties might have a potential application in chiral separation, and the PSEs with heteroaromatic rings play a promising role in heterogeneous asymmetric catalysis. In the following work, the Zhou group employed the CuH-catalyzed consecutive hydrosilylation/dehydrogenative cross-coupling polymerization of hydroxy ketones and dihydrosilanes to afford chiral PSEs.55
Conclusion and outlook
As a first approach, the Weber group employed transition metal catalysts (Ru/Rh salts) in the synthesis of PSEs using dicarbonyl and dihydrosilane, while they benefited from the work of the Ojima group. However, the experienced vigorous reaction conditions, and the low abundance and high cost of transition metal catalysts (Ru/Rh), pushed the chemists to develop new catalysts. In recent years, based on the hydrosilylation reaction, chemists have developed novel metal catalysts that are cheap and highly abundant. Zinc hydride complex, manganese–salen complex, zinc hydride–B(C6F5)3 complex, Pt complex, and copper complex were developed as catalysts in recent years. These catalysts allowed the synthesis of PSEs under relatively mild conditions. Of the catalysts, Mn and Pt complexes should be considered biocompatible catalysts and can be employed for the synthesis of PSEs from biorenewable feedstocks. Moreover, increasing interest in mild and metal-free techniques for the synthesis of small organic molecules and polymers led to the development of various metal-free catalysts. From this perspective, B(C6F5)3 as a metal-free catalyst has been widely used both for hydrosilylation coupling reactions and PSE synthesis under mild conditions.Since PSEs that contain Si–O–C bonds are more susceptible to acid- or base-catalyzed hydrolysis and methanolysis compared to polysiloxanes with Si–O–Si bonds, this feature makes them attractive materials for biomedical and environmental applications. Notably, the degradation behavior of PSEs, besides the thermomechanical properties, can be modified by varying the substituent groups on the Si and/or C atoms on the backbone of the PSEs and by copolymerization with other polymer chains. Thus, the hydrolytic lability of the Si–O–C linkage of PSEs makes them highly attractive for use in drug delivery systems. The PSEs achieved from biomass (furan monomers) can also undergo hydrolytic degradation and completely degrade under acidic conditions to yield the corresponding furan diol and silanediol.
Additionally, PSEs have found applications in the aerospace industry as CO2-philic polymer membranes, reprocessable and degradable thermosets, and for preparing dielectric or fluorescent materials and elastomeric materials. Enantiomerically enriched PSEs with good thermal stability are promising for applications in chiral separations as chiral stationary phases.
Conflicts of interest
There are no conflicts to declare.References
- I. Ojima, M. Nihonyanagi and Y. Nagai, J. Chem. Soc., Chem. Commun., 1972, 938a–938a RSC.
- R. Calas, J. Organomet. Chem., 1980, 200, 11–36 CrossRef CAS.
- R. Calas, E. Frainnet and J. Bonastre, C. R. Acad. Sci., 1960, 251, 2987–2989 CAS.
- I. Ojima, M. Nihonyanagi, T. Kogure, M. Kumagai, S. Horiuchi, K. Nakatsugawa and Y. Nagai, J. Organomet. Chem., 1975, 94, 449–461 CrossRef CAS.
- D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS.
- M. Oestreich, J. Hermeke and J. Mohr, Chem. Soc. Rev., 2015, 44, 2202–2220 RSC.
- S. Chandrasekhar, G. Chandrashekar, B. Nagendra Babu, K. Vijeender and K. Venkatram Reddy, Tetrahedron Lett., 2004, 45, 5497–5499 CrossRef CAS.
- M. P. Doyle, D. J. DeBruyn and D. A. Kooistra, J. Am. Chem. Soc., 1972, 94, 3659–3661 CrossRef CAS.
- C. T. West, S. J. Donnelly, D. A. Kooistra and M. P. Doyle, J. Org. Chem., 1973, 38, 2675–2681 CrossRef CAS.
- M. P. Doyle, D. J. DeBruyn, S. J. Donnelly, D. A. Kooistra, A. A. Odubela, C. T. West and S. M. Zonnebelt, J. Org. Chem., 1974, 39, 2740–2747 CrossRef CAS.
- J.-i. Kato, N. Iwasawa and T. Mukaiyama, Chem. Lett., 1985, 14, 743–746 CrossRef.
- M. B. Sassaman, K. D. Kotian, G. K. S. Prakash and G. A. Olah, J. Org. Chem., 1987, 52, 4314–4319 CrossRef CAS.
- T. Yokozawa and F. Nakamura, Makromol. Chem., Rapid Commun., 1993, 14, 167–172 CrossRef CAS.
- T. Yokozawa and K. Takenoya, React. Funct. Polym., 1996, 30, 251–260 CrossRef CAS.
- T. Miyai, Y. Onishi and A. Baba, Tetrahedron Lett., 1998, 39, 6291–6294 CrossRef CAS.
- T. Miyai, Y. Onishi and A. Baba, Tetrahedron, 1999, 55, 1017–1026 CrossRef CAS.
- T. Yokozawa and F. Nakamura, Macromolecules, 2002, 28, 4668–4674 CrossRef.
- B. A. Gellert, N. Kahlcke, M. Feurer and S. Roth, Chemistry, 2011, 17, 12203–12209 CrossRef CAS PubMed.
- N. Sakai, K. Nagasawa, R. Ikeda, Y. Nakaike and T. Konakahara, Tetrahedron Lett., 2011, 52, 3133–3136 CrossRef CAS.
- Y.-J. Zhang, W. Dayoub, G.-R. Chen and M. Lemaire, Tetrahedron, 2012, 68, 7400–7407 CrossRef CAS.
- N. Sakai, Y. Nonomura, R. Ikeda and T. Konakahara, Chem. Lett., 2013, 42, 489–491 CrossRef CAS.
- R. Savela and R. Leino, Synthesis, 2015, 47, 1749–1760 CrossRef CAS.
- Y. H. Lee and B. Morandi, Synlett, 2017, 28, 2425–2428 CrossRef CAS.
- C. Zhao, C. A. Sojdak, W. Myint and D. Seidel, J. Am. Chem. Soc., 2017, 139, 10224–10227 CrossRef CAS PubMed.
- S. Luleburgaz, G. Hizal, U. Tunca and H. Durmaz, Macromolecules, 2021, 54, 5106–5116 CrossRef CAS.
- S. Luleburgaz, G. Hizal, U. Tunca and H. Durmaz, Macromolecules, 2022, 55, 1533–1543 CrossRef CAS.
- E. Akar, D. Kandemir, S. Luleburgaz, V. Kumbaraci and H. Durmaz, Eur. Polym. J., 2022, 177, 111440 CrossRef CAS.
- M. A. Brook, J. B. Grande and F. Ganachaud, in Silicon Polymers, ed. A. M. Muzafarov, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 161–183 Search PubMed.
- V. Gevorgyan, M. Rubin, S. Benson, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 2000, 65, 6179–6186 CrossRef CAS PubMed.
- V. Gevorgyan, M. Rubin, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 2001, 66, 1672–1675 CrossRef CAS PubMed.
- S. Chandrasekhar, C. R. Reddy and B. N. Babu, J. Org. Chem., 2002, 67, 9080–9082 CrossRef CAS PubMed.
- J. E. Curry and J. D. Byrd, J. Appl. Polym. Sci., 1965, 9, 295–311 CrossRef CAS.
- Y. Nagasaki, F. Matsukura, M. Kato, H. Aoki and T. Tokuda, Macromolecules, 1996, 29, 5859–5863 CrossRef CAS.
- C. Cheng, A. Watts, M. A. Hillmyer and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 11872–11876 CrossRef CAS PubMed.
- C. Burger and F. H. Kreuzer, in Silicon in Polymer Synthesis, ed. H. R. Kricheldorf, Springer Berlin Heidelberg, Berlin, Heidelberg, 1996, pp. 113–222, DOI:10.1007/978-3-642-79175-8_3.
- N. S. Fedotov, V. L. Kozlikov and V. F. Mironov, Zh. Obshch. Khim., 1971, 40, 2589 Search PubMed . [J. Gen. Chem. USSR, 1971, 40, Engl. Transl.].
- H. Itoh, A. Kameyama and T. Nishikubo, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 3217–3225 CrossRef CAS.
- D.-J. Liaw, Polymer, 1997, 38, 5217–5219 CrossRef CAS.
- T. Nishikubo, A. Kameyama, Y. Kimura and T. Nakamura, Macromolecules, 1996, 29, 5529–5534 CrossRef CAS.
- J. K. Paulasaari and W. P. Weber, Macromolecules, 1998, 31, 7105–7107 CrossRef CAS.
- J. M. Mabry, M. K. Runyon and W. P. Weber, Macromolecules, 2001, 34, 7264–7268 CrossRef CAS.
- J. M. Mabry, M. K. Runyon and W. P. Weber, Macromolecules, 2002, 35, 2207–2211 CrossRef CAS.
- J. M. Mabry, J. K. Paulasaari and W. P. Weber, Polymer, 2000, 41, 4423–4428 CrossRef CAS.
- G. Lázaro, M. Iglesias, F. J. Fernández-Alvarez, P. J. Sanz Miguel, J. J. Pérez-Torrente and L. A. Oro, ChemCatChem, 2013, 5, 1133–1141 CrossRef.
- M. V. Jiménez, J. J. Pérez-Torrente, M. I. Bartolomé, V. Gierz, F. J. Lahoz and L. A. Oro, Organometallics, 2008, 27, 224–234 CrossRef.
- M. V. Jiménez, J. Fernández-Tornos, J. J. Pérez-Torrente, F. J. Modrego, S. Winterle, C. Cunchillos, F. J. Lahoz and L. A. Oro, Organometallics, 2011, 30, 5493–5508 CrossRef.
- G. Lázaro, F. J. Fernández-Alvarez, M. Iglesias, C. Horna, E. Vispe, R. Sancho, F. J. Lahoz, M. Iglesias, J. J. Pérez-Torrente and L. A. Oro, Catal. Sci. Technol., 2014, 4, 62–70 RSC.
- C. Li, X. Hua, Z. Mou, X. Liu and D. Cui, Macromol. Rapid Commun., 2017, 38, 1700590 CrossRef PubMed.
- S. Vijjamarri, V. K. Chidara and G. Du, ACS Omega, 2017, 2, 582–591 CrossRef CAS PubMed.
- S. Vijjami, S. Streed, E. M. Serum, M. P. Sibi and G. Du, ACS Sustainable Chem. Eng., 2018, 6, 2491–2497 CrossRef.
- C. Li, L. Wang, M. Wang, B. Liu, X. Liu and D. Cui, Angew. Chem., Int. Ed., 2019, 58, 11434–11438 CrossRef CAS PubMed.
- H. Fouilloux, M. N. Rager, P. Rios, S. Conejero and C. M. Thomas, Angew. Chem., Int. Ed., 2022, 61, e202113443 CrossRef CAS PubMed.
- C. S. Sample, S.-H. Lee, M. W. Bates, J. M. Ren, J. Lawrence, V. Lensch, J. A. Gerbec, C. M. Bates, S. Li and C. J. Hawker, Macromolecules, 2019, 52, 1993–1999 CrossRef CAS.
- X.-Q. Wang, X.-Y. Zhai, B. Wu, Y.-Q. Bai and Y.-G. Zhou, ACS Macro Lett., 2020, 9, 969–973 CrossRef CAS PubMed.
- X.-Q. Wang, B. Wu, Y.-Q. Bai, X.-Y. Zhai and Y.-G. Zhou, Eur. Polym. J., 2022, 177, 111474 CrossRef CAS.
- Y. Li, M. Seino and Y. Kawakami, Macromolecules, 2000, 33, 5311–5314 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |