
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUsing a diphenyl-bi-(1,2,4-triazole) tricarbonylrhenium(I) complex with intramolecular π–π stacking interaction for efficient solid-state luminescence enhancement†
Alexandre
Poirot
 a,
Corinne
Vanucci-Bacqué
a,
Béatrice
Delavaux-Nicot
b,
Clarisse
Meslien
a,
Nathalie
Saffon-Merceron
c,
Charles-Louis
Serpentini
d,
Florence
Bedos-Belval
a,
Eric
Benoist
a and
Suzanne
Fery-Forgues
*a
a,
Corinne
Vanucci-Bacqué
a,
Béatrice
Delavaux-Nicot
b,
Clarisse
Meslien
a,
Nathalie
Saffon-Merceron
c,
Charles-Louis
Serpentini
d,
Florence
Bedos-Belval
a,
Eric
Benoist
a and
Suzanne
Fery-Forgues
*a
aSPCMIB, CNRS UMR 5068, Université de Toulouse III Paul Sabatier, 118 route de Narbonne, 31062 Toulouse cedex 9, France. E-mail: suzanne.fery-forgues@univ-tlse3.fr
bLaboratoire de Chimie de Coordination, CNRS (UPR 8241), Université de Toulouse (UPS, INPT), 205 route de Narbonne, 31077 Toulouse Cedex 4, France
cService Diffraction des Rayons X, Institut de Chimie de Toulouse, ICT- UAR 2599, Université de Toulouse III Paul Sabatier, 118 route de Narbonne, 31062 Toulouse cedex 9, France
dLaboratoire IMRCP, CNRS UMR 5623, Université de Toulouse III Paul Sabatier, 118 route de Narbonne, 31062 Toulouse cedex 9, France
First published on 7th March 2023
Abstract
Since intramolecular π–π stacking interactions can modify the geometry, crystal packing mode, or even the electronic properties of transition metal complexes, they are also likely to influence the solid-state luminescence properties. Following this concept, a new tricarbonylrhenium(I) complex (Re-BPTA) was designed, based on a simple symmetrical 5,5′-dimethyl-4,4′-diphenyl-3,3′-bi-(1,2,4-triazole) organic ligand. The complex was prepared in good yield using a three-step procedure. The crystallographic study revealed that both phenyl rings are located on the same side of the molecule, and twisted by 71° and 62°, respectively, with respect to the bi-(1,2,4-triazole) unit. They overlap significantly, although they are slipped parallel to each other to minimize the intramolecular interaction energy. The π–π stacking interaction was also revealed by 1H NMR spectroscopy, in good agreement with the results of theoretical calculations. In organic solutions, a peculiar electrochemical signature was observed compared to closely-related pyridyl-triazole (pyta)-based complexes. With regard to the optical properties, the stiffness of the Re-BPTA complex led to the stabilization of the 3MLCT state, and thus to an enhancement of the red phosphorescence emission compared to the more flexible pyta complexes. However, an increased sensitivity to quenching by oxygen appeared. In the microcrystalline phase, the Re-BPTA complex showed strong photoluminescence (PL) emission in the green-yellow wavelength range (λPL = 548 nm, ΦPL = 0.52, 〈τPL〉 = 713 ns), and thus a dramatic solid-state luminescence enhancement (SLE) effect. These attractive emission properties can be attributed to the fact that the molecule undergoes little distortion between the ground state and the triplet excited state, as well as to a favorable intermolecular arrangement that minimizes detrimental interactions in the crystal lattice. The aggregation-induced phosphorescence emission (AIPE) effect was clear, with a 7-fold increase in emission intensity at 546 nm, although the aggregates formed in aqueous medium were much less emissive than the native microcrystalline powder. In this work, the rigidity of the Re-BPTA complex is reinforced by the intramolecular π–π stacking interaction of the phenyl rings. This original concept provides a rhenium tricarbonyl compound with very good SLE properties, and could be used more widely to successfully develop this area of research.
Introduction
In recent years, transition metal complexes that exhibit aggregation-induced phosphorescence emission (AIPE) or more generally solid-state luminescence enhancement (SLE),1i.e. whose emission is significantly more intense in the various solid states than in solution, have become increasingly attractive.2–5 Their good photoluminescence (PL) efficiency, ease of emission wavelength tuning, photo- and thermal stabilities, relatively long excited-state lifetime and possibility to generate active oxygen species make them promising candidates for various applications such as chemosensors,6 smart materials,7 emissive materials in optoelectronic devices,8 as well as nanoparticles for biological imaging and theranostic systems.9–11 However, their development is a delicate task because their optical behavior depends on many factors (i.e. electronic properties, geometry, packing mode, intermolecular interactions) that are difficult to disentangle. Intramolecular π–π stacking could be a powerful tool to act on all these factors. In the early part of the last decade, this phenomenon has been used to improve the emission properties of iridium complexes. Indeed, the stacking between aromatic rings of cyclometalated ligands blocks nucleophilic attacks on the complex cores, thus reducing the formation of degradation products capable of quenching luminescence,12–17 and the direct effect on the electronic levels stabilizes the complexes.17 The result is an increased lifetime of light-emitting electrochemical cells (LECs),12–18 which is observed as long as the stacking does not induce an exaggerated distortion of the complexes.19,20 Intramolecular π–π stacking has also been shown to improve the emission efficiency in solution and in polymer films.18,21 Noticeably, it is associated to solid-state luminescent and piezochromic materials.17 Recently, the encapsulation of an iridium complex with intramolecularly stackable and bulky substituents has been reported. It leads to enhanced photoluminescence both in solution and in the crystalline state.22 For rhenium(I) complexes, intramolecular π–π interactions between the bipyridine (bpy) ligand and the phenyl rings of the phosphorous ligand of fac-[Re(bpy)(CO)3(PR3)]+ (R = p-MeOPh, p-MePh, Ph, p-FPh, OPh) complexes have been shown to affect their structure, electrochemical behavior and some spectroscopic properties in solution. However, no effect on possible solid-state emission properties has been reported.23,24 Practical implementation of the aforementioned concept is therefore virtually unprecedented in the field of SLE/AIPE-active transition metal complexes, including for rhenium(I) complexes.Over the past few years, our group has developed the synthesis of tricarbonylrhenium(I) complexes that are well suited to various substitution schemes and, for most of them, exhibit remarkable solid-state emission properties.25–29 The SLE/AIPE behavior was shown to be related to the presence of an aromatically-substituted 3-(2-pyridyl)-1,2,4-triazole (pyta) ligand.25 More specifically, it is promoted by a large phenyl-pyta angle,25 a significant steric hindrance at the phenyl ring,26,28 and the presence of a chloride ancillary ligand,29 as illustrated in Fig. 1. Keeping these results in mind and prioritizing simplicity of synthesis, a new Re-BPTA complex (Fig. 1) was designed to investigate the influence of an intramolecular π–π stacking interaction on AIPE/SLE properties. The organic pyta ligand is now replaced by a symmetrical bi-(1,2,4-triazole) ligand,30i.e. an aromatic molecule with six potentially coordinating N atoms, known to exhibit various geometrical configurations and coordination modes with transition metal ions such as Mn(II), Co(II), Cu(II), Cd(II) and Zn(II).31–33 In the present case, density functional theory (DFT) calculations allowed anticipating that the incorporation of a phenyl ring at the 4 and 4′-positions of this ligand would lead to a rigid complex where the phenyl rings are close to perpendicular with respect to the bi-triazole moiety, and therefore, significantly stacked (Fig. S9†). Knowing that face-to-face π–π staking of phenyl rings is unfavorable from an energetic viewpoint,34,35 it seemed instructive to know what type of structure is actually obtained and what is the effect on the electronic properties. We report here the synthesis, and the complete theoretical and experimental study of the original complex Re-BPTA, with particular emphasis on its SLE and AIPE behavior.
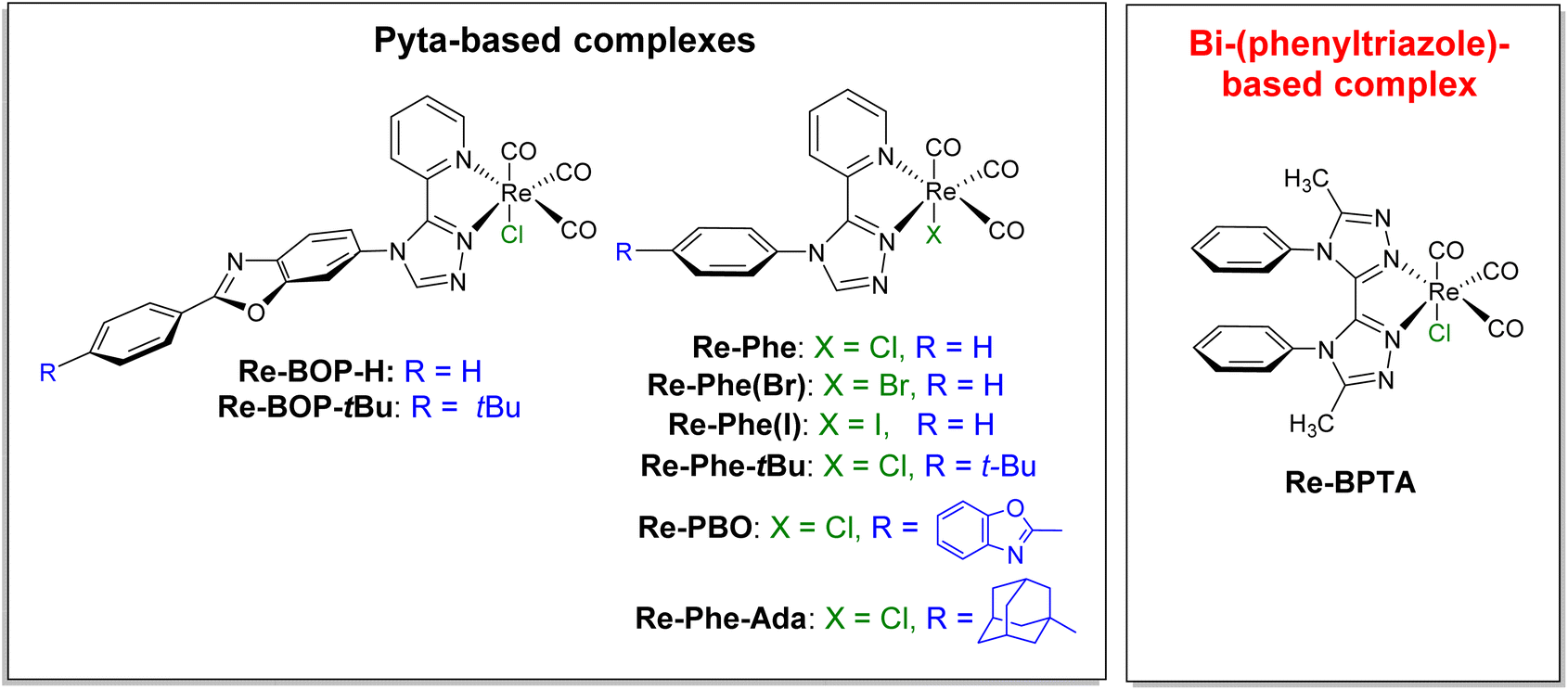 | ||
| Fig. 1 Chemical structures of the SLE-active tricarbonylrhenium(I) complexes previously studied (ref. 25–29) and of the new Re-BPTA complex. | ||
Results and discussion
Synthesis and characterization
Complex Re-BPTA was easily obtained by an efficient three-step synthesis (Scheme 1). The intermediate 5,5′-dimethyl-2,2′-bi-(1,3,4-oxadiazole) (1) was initially prepared from 5-methyltetrazole and oxalyl chloride,36 and then it was condensed on aniline in the presence of catalytic p-TsOH to obtain 5,5′-dimethyl-4,4′-diphenyl-3,3′-bi-(1,2,4-triazole) (BPTA) in 92% yield. The classical complexation conditions (slight excess of [ReCl(CO)5] in refluxing methanol) did not provide the corresponding complex. The complexation of BPTA was then performed at higher temperature in refluxing butan-2-ol. Complex Re-BPTA, which precipitates in the medium, was isolated in 73% yield. The overall yield of the synthesis was 46%.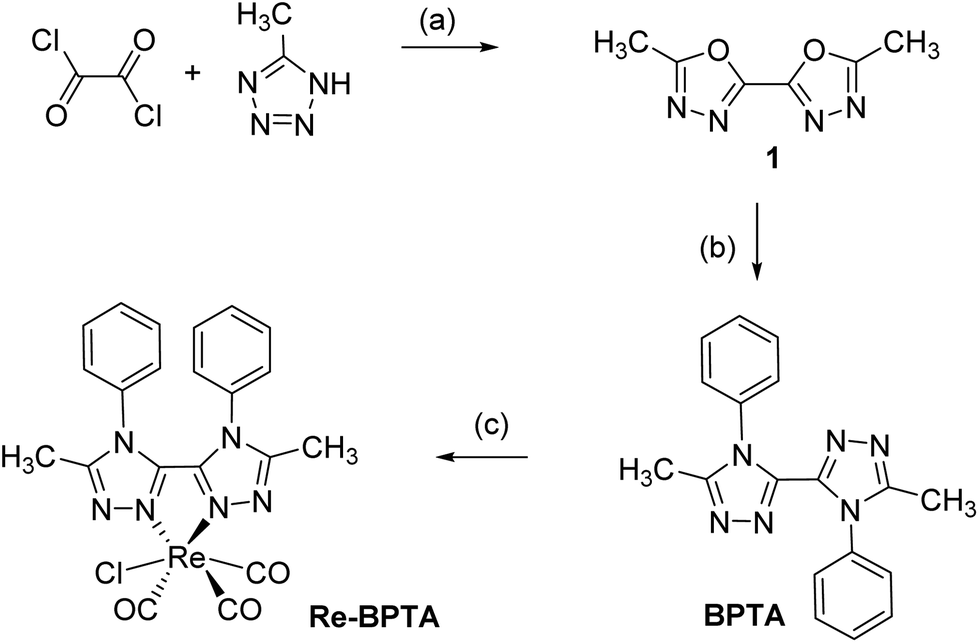 | ||
| Scheme 1 Synthesis of the tricarbonylrhenium(I) complex Re-BPTA. (a) Xylenes, N2, 2 h, 120 °C, 69%; (b) Xylenes, aniline (3.6 eq.), p-TsOH.H2O (0.3 eq.), 18 h, 140 °C, 92%; (c) [ReCl(CO)5] (1.3 eq.), butan-2-ol, 100 °C, 18 h, 73%. | ||
Complex Re-BPTA was unambiguously identified by 1H and 13C NMR (Fig. S1–S4, ESI†) and high resolution mass spectrometry (Fig. S5†). Its purity was checked by microanalysis. The Fourier transform infrared (FTIR) spectrum in dichloromethane (DCM) solutions showed stretching bands at 2017, 1914 and 1888 cm−1 as the characteristic signature of the three CO groups in a fac-[Re(CO)3] arrangement. The average value of these bands (1940 cm−1), which reflects the electron density at the metal center,37 was slightly lower than that of 1,2,4-pyta-based complexes (1947–1950 cm−1),26,28 suggesting that ligand BPTA is a slightly better electron donor than the pyta ligand.
The 1H NMR spectrum of complex Re-BPTA at room temperature (298 K) (Fig. S2,† with proton numbering available in Fig. S1, ESI†) shows that the aromatic protons H8 and H12 at ortho positions of the phenyl ring, on the one hand, and H9 and H11 at meta positions of the phenyl ring, on the other hand, are magnetically non-equivalent, meaning that the compound displays a rigid conformation on the NMR time scale. Indeed, this is confirmed by the variable temperature (VT) NMR performed in deuterated 1,1,2,2-tetrachloroethane (Fig. S6†). At 298 K, the signals of H8 and H12 look like two distinct apparent doublets of doublet at 6.85 and 6.77 ppm. When increasing the temperature, these signals progressively coalesce, and give rise to a single apparent large doublet situated at 6.86 ppm at 368 K. Similarly, the signals corresponding to H9 and H11, which appear as a split quartet at 7.19 ppm at room temperature, coalesce to give an apparent triplet centered at 7.23 ppm at 368 K. The whole phenomenon is reversible when coming back to room temperature. These observations indicate that the rotation of the phenyl groups is virtually prevented at room temperature and only allowed when heating, meaning that the intramolecular π–π interaction actively contributes to the stabilization of the complex.
Crystal structures
X-Ray quality crystals of ligand BPTA and complex Re-BPTA were grown by slow evaporation of chloroform solutions. Selected crystallographic data are collected in the experimental section and in Tables S1 and S2.† Ligand BPTA shows a symmetrical structure (Fig. 2). The bi-triazole moiety is planar, and the phenyl rings are displayed almost perpendicular to it (∼83°), at opposite sides of the molecule. In the network, the molecules exhibit a classical herringbone packing pattern, without significant overlap between the aromatic moieties (Fig. S7†). Regarding complex Re-BPTA, the coordination sphere exhibits a slightly distorted octahedral geometry (Fig. 2). The rhenium atom is coordinated to three carbonyl groups in a fac configuration, two nitrogen atoms of the organic ligand and one chlorine atom. The bi-triazole moiety is slightly twisted (dihedral angles N3–C4–C7–N6 = 23.6(8)° and N4–C7–C4–N2 = 13.5(5)°). Above all, the two phenyl rings are located on the same side of the molecule. They swing in the same direction, and their planes form an angle close to 71° and 62° with respect to the corresponding triazoles (Fig. 3). Their aromatic systems show a wide overlap. The centroid–centroid distance is 3.5 Å, indicating a strong π–π stacking interaction. By comparing the complex and the ligand structures, it is obvious that complexation is the main driving force that induces the conformational change and makes the phenyl rings closer to each other. The phenyl rings are now slipped parallel to each other to minimize energy. Indeed, among the three stacking patterns currently described for phenyl-phenyl π–π stacking interactions, two (i.e. edge-to-face (T-shaped), and offset face-to-face (parallel-displaced)) are stable in energy, while the third one, i.e. face-to-face (sandwich), is unstable and rarely observed because of the repulsion between the two stacking phenyl rings,34,35,38 including for metal complexes with aromatic nitrogen-containing ligands.39 The complex molecules are arranged as herringbone (Fig. S8a†). The network is structured by strong interactions that take place between the N1/N5 triazole nitrogen atom or the chlorine atom of one molecule, and one methyl hydrogen atom of a neighboring molecule (D ∼ 2.6 Å and 2.9 Å, respectively) (Fig. S8b and c†). Remarkably, there is virtually no overlap between the aromatic systems of two phenyl rings belonging to neighboring molecules (centroid-to-centroid distance ∼4.2 Å) (Fig. 3), and the closest superimposed triazoles are distant by about 4.7 Å (Fig. S8d†).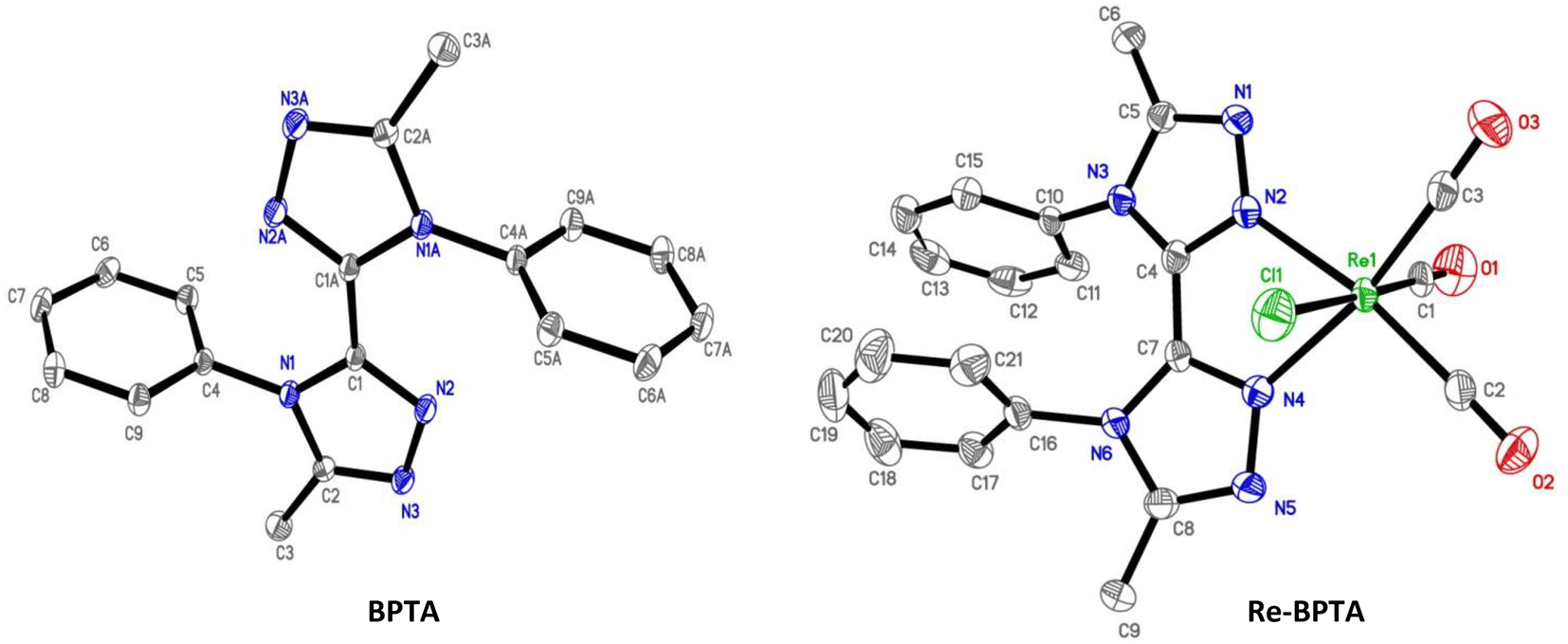 | ||
| Fig. 2 Molecular views of ligand BPTA and complex Re-BPTA. Hydrogen atoms are not represented for the sake of clarity. Displacement ellipsoids are drawn at 30% probability. | ||
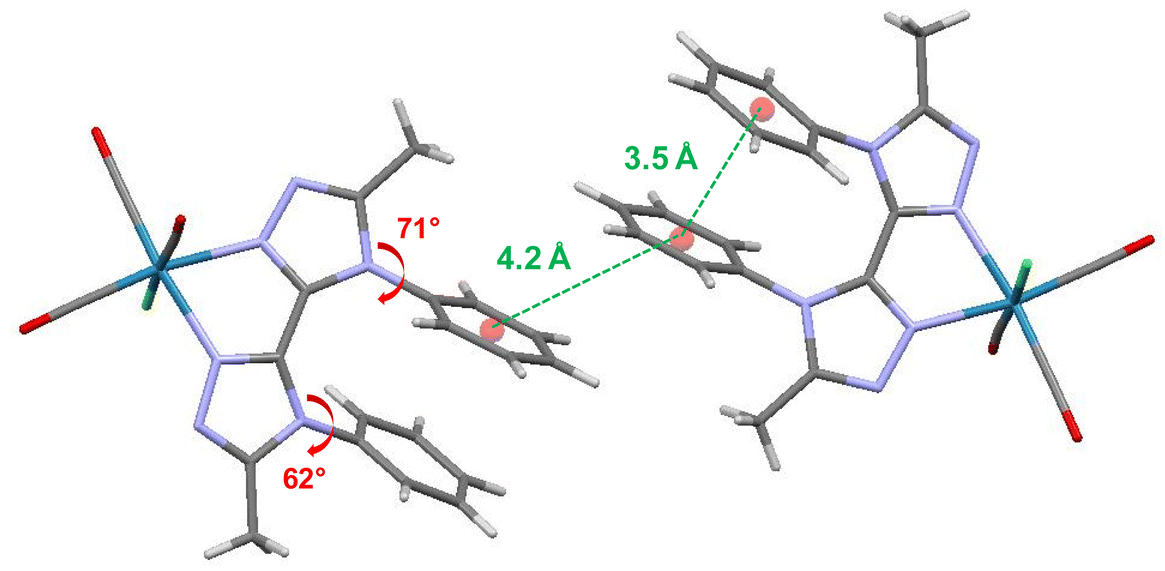 | ||
| Fig. 3 Crystallographic arrangement of complex Re-BPTA showing the centroid-to-centroid distances between phenyl rings (green ink) and the phenyl–triazole dihedral angles (red ink). | ||
Electronic properties
Computational studies were made using the density functional theory (DFT) and time-dependent DFT (TD-DFT) methods considering complex Re-BPTA in dichloromethane continuum. The composition of the frontier molecular orbitals and the main electronic transitions are described in Tables S3 and S4.† As shown in Fig. 4, the three highest occupied molecular orbitals (HOMO, HOMO − 1 and HOMO − 2) are localized on the rhenium atom, the carbonyl ligands and the chlorine atom, as is commonly the case for tricarbonylrhenium(I) complexes. Regarding the lowest unoccupied molecular orbitals, the LUMO is mainly localized on the bi-triazole moiety, and the LUMO + 1 on both phenyl rings. This feature is characteristic of the Re-BPTA complex compared to the pyta-based Re-Phe complex, where the phenyl contribution is only significant at the LUMO + 3 level (at −1.03 eV). Lastly, the LUMO + 2 is mainly localized on the carbonyl ligands, and higher orbitals (LUMO + 3 to LUMO + 5) partly on the phenyl rings. Compared to the pyta-based complexes, the HOMO and HOMO − 1 of Re-BPTA are almost at the same energy level (±0.03 eV) than the corresponding orbitals in Re-Phe and Re-PBO, and a difference of +0.2 eV is observed for the LUMO with respect to Re-Phe. Noticeably, the LUMO + 1–LUMO energy difference of +1.32 eV observed for Re-BPTA is the largest one among these three compounds.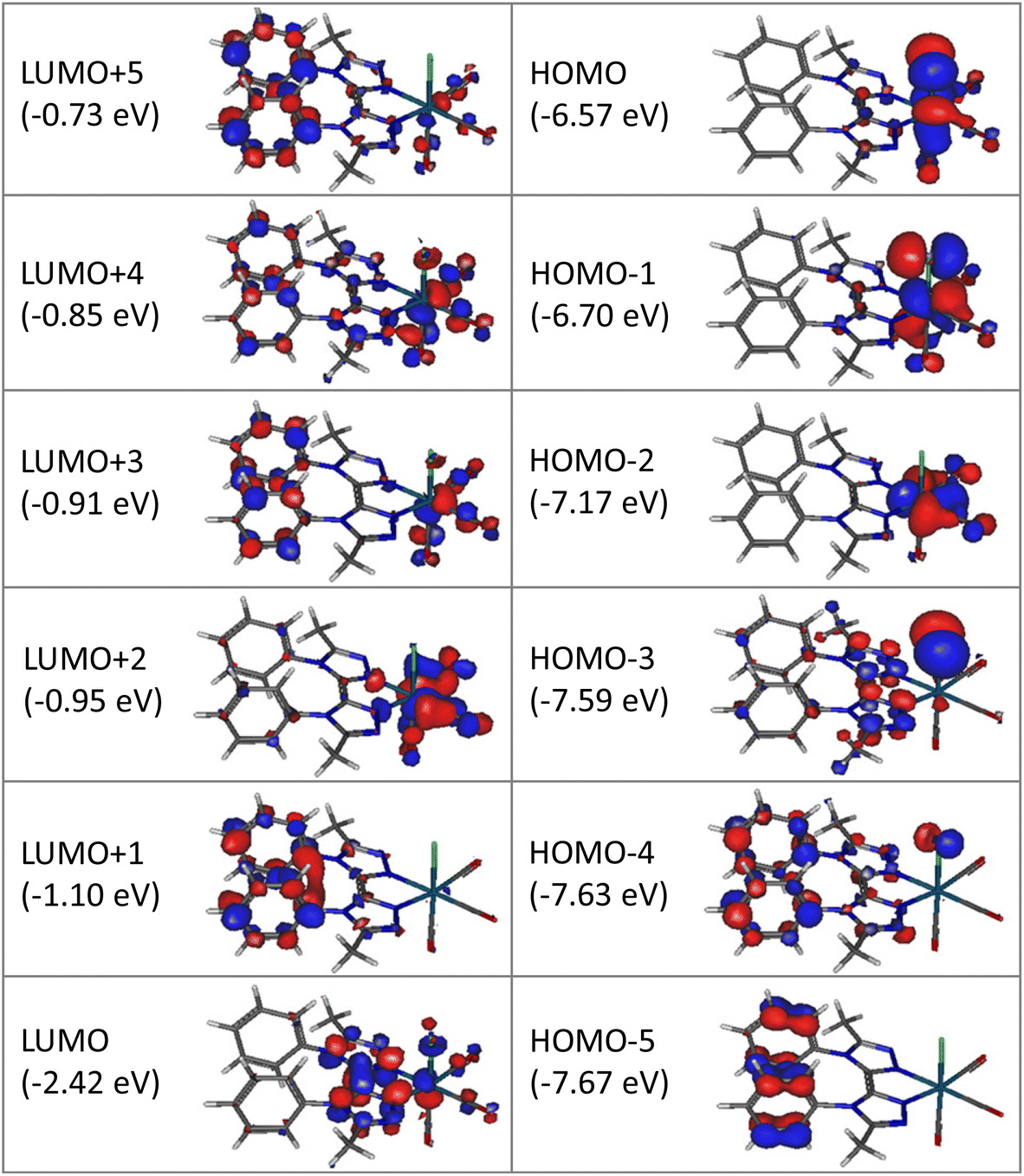 | ||
| Fig. 4 Isodensity plots (isovalue = 0.03 e bohr−3) and energy levels of the first frontier molecular orbitals, for complex Re-BPTA in dichloromethane, according to DFT calculations at the PBE0/LANL2DZ level of theory. | ||
The lowest energy transition associated with high oscillation strength is a HOMO − 1 → LUMO transition with preponderant metal-to-ligand charge transfer (MLCT) character, and a minor halide-to-ligand and ligand-to-ligand charge transfer (XLCT and LLCT, respectively) character. This transition is predicted at 392.7 nm. High energy transitions, predicted between 290.6 and 249.8 nm mainly involve low-lying HOMO orbitals (i.e. H-6, H-8 and H-9) and the LUMO, and they have mixed MLCT and LLCT character.
Electrochemical properties
The electrochemical behavior of the BPTA ligand and Re-BPTA complex was studied by cyclic voltammetry (CV) and Osteryoung square wave voltammetry (OSWV) measurements in DCM at room temperature (Fig. 5 and Fig. S12–S17†). In the OSWV anodic part, the first oxidation potential of the complex situated at 1.37 V can be mainly assigned to an irreversible Re(I)/Re(II) oxidation process.40,41 This oxidation potential is around 90 mV lower than that of the Re-Phe complex and its derivatives, thus indicating a better electron donating capability of the new species. In contrast, in the cathodic region, the first reduction potential observed at −1.51 V is around 0.2 V more negative than that of the first characteristic reduction potential of the 3-(2-pyridyl)-1,2,4-triazole complexes generally detected at ≈−1.3 V (Table S6†). This reduction is ligand-centered and probably first occurs on both triazole moieties. A second reduction process of minor intensity is also observed as a shoulder at −1.66 V. Regarding the CV experiments, the latter two processes correspond to a complex and broad reduction wave of the whole ligand (Fig. 5). Contrary to our previous pyta derivatives, no quasi-reversible reduction process was detected, even by thorough examination at different reduction potentials and different scan rates. In addition, the intensity of this whole reduction process is not in a 1/1 ratio when compared to the first one-oxidation process, as only observed for Re-Phe(I). Furthermore, the first reduction potential of Re-BPTA is close to that of dichloromethane presenting a first reduction process of very weak intensity at −1.60 V under the same conditions, which limits the electrochemical analysis. A particular electrochemical behavior, such as a decomposition process or reaction with the solvent could be envisaged, thus influencing the above observations related to the broad reduction wave in CV. However, careful examination of the reduction wave after accumulation of several successive scans under different conditions does not really support this hypothesis since no significant changes were detected. All of these observations in the cathodic region are probably mainly related to the different intrinsic nature of the LUMO–LUMO + 3 frontier molecular orbitals of the Re-BPTA compound (see above) leading to this peculiar electrochemical signature. Coordination of the BPTA ligand induces a substantial decrease of the value of its reduction potential, as usually observed for the substituted triazole ring.42,43 The resulting new bi-(phenyltriazole) complex formed is more prone to oxidation and more difficult to reduce than the pyta counterpart Re-Phe. However, the value of its electrochemical gap (2.70 eV) is very close to that of Re-Phe (2.65 eV) (Table S7†) and its derivatives.28 Finally, these electrochemical data are in good agreement with the theoretical study and the UV-visible absorption data.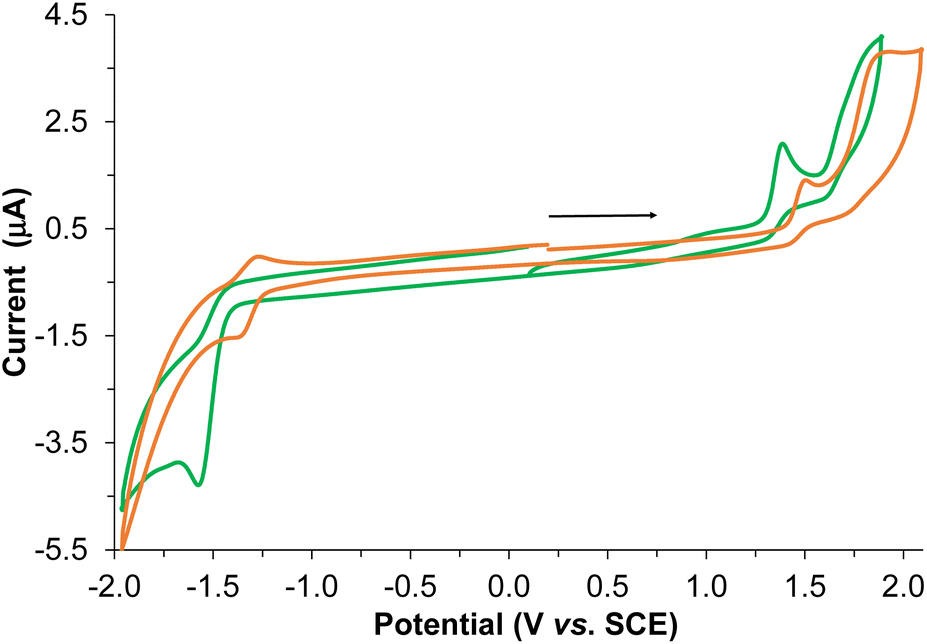 | ||
| Fig. 5 Cyclic voltammograms of Re-BPTA (green line) and Re-Phe (orange line) on a Pt working electrode in CH2Cl2 + 0.1 M n[Bu4N][BF4] at room temperature and at a scan rate of 0.2 V s−1 toward anodic potentials. | ||
UV-visible absorption and emission properties
The optical properties were studied on freshly prepared compound. In fact, it was noticed that ageing of the samples for several months leads to a significant decrease of the emission efficiency, especially for solid-state samples, where energy transfers are particularly strong,44 making them very sensitive to the presence of quenching impurities. The UV-visible absorption spectra of complex Re-BPTA were recorded in methanol, acetonitrile and DCM (Fig. 6 and Table 1). They showed intense bands below 300 nm and a moderately intense band at long wavelengths, the maximum of which varies significantly between 340 and 358 nm. The experimental spectrum in DCM was situated at higher energy than that predicted by the calculation (Fig. S10†). Such a discrepancy ∼0.30 eV, at the limit of what is acceptable when using TD-DFT,45 is attributable to intramolecular motions that are not considered by calculations.| Complex | Solvent | λ abs [nm] | ε [M−1 cm−1] | λ P [nm] | Φ P | Φ P | τ [ns] | χ 2 | k r [s−1] | k nr [s−1] |
|---|---|---|---|---|---|---|---|---|---|---|
| When not specified, the medium was air-equilibrated.a Argon-bubbled medium.b λex = 360 nm for steady-state emission and λex = 371 nm for decay measurements.c λem = 600 nm.d λem = 570 nm.e From ref. 28.f From ref. 26. | ||||||||||
Re-BPTA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
Methanol | 238 | 11700 |
590 | 0.018 | 0.029 | 71c | 1.20 | 2.5 × 105 | 1.4 × 107 |
| 264 | 12900 |
|||||||||
| 342 | 5900 | |||||||||
| Acetonitrile | 236 | 12700 |
586 | 0.024 | 0.078 | 97c | 1.06 | 2.5 × 105 | 1.0 × 107 | |
| 262 | 11700 |
|||||||||
| 342 | 4600 | |||||||||
| Dichloromethane | 242 | 13800 |
592 | 0.042 | 0.080 | 154d | 1.06 | 2.7 × 105 | 6.2 × 106 | |
| 264 | 13200 |
|||||||||
| 358 | 5700 | |||||||||
|
Re-Phee |
Dichloromethane | 240 | 15100 |
626 | 0.020 | 0.029 | 74.7 | 1.29 | 2.7 × 105 | 1.3 × 107 |
| 282 | 11600 |
|||||||||
| 382 | 4000 | |||||||||
|
Re-PBOf |
Dichloromethane | 310 | 29600 |
632 | 0.012 | 0.015 | 80 | 1.19 | 1.5 × 105 | 1.2 × 107 |
| 388 | 5100 | |||||||||
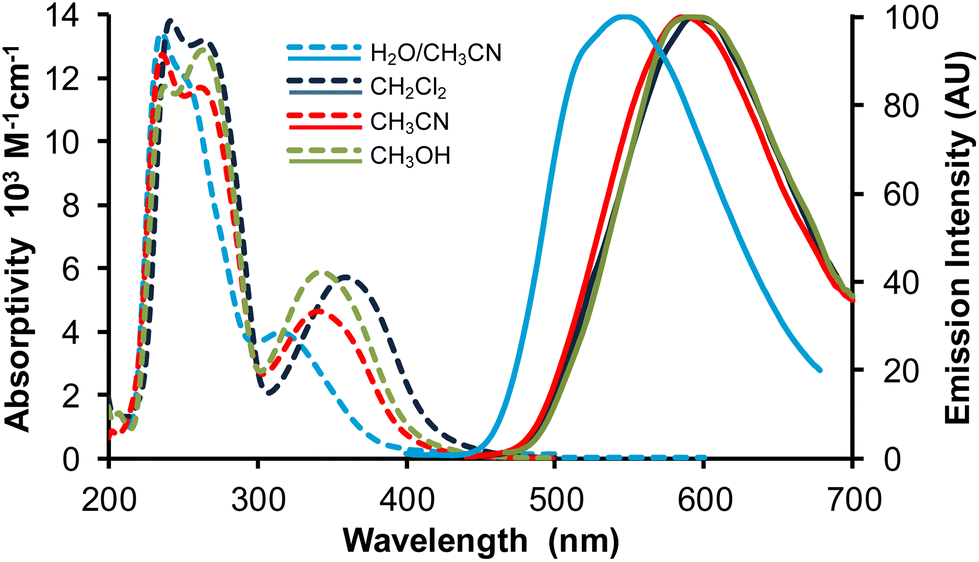 | ||
| Fig. 6 Absorption (broken lines) and normalized emission spectra (full lines) of complex Re-BPTA in three organic solvents and in the water/acetonitrile 90:10 v/v mixture. λex = 360 nm. | ||
The emission spectra of Re-BPTA in the three organic solvents were quite close. They displayed only one unresolved band, with a maximum between 586 and 592 nm. The emission quantum yields were low in every case, especially when the polarity and proticity of the solvent increase. A striking difference was found between air-equilibrated and deaerated solutions. After bubbling with argon, the quantum yields were increased by a factor 1.6, 1.9 and 3.2 in methanol, dichloromethane and acetonitrile, respectively, in the same order than the amount of oxygen dissolved by the solvent,46,47 and this effect was perfectly reversible. The luminescence decays measured in solutions were monoexponential, varying from 71 to 154 ns, which confirms that the emission detected is mainly phosphorescence. A further argument in favor of this attribution is that the experimental emission maximum in DCM is in rather good agreement with that calculated for luminescence arising from the lowest excited triplet state (614 nm). It must be noticed that our theoretical calculations consider that the lowest energy transition is a LUMO → HOMO, but a LUMO → HOMO − 1 transition of very close energy is also very likely to occur. The molecule in its lowest MLCT triplet excited state is shown in Fig. 7 and Fig. S11.† The spin density distribution is almost exclusively localized on the coordination sphere, as is also the case for pyta-based complexes. This suggests that, while the phenyl rings contribute to the overall rigidity of complex Re-BPTA, they are not directly involved in the emissive phosphorescence process.
 | ||
| Fig. 7 Spin density distribution for the lowest MLCT triplet excited state of Re-BPTA (isovalue = 0.03 e bohr−3) according to DFT calculations at the PBE0/LANL2DZ level of theory. | ||
At first glance, the spectroscopic features of Re-PBO, Re-Phe and Re-BPTA in DCM are relatively close (Table 1). All of them absorb in the UV range and emit orange-red light. The shorter absorption and emission wavelengths of Re-BPTA can be mainly attributed to the change in the chelating unit. However, a closer examination reveals that Re-BPTA exhibits better luminescence efficiency and a significantly longer lifetime than the pyta-based complexes. The calculation of the photophysical rate constants shows that the non-radiative constant knr of Re-BPTA is divided by two with respect to the pyta complexes (Table 1). An explanation could be that, in the rigid coordination sphere of Re-BPTA, vibrations and distortions responsible for the waste of excitation energy are limited, making the emitting 3MLCT state more stable than for the pyta-based complexes. The other side of the coin is that complex Re-BPTA is more sensitive to the effect of oxygen because of its longer lifetime.
The solid-state spectroscopic data are gathered in Table 2 and Table S8.† Two microcrystalline samples of complex Re-BPTA were investigated. One is the powder obtained by precipitation in butan-2-ol. The second one results from recrystallization in chloroform, and corresponds to the crystals analyzed by XRD. Both powders emitted bright greenish-yellow light when illuminated by a hand-held UV lamp (365 nm) (an example is given in Fig. 8a, inset). Their excitation spectra revealed the presence of a strong band around 374 nm, and a weaker one around 470 nm, tailing till 500 nm. It was checked that the powders were emissive when excited in the blue region of the visible spectrum. The emission spectrum of the powder obtained from butan-2-ol showed one unresolved band peaking at 548 nm (Fig. 8a). The photoluminescence quantum yield (PLQY) was 0.52. For the compound recrystallized from chloroform, the emission spectrum was shifted to 554 nm, with a slightly different shape (Fig. 8a), and the PLQY was only 0.27. Such an influence of the crystallization process on the PL properties has already been noticed for Re-Phe(I).29 In fact, the PL properties are highly dependent on molecular geometry, intermolecular interactions and crystal defects.1 Reabsorption phenomena related to the crystal size can also strongly alter the shape and position of the PL spectra.48 These parameters may vary more or less depending on the crystallization method. For both samples, a very strong SLE effect was observed with respect to solutions.
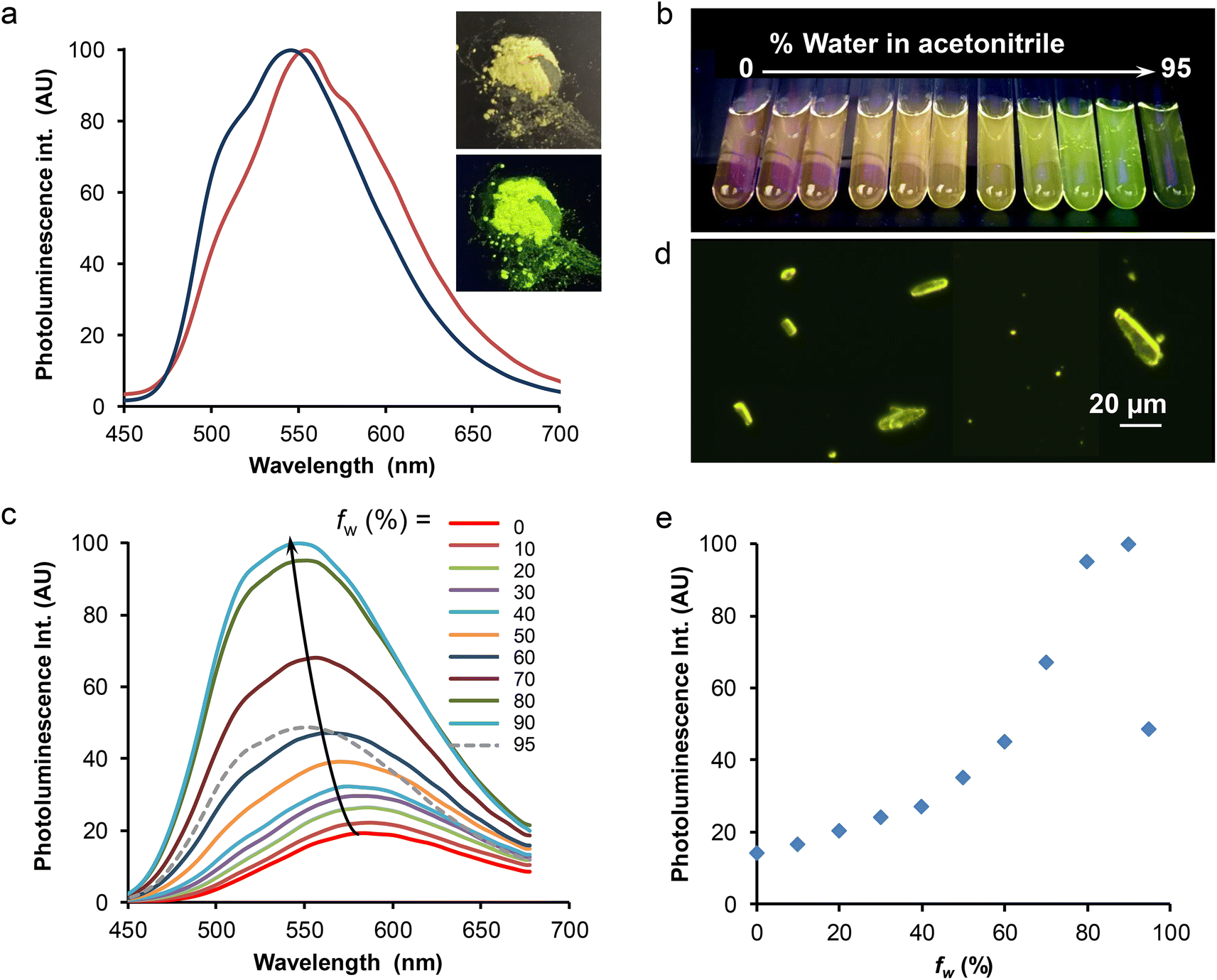 | ||
| Fig. 8 (a) Normalized PL spectra of Re-BPTA as powders obtained from butan-2-ol (blue line) and chloroform (red line), λex = 380 nm. Inset: powder obtained from butan-2-ol observed in daylight (top) and under illumination at 365 nm (bottom). (b) Samples of Re-BPTA at 3.3 × 10−5 M in acetonitrile solutions containing from 0 to 95% water (from left to right), illuminated at 365 nm. (c) Corresponding emission spectra, λex = 350 nm. (d) Suspension with fw = 80% observed under the fluorescence microscope. (e) Evolution of the PL intensity at 546 nm as a function of the water/acetonitrile ratio. All samples for AIPE measurements were observed 2 h after preparation. | ||
:10 v/v mixture (AIPE experiment), and after dispersion of the powder in water by sonication. Comparison with the pristine microcrystalline powders of pyta-based complexes Re-Phe, Re-Phe-Ada and Re-PBO. Photoluminescence emission maximum (λPL) and quantum yield (ΦPL), lifetime (τ) with fractions of intensity (fi), weighted average lifetimes 〈τ〉, and chi square (χ2) values
| Compound | λ PL [nm]a |
Φ
PLa |
τ PL [ns] | 〈τPL〉 [ns] | χ 2 | |
|---|---|---|---|---|---|---|
| a λ ex ∼ 382 nm. b This work. λex = 370 nm and λem = 550 nm. c Air-equilibrated. d From ref. 28. e From ref. 27. | ||||||
| Re-BPTA | Microcrystalline powder | 546 | 0.52 | 60 (0.02), 730 (0.97)b | 713 | 1.11 |
| Suspension in water/acetonitrile 90:10 v/vc |
548 | 0.09 | 58 (0.05), 395 (0.95)b | 377 | 1.09 | |
| Dispersion in waterc | 538 | — | 49 (0.01), 799 (0.99)b | 791 | 1.05 | |
| Re-Phe | Microcrystalline powder | 550d | 0.42d | 60 (0.02), 639 (0.98)b | 624 | 1.07 |
| Re-Phe-Ada | Microcrystalline powder | 550d | 0.59d | 60 (0.02), 646 (0.97)b | 631 | 1.07 |
|
Re-PBOe |
Microcrystalline powder | 542 | 0.55 | 93 (0.12), 563 (0.85) | 490 | — |
The PL decay was measured on the powder precipitated from butan-2-ol. It was found to be multiexponential, although it essentially consists of one long lifetime with a fraction of intensity of 97%. Minor contributions associated to short lifetimes probably arise from molecules located in different microenvironments. The mean lifetime was around 713 ns, drastically longer than for solutions. The spectroscopic differences, i.e. marked blue shift, increased PLQY and longer lifetime, observed for the microcrystalline powders with respect to solutions, stem from environmental effects on the molecules. On the one hand, intermolecular interactions that take place in the solid state influence the energy of the ground and excited states. On the other hand, intramolecular motions are much more limited in crystals than in solution, which prevents vibrational deactivation of the excited states. The crystal packing mode, which does not show any detrimental intermolecular interaction, favors efficient light emission. Besides, the access of oxygen is limited in the crystal phase. All these parameters contribute to the good solid-state emission of Re-BPTA.
Let us now compare the performance of Re-BPTA (precipitated from butan-2-ol) with that of the pyta-based complexes as microcrystalline powders. The PLQY of Re-BPTA is slightly below that of the most emissive complexes, i.e.Re-Phe-Ada and Re-PBO, and much better than that of Re-Phe, whose structure is the most closely related. Its lifetime is also much longer than that of all pyta-based complexes investigated so far. The lowest triplet excited state, responsible for the phosphorescence of Re-BPTA, therefore seems particularly well stabilized in the crystalline phase. Probably, the main reason is that the overall geometry of the molecules does not vary much between the ground state and the emitting triplet state, according to the results of theoretical calculations (Fig. S9 and Table S5†). Besides, these calculated geometries are close to that of the molecule in the crystal, experimentally determined by XRD analysis (Fig. 3 and Table S2†). When the molecules reach the 3MLCT state, both phenyl rings undergo a little rotation in the same direction and the dihedral angle that they form with the triazole unit is reduced by about 27°. But, the π–π stacking interaction appears to be maintained with a distance of 3.82 Å between the centroids of the both phenyl rings. Therefore, in the excited state, even though they are not directly involved in the emission process, the two phenyl rings help to minimize the molecular deformations in the coordination sphere, thus allowing the emissive part of the molecule to be effective.
Subsequently, mechanoresponsive luminescence (MRL) properties were investigated (Table S8†). For the powder crystallized from butan-2-ol, the emission wavelength was only red-shifted by 2 nm upon grinding the pristine powder with a pestle and a mortar, and the quantum yield was decreased (0.38). The spectral characteristics of the pristine powder were fully recovered after fuming with tetrahydrofuran for 48 h. The sample crystallized in chloroform did not show any particular spectral change upon grinding. The insignificant MRL behavior of Re-BPTA is therefore very different from that of the pyta-based complexes investigated so far, and particularly Re-PBO whose emission spectrum is dramatically red-shifted by 58 nm after mechanical stimulus.27 This could be explained by the stiffness of Re-BPTA: the relaxation of the packing constraints due to a more or less advanced amorphization does not lead to major geometrical changes in the molecule.
As the comparison between the solution and solid-state emission quantum yields shows a clear solid-state luminescence enhancement (SLE) effect, the effect of aggregation in aqueous medium was also investigated. The popular procedure that consists in increasing the water proportion in an organic solution of the complex was used. Fig. 8b–e illustrates this effect starting from pure acetonitrile, which was chosen for solubility reasons although the complex is more emissive in this solvent than in methanol. The photoluminescence intensity was increased steadily when the water proportion in acetonitrile passed from 0 to 90%, and then the intensity abruptly decreased for 95% water. This intensity change was accompanied by a significant (∼40 nm) blue shift of the spectrum, which peaked at around 546–550 nm, much closer to the microcrystalline powder than to solutions (Fig. 6). The mean emission lifetime of the sample containing 90% water was 377 ns, almost four times longer than in acetonitrile. It is characteristic of molecules in a solid phase. Besides, the observation of the suspensions with the fluorescence microscope showed the presence of tiny microcrystals that strongly emit in the yellow (Fig. 8d), and a diffuse background of small, weakly emissive particles. Microcrystals seemed to be fewer in the sample that contains the largest amount of water, where amorphous particles may form very quickly, which could explain the weaker PL signal. A slight increase of the emission intensity of the water-containing samples, and an evolution of their respective intensities, were also observed after 24 h, probably related to slow growth of the particles (Fig. S18†). At 546 nm, the intensity of the PL signal was increased more than seven times after 2 h, and ten times after 24 h. However, the AIPE experiment does not reflect the strong difference observed between the PLQY of acetonitrile solution and microcrystalline powders of Re-BPTA. The comparison between air-equilibrated and argon-bubbled samples showed that the effect of dissolved oxygen does not explain this discrepancy: for the sample containing 90% water, the PLQY only varied from 0.09 to 0.11. It can then be imagined that the microcrystals formed are sensitive to quenching by water. To test this hypothesis, a small amount of microcrystalline Re-BPTA was dispersed by sonication (5 min) in water, where it is insoluble. The sample was strongly emissive under the UV lamp, the shape of the emission spectrum and the long lifetime (i.e. 791 ns) resembled those of the native powder, showing that the aqueous surrounding has a weak effect on microcrystal emission. The comparison with the suspension in H2O/CH3CN 90:10 v/v (Fig. S19†) confirms that molecules in the solid state are the emitting species in both samples, but their nature is probably different. The lower quantum yield and shorter lifetime of the suspensions formed during the AIPE experiment could be attributed to the predominant formation of a solid, i.e. amorphous aggregates, hydrates, or very small crystals containing a lot of surface defects, much less emissive than the microcrystals prepared from an organic solution. Finally, it must be added that a phosphorescence decrease was observed after several days for the water-containing samples, suggesting a slow degradation of Re-BPTA in these conditions, but this phenomenon was not noticeable given the duration of the AIPE experiment.
Conclusions
Although this approach has been known for a long time, until now limited efforts have been devoted to the design and synthesis of transition metal complexes with intramolecular π–π stacking for solid-state luminescence enhancement. The fact that π–π stacking intramolecular interactions improve the molecular rigidity, and thus lead to increased PL, has already been reported in the literature e.g. for heteroleptic mono- and di-nuclear Cu(II) complexes49 and for Ir(I) complexes with very elaborated structures.22 In the present work, the intramolecular interaction does not require very bulky ligands12–18,21,22 or polyphenyl units,23,24 specially designed to form a protecting cage around the metal ion. Instead, we designed a minimalist ligand. A symmetrical bi-(1,2,4-triazole) unit (less popular than the bi-(1,2,3-triazole) unit that is easily obtained by click chemistry50–52), provided with two appended phenyl rings, was efficiently prepared. The resulting ligand, i.e.BPTA, exhibits excellent chelating properties and leads to complex Re-BPTA, the rigidity of which is reinforced by the π–π stacking interaction between the phenyl rings. One consequence of the molecular stiffness is that, for molecules in solution, the emissive 3MLCT state is stabilized with respect to our previously studied pyta-based complexes in which the phenyl ring rotates more freely. The phosphorescence quantum yield and lifetime are thus improved, but this effect is partly counterbalanced by an increased sensitivity to dissolved oxygen. Consequently, the overall emission efficiency remains weak. In the microcrystalline phase, Re-BPTA shows high PLQY and competes well with the most emissive pyta-based complexes. The particularly long emission lifetime reflects the good stability of the 3MLCT state. In addition, evidence was given for clear AIPE properties, most of the emission enhancement being due to the formation of microcrystals.To the best of our knowledge, our work presents the first example of Re(I) complex exploiting the concept of intramolecular π–π stacking for solid state luminescent enhancement. Moreover, this simple Re-BPTA complex can be seen as a new starting block for the preparation of valuable families of SLE-active photoluminescent materials. Indeed, preliminary experiments have shown that the Re-BPTA complex can be substituted with a linear alkyl chain in the para-position of each phenyl rings, while retaining almost the same optical properties. The advantage of this symmetrical molecule is that it can easily carry a double recognition unit, thus increasing its affinity towards a given target, potentially making the substituted complex a better candidate than the parent compound for AIPE-based biological applications. More generally, substitution on the phenyl rings with different groups could also allow the molecular framework to be modified. All these possibilities offer the means to better understand the role of intramolecular π–π stacking interactions on the SLE properties of Re(I) complexes, and to modulate them through an original approach.
Experimental section
General methods
All purchased chemicals were of the highest purity commercially available and used without further purification. Unless otherwise noted, all experiments were carried out under a nitrogen atmosphere. All reactions were monitored by TLC on silica gel Alugram® Xtra SIL G/UV254. Column chromatography was performed on Machery–Nagel silica gel. NMR spectra were recorded with Bruker Avance 300, 400 and 500 instruments. Chemical shifts are given in ppm and are referenced by using the residual signals of the solvent as internal standard. Signals are described as follow: s, singlet; d, doublet; t, triplet; m, multiplet. HRMS data were recorded on a Xero G2 QTOF (Waters) instrument. Infrared spectra were obtained on a Nexus Thermonicolet apparatus with DTGS as the detector. The microanalysis was made in the “Service d'analyse” of LCC using a PerkinElmer 2400 series II analyzer.5,5′-Dimethyl-2,2′-bi(1,3,4-oxadiazole) (1)
Oxalyl chloride (200 μL, 2.33 mmol) was slowly introduced into a solution of 5-methyltetrazole (567 mg, 6.74 mmol) in anhydrous xylenes (10 mL) under nitrogen atmosphere. The mixture was heated for 2 h at 120 °C. The solvent was removed by evaporation under vacuum and the resulting solid was purified on a silica gel column chromatography (DCM/MeOH from 100:0 to 98:2 v/v) to yield compound 1 as a white solid (267 mg, 69%).36
1H NMR (300 MHz, CDCl3): δ (ppm) = 2.71 (s, 6H, H6). 13C NMR (75 MHz, CDCl3): δ (ppm) = 165.9 (C2), 153.5 (C5), 11.2 (C6).
5,5′-Dimethyl-4,4′-diphenyl-4H,4′H-3,3′-bi(1,2,4-triazole) (BPTA)
A mixture of compound 1 (148 mg, 0.9 mmol), p-toluenesulfonic acid hydrate (51 mg, 0.27 mmol) and aniline (0.3 mL, 3.2 mmol) in xylenes (8 mL) was heated at 140 °C under nitrogen atmosphere overnight. After cooling to room temperature, the solvent was removed under vacuum and the residue was purified on a silica gel column chromatography (DCM/MeOH 97:3) to yield compound BPTA as a white solid (261 mg, 92%).
1H NMR (300 MHz, CDCl3): δ (ppm) = 7.56–7.34 (m, 6H, H9,10,11), 7.14–6.99 (m, 4H, H8,12), 2.28 (s, 6H, H6). 13C NMR (75 MHz, CDCl3): δ (ppm) = 153.1 (C2), 143.6 (C5), 133.6 (C7), 129.91 (C10), 129. 86 (C9,11), 127.0 (C8,12), 11.3 (C6). HRMS (ESI+): m/z = 317.1515 ([M + H]+ calcd for C18H17N6: 317.1506).
1H NMR (500 MHz, CDCl3): δ (ppm) = 7.35 (tt, J = 7.6, 1.1 Hz, 2H, H10), 7.20 (m, 4H, H9,11), 6.90 (d, J = 7.9 Hz, 2H, H8), 6.86 (d, J = 8.0 Hz, 2H, H12), 2.20 (s, 6H, H6). 13C NMR (125 MHz, CDCl3): δ (ppm) = 196.3 (COax), 188.1 (COeq), 156.2 (C2), 147.2 (C5), 132.3 (C7), 131.2 (C10), 130.9 (C11), 130.6 (C9), 127.0 (C12), 126.4 (C8), 11.2 (C6). HRMS (ESI+): m/z = 585.0820 ([M − Cl]+ calcd for C21H16N6O3185Re: 585.0813). IR (ATR): ν(CO) = 2017, 1914, 1888 cm−1. Anal. Calcd (%) for C21H16N6O3ReCl: C 40.55, H 2.59, N, 13.51; found C 40.04, H 2.24, N, 13.28.
Crystallography
Crystal data were collected at 193K using MoKα radiation (wavelength = 0.71073 Å) on a Bruker AXS D8-Venture diffractometer equipped with a Mo Kα sealed tube (λ = 0.71073 Å), a multilayer TRIUMPH X-ray mirror, a Photon III-C14 detector and an Oxford Instruments Cryostream 700+ Series low-temperature device. Phi- and omega-scans were used. The space group was determined on the basis of systematic absences and intensity statistics. Semi-empirical absorption correction was employed.53 The structures were solved using an intrinsic phasing method (ShelXT).54 All non-hydrogen atoms were refined anisotropically using the least-square method on F2.55 Hydrogen atoms were refined isotropically at calculated positions using a riding model with their isotropic displacement parameters constrained to be equal to 1.5 times the equivalent isotropic displacement parameters of their pivot atoms for terminal sp3 carbon and 1.2 times for all other carbon atoms. CCDC 2212965 (BPTA) and CCDC 2212966 (Re-BPTA)† contain the supplementary crystallographic data for this paper. Selected crystallographic data are collected in Table 3.| BPTA | Re-BPTA | |
|---|---|---|
| Empirical formula | C18 H16 N6 | C21H16N6O3ClRe |
| Formula weight | 316.37 | 622.05 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c |
| Unit cell dimensions | ||
| a (Å) | 9.5185(5) | 8.3018(5) |
| b (Å) | 7.0893(4) | 22.7231(10) |
| c (Å) | 11.7987(6) | 11.6873(7) |
| α (°) | 90 | 90 |
| β (°) | 98.989(2) | 99.998(2) |
| γ (°) | 90 | 90 |
| Volume (Å3) | 786.39(7) | 2171.2(2) |
| Z | 2 | 4 |
| Density (calculated) (Mg m−3) | 1.336 | 1.903 |
| Crystal size (mm3) | 0.300 × 0.150 × 0.100 | 0.200 × 0.080 × 0.060 |
| Reflections collected | 32207 |
69475 |
| Independent reflections | 2389 [R(int) = 0.0310] | 5397 [R(int) = 0.0637] |
| Restraints/parameters | 0/110 | 0/291 |
| Final R1 index I > 2σ (I) | 0.0476 | 0.0306 |
| wR2 (all data) | 0.1303 | 0.0648 |
| Largest diff. peak and hole (e Å−3) | 0.409 and −0.211 | 1.768 and −1.581 |
| CCDC | 2212965† | 2212966† |
Spectroscopy
Spectroscopic measurements in solutions were conducted at 20 °C in a temperature-controlled cell. UV-visible absorption spectra and emission spectra in solutions were measured with a Xenius SAFAS spectrofluorometer using cells of 1 cm optical pathway. All emission spectra were corrected. The emission quantum yields in solution (Φ) were determined using the classical formula:| Φx = (As × Ix × nx2 × Φs)/(Ax × Is × ns2) | (1) |
For AIPE measurements, a small volume (30 μL) of a concentrated solution of Re-BPTA in acetonitrile was injected in 2.97 mL of various acetonitrile/water mixtures. The samples were left to stand under stirring in the dark, and then they were sonicated for 5 min before optical measurement, so that they were as homogeneous as possible. Absorbance variations due to scattering by microparticles, and in particular the baseline deviation, were taken into account for measuring the extinction coefficient value, the fluorescence quantum yields of the suspension in the water/acetonitrile 90:10 v/v mixture, and the magnitude of the AIPE effect.
Solid state spectra were recorded on the same Xenius SAFAS spectrofluorometer using an integrating sphere. They were corrected using a home-made correction curve. Solid samples were deposited on a metal support. The absolute photoluminescence quantum yield values (ΦP) were determined by a method based on the one developed by De Mello et al.,57 as described elsewhere.28 The error was estimated to be about 20%.
The emission decay curves were recorded using the time-correlated single-photon counting method (TCSPC) on a HORIBA Fluorolog 3-2(iHR320) spectrofluorometer equipped with a nanoled-370 (λex = 371 nm). The absorbance of solutions and suspensions at λex was lower than 0.1. The solid state sample was deposited on a quartz holder. Photons were detected at 90° through a monochromator by means of a Hamamatsu R928 photomultiplier. Emission was recorded near the maximum with a bandpass of 10–15 nm. The instrumental response was recorded at 371 nm. All analyses were recorded using the Datastation v2.7 software. The decay curves (Fig. S20–S27†) were analyzed with reconvolution and global non-linear least-squares minimization method using DAS6 v6.8 software.
The rate constants for radiative (kr) and nonradiative (knr) deactivations were calculated using the following equation:
| kr = Φ/τ and knr = (1 − Φ)/τ | (2) |
Fluorescence microscopy was performed with a Leitz Laborlux D fluorescence microscope equipped with an Andor Luca camera (λex ∼ 450–490 nm, λem > 500 nm).
Computational details
The ORCA software was employed for all calculations (the geometry optimization, the ground-state and excited-state electronic structures, and optical spectra) with the aid of the Gabedit visualization program.58 Density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations were performed with the PBE0 functional.59 The ground state (S0) and the lowest triplet state (T1) geometries were fully optimized with the DFT method using the Perdew–Burke–Ernzerhof PBE0 functional without symmetry constraints.60 The “double-ζ” quality basis set LANL2DZ with Hay and Wadt's relative effective core potential ECP (outer-core [(5s25p6)] electrons and the (5d6) valence electrons)61,62 was employed for the Re+ cation. The solvent effect (DCM, ε = 9.08) was simulated using the Conductor-like Polarizable Continuum Model (CPCM).63,64 The vibrational frequency calculations were performed using the optimized structural parameters of the complex, to confirm that the optimized structure represents a local minimum on the potential energy surface. On the basis of the optimized ground state geometry, the absorption properties were calculated by the TD-DFT method at the PBE0/LANL2DZ level. The emission has been calculated by DFT considering the difference of energy between the optimized triplet state and the singlet state at the same geometry. Orbital compositions have been calculated using Multifwn software65 with the orbital composition analysis (Mulliken partition) function.66Electrochemistry
The electrochemical properties of BPTA and Re-BPTA were determined by cyclic voltammetry (CV) and Osteryoung square wave voltammetry (OSWV) in DCM. The solutions used during the electrochemical studies were typically 1 × 10−3 M in complex, and 0.1 M in supporting electrolyte. The supporting electrolyte (nBu4N)(BF4) (Fluka, 99% electrochemical grade) was used as received and simply degassed under Ar. DCM was dried using an MB SPS-800 solvent purification system just prior to use. The measurements were carried out with an Autolab PGSTAT100 potentiostat controlled by GPES 4.09 software. Experiments were performed at room temperature (r.t.) in a homemade airtight three-electrode cell connected to a vacuum/Ar line. The reference electrode consisted of a saturated calomel electrode (SCE) separated from the solution by a bridge compartment. The counter electrode was a Pt wire of ca. 1 cm2 apparent surface. The working electrode was a Pt microdisk (0.5 mm diameter). Before each measurement, the solutions were degassed by bubbling Ar and the working electrode was polished with a polishing machine (Presi P230). Under these experimental conditions, Fc+/Fc is observed at +0.55 ± 0.01 V vs. SCE. OSWVs were obtained using an amplitude of 20 mV, a frequency of 20 Hz, and a step potential of 5 mV.Conflicts of interest
There are no conflicts to declare.Acknowledgements
ANR is gratefully acknowledged for funding (PANTHERA project # ANR-22-CE29-0008-1). We thank Dr Alix Sournia-Saquet and Mr Alain Moreau (LCC) for their help in electrochemical measurements and Mr Anthony Augé for repreparing the Re-BPTA complex.References
- J. Gierschner, J. Shi, B. Milián-Medina, D. Roca-Sanjuán, S. Varghese and S. Y. Park, Adv. Opt. Mater., 2021, 2002251 CrossRef CAS.
- P. Alam, C. Climent, P. Alemany and I. R. Laskar, J. Photochem. Photobiol., C, 2019, 41, 100317 CrossRef CAS.
- L. Ravotto and P. Ceroni, Coord. Chem. Rev., 2017, 346, 62–76 CrossRef CAS.
- V. Sathish, A. Ramdass, P. Thanasekaran, K.-L. Lu and S. Rajagopal, J. Photochem. Photobiol., C, 2015, 23, 25–44 CrossRef CAS.
- M. Mauro and C. Cebrián, Isr. J. Chem., 2018, 58, 901–914 CrossRef CAS.
- A. Ramdass, V. Sathish and P. Thanasekaran, Results Chem., 2022, 4, 100337 CrossRef CAS.
- Z. Song, R. Liu, H. Zhu, Y. Lu, X. Li and H. Zhu, Sens. Actuators, B, 2019, 279, 385–392 CrossRef CAS.
- H. Yang, H. Li, L. Yue, X. Chen, D. Song, X. Yang, Y. Sun, G. Zhou and Z. Wu, J. Mater. Chem. C, 2021, 9, 2334–2349 RSC.
- L. Ma, Y. Wang, X. Wang, Q. Zhu, Y. Wang, L. Li, H.-B. Cheng, J. Zhang and X.-J. Liang, Coord. Chem. Rev., 2022, 473, 214822 CrossRef CAS.
- H. Shen, C. Xu, F. Sun, M. Zhao, Q. Wu, J. Zhang, S. Li, J. Zhang, J. W. Y. Lam and B. Z. Tang, ChemMedChem, 2022, 17, e202100578 CrossRef CAS PubMed.
- X. Lin, W. Li, Y. Wen, L. Su and X. Zhang, Biomaterials, 2022, 287, 121603 CrossRef CAS PubMed.
- S. Graber, K. Doyle, M. Neuburger, C. E. Housecroft, E. C. Constable, R. D. Costa, E. Ortí, D. Repetto and H. J. Bolink, J. Am. Chem. Soc., 2008, 130, 14944–14945 CrossRef CAS PubMed.
- R. D. Costa, E. Ortí, H. J. Bolink, S. Graber, C. E. Housecroft and E. C. Constable, Chem. Commun., 2011, 47, 3207–3209 RSC.
- R. D. Costa, E. Ortí, H. J. Bolink, S. Graber, C. E. Housecroft and E. C. Constable, J. Am. Chem. Soc., 2010, 132, 5978–5980 CrossRef CAS PubMed.
- R. D. Costa, E. Ortí, H. J. Bolink, S. Graber, C. E. Housecroft and E. C. Constable, Adv. Funct. Mater., 2010, 20, 1511–1520 CrossRef CAS.
- H. J. Bolink, E. Coronado, R. D. Costa, E. Ortí, M. Sessolo, S. Graber, K. Doyle, M. Neuburger, C. E. Housecroft and E. C. Constable, Adv. Mater., 2008, 20, 3910–3913 CrossRef CAS.
- G.-G. Shan, H.-B. Li, D.-X. Zhu, Z.-M. Su and Y. Liao, J. Mater. Chem., 2012, 22, 12736–12744 RSC.
- L. He, L. Duan, J. Qiao, D. Q. Zhang, L. D. Wang and Y. Qiu, Chem. Commun., 2011, 47, 6467–6469 RSC.
- R. D. Costa, E. Ortí, H. J. Bolink, S. Graber, C. E. Housecroft, M. Neuburger, S. Schaffner and E. C. Constable, Chem. Commun., 2009, 2029–2031 RSC.
- R. D. Costa, F. Monti, G. Accorsi, A. Barbieri, H. J. Bolink, E. Ortí and N. Armaroli, Inorg. Chem., 2011, 50, 7229–7238 CrossRef CAS PubMed.
- L. He, D. Ma, L. Duan, Y. Wei, J. Qiao, D. Zhang, G. Dong, L. Wang and Y. Qiu, Inorg. Chem., 2012, 51, 4502–4510 CrossRef CAS PubMed.
- K. Traskovskis, V. Kokars, S. Belyakov, N. Lesina, I. Mihailovs and A. Vembris, Inorg. Chem., 2019, 58, 4214–4222 CrossRef CAS PubMed.
- H. Tsubaki, S. Tohyama, K. Koike, H. Saitoh and O. Ishitani, Dalton Trans., 2005, 385–395 RSC.
- H. Tsubaki, A. Sekine, Y. Ohashi, K. Koike, H. Takeda and O. Ishitani, J. Am. Chem. Soc., 2005, 127, 15544–15555 CrossRef CAS PubMed.
- J. Wang, B. Delavaux-Nicot, M. Wolff, S. Mallet-Ladeira, R. Métivier, E. Benoist and S. Fery-Forgues, Dalton Trans., 2018, 47, 8087–8099 RSC.
- J. Wang, A. Poirot, B. Delavaux-Nicot, M. Wolff, S. Mallet-Ladeira, J. P. Calupitan, C. Allain, E. Benoist and S. Fery-Forgues, Dalton Trans., 2019, 48, 15906–15916 RSC.
- J. P. Calupitan, A. Poirot, J. Wang, B. Delavaux-Nicot, M. Wolff, M. Jaworska, R. Métivier, E. Benoist, C. Allain and S. Fery-Forgues, Chem. – Eur. J., 2021, 27, 4191–4196 CrossRef CAS PubMed.
- A. Poirot, C. Vanucci-Bacqué, B. Delavaux-Nicot, N. Leygue, N. Saffon-Merceron, F. Alary, F. Bedos-Belval, E. Benoist and S. Fery-Forgues, Dalton Trans., 2021, 50, 13686–13698 RSC.
- A. Poirot, C. Vanucci-Bacqué, B. Delavaux-Nicot, N. Saffon-Merceron, C.-L. Serpentini, N. Leygue, F. Bedos-Belval, E. Benoist and S. Fery-Forgues, Photochem. Photobiol. Sci., 2023, 22, 1110–1119 Search PubMed.
- K. M. Dawood, B. F. Abdel-Wahab and M. A. Raslanc, Arkivoc, 2018, part i, 179–215 Search PubMed.
- L.-Y. Du, H. Wang, G. Liu, D. Xie, F.-S. Guo, L. Hou and Y.-Y. Wang, Dalton Trans., 2015, 44, 1110–1119 RSC.
- L.-H. Jia, A.-C. Liu, B.-W. Wang, Z.-M. Wang and S. Gao, Polyhedron, 2011, 30, 3112–3115 CrossRef CAS.
- E. V. Govor, A. B. Lysenko and K. V. Domasevitch, Acta Crystallogr., 2008, C64, m201–m204 Search PubMed.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- M. O. Sinnokrot and C. D. Sherrill, J. Phys. Chem. A, 2006, 110, 10656–10668 CrossRef CAS PubMed.
- A. V. Kuttatheyil, M. Handke, J. Bergmann, D. Lässig, J. Lincke, J. Haase, M. Bertmer and H. Krautscheid, Eur. J. Chem., 2015, 21, 1118–1124 CrossRef CAS PubMed.
- L. Suntrup, S. Klenk, J. Klein, S. Sobottka and B. Sarkar, Inorg. Chem., 2017, 56, 5771–5783 CrossRef CAS PubMed.
- G. R. Desiraju and A. Gavezzotti, Acta Crystallogr., 1989, B45, 473–482 CAS.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- R. Eychenne, S. Guizani, J.-H. Wang, C. Picard, N. Malek, P.-L. Fabre, M. Wolff, B. Machura, N. Saffon, N. Lepareur and E. Benoist, Eur. J. Inorg. Chem., 2017, 69–81 CrossRef CAS.
- T. Y. Kim, A. B. S. Elliott, K. J. Shaffer, C. J. McAdam, K. C. Gordon and J. D. Crowley, Polyhedron, 2013, 52, 1391–1398 CrossRef CAS.
- W. K. C. Lo, G. S. Huff, J. R. Cubanski, A. D. W. Kennedy, C. J. McAdam, D. A. McMorran, K. C. Gordon and J. D. Crowley, Inorg. Chem., 2015, 54, 1572–1587 CrossRef CAS PubMed.
- P. Datta, D. Sarkar, A. P. Mukhopadhyay, E. Lopez-Torrez, C. J. Pastor and C. Sinha, J. Organomet. Chem., 2011, 696, 488–495 CrossRef.
- J. Su, T. Fukaminato, J.-P. Placial, T. Onodera, R. Suzuki, H. Oikawa, A. Brosseau, F. Brisset, R. Pansu, K. Nakatani and R. Métivier, Angew. Chem., Int. Ed., 2016, 55, 3662–3666 CrossRef CAS PubMed.
- A. D. Laurent and D. Jacquemin, Int. J. Quantum Chem., 2013, 113, 2019–2039 CrossRef CAS.
- T. Sato, Y. Hamada, M. Sumikawa, S. Araki and H. Yamamoto, Ind. Eng. Chem. Res., 2014, 53, 19331–19337 CrossRef CAS.
- C. Franco and J. Olmsted 3rd, Talanta, 1990, 37, 905–909 CrossRef CAS PubMed.
- C. Carayon, A. Ghodbane, N. Leygue, J. Wang, N. Saffon-Merceron, R. Brown and S. Fery-Forgues, ChemPhotoChem, 2019, 3, 545–553 CrossRef CAS.
- E. Leoni, J. Mohanraj, M. Holler, M. Mohankumar, I. Nierengarten, F. Monti, A. Sournia-Saquet, B. Delavaux-Nicot, J.-F. Nierengarten and N. Armaroli, Inorg. Chem., 2018, 57, 15537–15549 CrossRef CAS PubMed.
- S. Hohloch, L. Suntrup and B. Sarkar, Organometallics, 2013, 32, 7376–7385 CrossRef CAS.
- C. E. Welby, S. Grkinic, A. Zahid, B. S. Uppal, E. A. Gibson, C. R. Rice and P. I. P. Elliott, Dalton Trans., 2012, 41, 7637–7646 RSC.
- A. Mattiuzzi, I. Jabin, C. Moucheron and A. Kirsch-De Mesmaeker, Dalton Trans., 2011, 40, 7395–7402 RSC.
- SADABS , Program for data correction, Bruker-AXS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC.
- J. C. De Mello, H. F. Wittmann and R. H. Friend, Adv. Mater., 1997, 9, 230–232 CrossRef CAS.
- A.-R. Allouche, J. Comput. Chem., 2011, 32, 174–182 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151–2158 CrossRef CAS.
- M. Cossi, V. Barone, B. Mennucci and J. Tomasi, Chem. Phys. Lett., 1998, 286, 253–260 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. Lu and F. Chen, Acta Chim. Sin., 2011, 69, 2393–2406 CAS (https://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract340458.shtml).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including proton numbering for NMR, NMR spectra, molecular views and crystallographic data, electrochemical experiments, and photoluminescence spectra and decays. CCDC 2212965 and 2212966. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03573a |
| This journal is © The Royal Society of Chemistry 2023 |