
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOs(II/III) complex supports pH-insensitive electrochemical DNA-based sensing with superior operational stability than the benchmark methylene blue reporter†
Miguel
Aller Pellitero
 a,
Nandini
Kundu
b,
Jonathan
Sczepanski
b and
Netzahualcóyotl
Arroyo-Currás
*ac
a,
Nandini
Kundu
b,
Jonathan
Sczepanski
b and
Netzahualcóyotl
Arroyo-Currás
*ac
aDepartment of Pharmacology and Molecular Sciences, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, USA. E-mail: netzarroyo@jhmi.edu; Tel: +1 (443) 287-4798
bDepartment of Chemistry, Texas A&M University, College Station, TX 77843, USA
cDepartment of Chemical and Biomolecular Engineering, Johns Hopkins University, Baltimore, Maryland 21218, USA
First published on 5th January 2023
Abstract
DNA-based electrochemical sensors use redox reporters to transduce affinity events into electrical currents. Ideally, such reporters must be electrochemically reversible, chemically stable for thousands of redox cycles, and tolerant to changing chemical environments. Here we report the first use of an Os(II/III) complex in DNA-based sensors, which undergoes pH-insensitive electron transfer with 35% better operational stability relative to the benchmark methylene blue, making it a promising reporter for continuous molecular monitoring applications where pH fluctuates with time.
Introduction
Electrochemical, DNA-based (E-DNA) sensors rely on binding/hybridization-induced conformational changes to generate an electrical signal, enabling single-point1 or continuous2 molecular measurements. Electrochemical, aptamer-based (E-AB) sensors represent a specific type of E-DNA sensors that enable the continuous and real-time monitoring of molecular targets.3 For simplicity, and given their ability to support continuous measurements, this work focuses on E-AB sensors as a model system. E-AB sensors consist of mixed monolayers of blocking alkylthiols and alkylthiol-modified aptamers self-assembled onto gold electrodes, and a redox reporter attached to the terminal end of the aptamers (Fig. 1A). These aptamers are designed to undergo rapid and reversible binding-induced conformational changes that affect electron transfer kinetics from the redox reporter in a way easily measurable via electrochemical methods (Fig. 1B),4 thus enabling the continuous monitoring of targets. Moreover, the target-induced conformational switching in E-AB sensors readily allows sensing in vivo,5 while also minimizing the effect of spurious non-specific binding to the aptamers themselves. Given these characteristics, E-AB sensors represent an unparalleled platform for continuous molecular sensing.6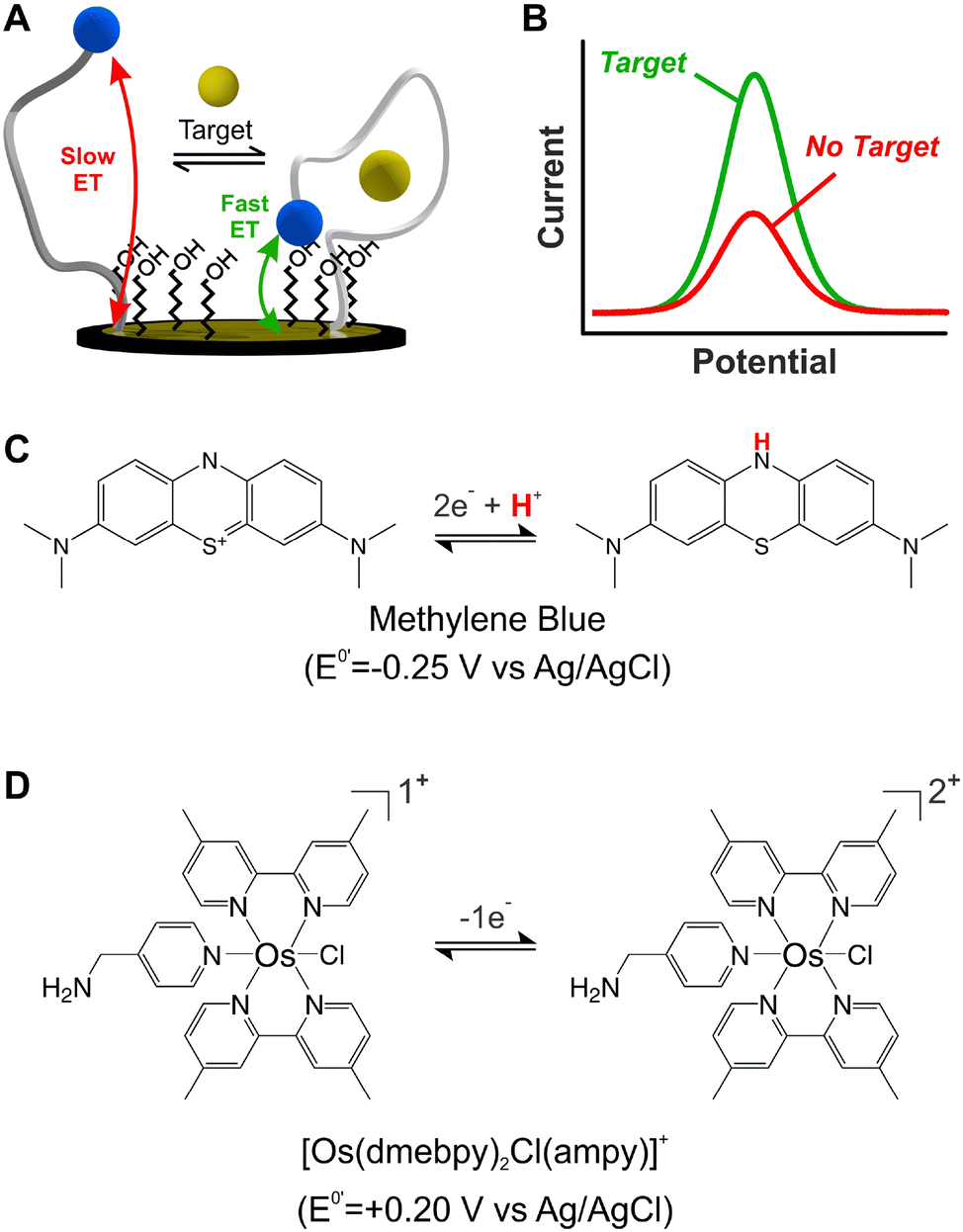 | ||
| Fig. 1 DNA-based electrochemical sensors use redox reporters to convert affinity events to electrical currents. (A) Anatomy and working principle of electrochemical, aptamer-based (E-AB) sensors, which are used in this study as a model system. (B) In the presence of target, aptamers undergo binding-induced conformational changes that alter the electron transfer rate of the redox reporter with the electrode. These changes can be monitored via square wave voltammetry. (C) State-of-the-art E-AB sensors rely on methylene blue (MB) as redox reporter, which undergoes the exchange of 2 electrons and 1 proton. (D) In contrast, [Os(dmebpy)2Cl(ampy)]+ only exchanges 1 electron and is not proton concentration-dependent. | ||
Despite their unique characteristics, however, the field of application of E-AB sensors is limited by two main drawbacks. First, E-AB sensors undergo rapid signal loss upon continuous electrochemical interrogation, especially when deployed in biological fluids.7 And second, the strong dependency of their signaling output on pH limits their use to pH-regulated environments. Seeking to address the first problem, several studies have focused on different elements of the sensing layer like the chemical nature of the blocking alkylthiol8,9 or the DNA strands,10 the technique used for sensor interrogation,11,12 and the electrode material.13 However, the second problem has received significantly less attention. Seventeen years after the first report on this technology,3 most published E-AB sensors still rely on the use of a single redox reporter which undergoes pH-dependent electron transfer: methylene blue (MB).14,15
Alternative redox reporters such as anthraquinone or ferrocene have been used for the fabrication of E-AB sensors; however, these reporters are not suitable for continuous monitoring applications. On the one hand, anthraquinone presents a formal redox potential of 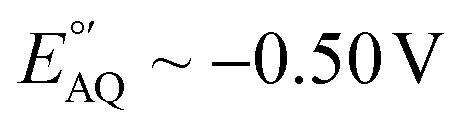 vs. Ag/AgCl,16,17 which overlaps with the reduction of molecular oxygen, making the electrochemical response of this reporter oxygen concentration-dependent. In addition, at such voltages the hydrogen peroxide generated by the electrochemical reduction of oxygen18 can compromise the stability of the monolayer. On the other hand, the product of the oxidation of ferrocene (
vs. Ag/AgCl,16,17 which overlaps with the reduction of molecular oxygen, making the electrochemical response of this reporter oxygen concentration-dependent. In addition, at such voltages the hydrogen peroxide generated by the electrochemical reduction of oxygen18 can compromise the stability of the monolayer. On the other hand, the product of the oxidation of ferrocene (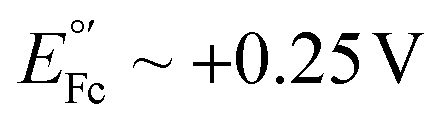 ), the ferrocinium ion, undergoes ligand exchange reactions with chloride ions naturally present in biological media, leading to fast current loss.19,20 Moreover, the positive voltages required for ferrocene oxidation overlap with the beginning of surface oxidation of gold, also compromising the monolayer. Only recently, Li et al. published a novel reporter based on tetrathiafulvalene that has a positive formal reduction potential,
), the ferrocinium ion, undergoes ligand exchange reactions with chloride ions naturally present in biological media, leading to fast current loss.19,20 Moreover, the positive voltages required for ferrocene oxidation overlap with the beginning of surface oxidation of gold, also compromising the monolayer. Only recently, Li et al. published a novel reporter based on tetrathiafulvalene that has a positive formal reduction potential, 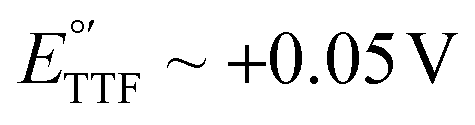 , and undergoes pH-independent electron transfer.21 However, the radical-based mechanism of electron transfer in this molecule may promote secondary chemical reactions with species present in commonly used buffers as well as biomolecules present in biological fluids such as serum or blood, limiting its ability to support long-term sensor operation. Further studies are needed to conclusively determine the value of this reporter for continuous molecular monitoring.
, and undergoes pH-independent electron transfer.21 However, the radical-based mechanism of electron transfer in this molecule may promote secondary chemical reactions with species present in commonly used buffers as well as biomolecules present in biological fluids such as serum or blood, limiting its ability to support long-term sensor operation. Further studies are needed to conclusively determine the value of this reporter for continuous molecular monitoring.
The sole availability of MB as a stable reporter has prevented potential applications of E-AB sensors in biomedical platforms. For example, the electrochemistry of MB is strongly pH-dependent (Fig. 1C), not allowing reliable use in unbuffered matrices such as sweat, saliva, or other time-changing, non-biological environments.6 In addition, and similarly to anthraquinone-based sensors, the negative voltage window necessary to enable MB electron transfer (−0.1 V to −0.5 V vs. Ag/AgCl) promotes the loss of thiol-based monolayer elements from gold surfaces.22 Seeking to address these issues, here we assess the suitability of an Os(II/III) organometallic complex, [Os(dmebpy)2Cl(ampy)]+ (Fig. 1D), to act as redox reporter for E-AB and other E-DNA platforms. While other Os complexes have been used for decades in enzyme-based sensors,23–26 and some of them are commercially used for continuous glucose monitoring,27 [Os(dmebpy)2Cl(ampy)]+ has never been used in E-DNA platforms. Additionally, previous examples of Os-based complexes in DNA-based systems use chemically labile and toxic compounds.28,29 In contrast, our reporter is not labile and uses Os(II/III) oxidation states, which are nontoxic and already commercially approved for use in glucose monitoring, thus offering a clear translational path for incorporation into molecular monitors.
Materials and methods
Chemicals and materials
Ammonium hexachloroosmate(IV) ((NH4)2OsCl6), ammonium hexafluorophosphate (NH4PF6), 4-(aminomethyl)pyridine (ampy), 4,4′-dimethyl-2,2-dipyridil (dmebpy), tris(2-carboxyethyl)phosphine (TCEP), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), guanidine hydrochloride (GC), procaine hydrochloride, N,N-dimethylformadide (DMF), ethylene glycol (EG), diethyl ether, methanol, ethanol, 11-mercaptoundecanoic acid (MUA), and 6-mercapto-1-hexanol (MCH) were purchased from Sigma-Aldrich (St Louis, MO, USA). Phosphate buffer saline (PBS, 11.9 mM HPO32−, 137 mM NaCl, 2.7 mM KCl; pH 7.4), magnesium chloride hexahydrate (MgCl2·6H2O), sodium chloride (NaCl) sulfuric acid (H2SO4), sodium hydroxide (NaOH), and new methylene blue (NMB) were purchased from Fisher Scientific (Waltham, MA, USA). Tobramycin sulfate was purchased from Spectrum Chemical (New Brunswick, NJ, USA). Gold working (cat. #002314, Ø = 1.6 mm) and platinum counter electrodes (cat. #012961) were purchased from ALS (Tokyo, Japan). Ag/AgCl (1 M KCl) reference electrodes (CHI111) and a multichannel potentiostat (CHI1040C) were purchased from CH Instruments (Austin, TX, USA). Silicon carbide grinding paper (36-08-1200) was purchased from Buehler (Lake Bluff, IL, USA). Cloth pads (MF-1040) and alumina slurry (CF-1050) were purchased from BASi (West Lafayette, IN, USA). The DNA sequences used for the fabrication of E-AB sensors are listed below:| Name | Sequence | Manufacturer |
| Tobramycin–COOH | 5′ [COOH-C10] GG GAC TTG GTT TAG GTA ATG AGT CCC [C6-S-S] 3′ | Biosyn (Lewisville, TX, USA) |
| Procaine–COOH | 5′ [COOH-C10] GAC AAG GAA ATC CTT CAA CGA AGT GGG TC [C6-S-S] 3′ | Biosyn (Lewisville, TX, USA) |
| MB-5′–tobramycin | 5′ [MB] GG GAC TTG GTT TAG GTA ATG AGT CCC [C6-S-S] 3′ | Sigma-Aldrich (St Louis, MO, USA) |
| Tobramycin-3′–MB | 5′ [C6-S-S] GG GAC TTG GTT TAG GTA ATG AGT CCC [MB] 3′ | Sigma-Aldrich (St Louis, MO, USA) |
| Tobramycin complement | 5′ GGA CTC ATT ACC TAA ACC AAG TCC 3′ | Sigma-Aldrich (St Louis, MO, USA) |
Synthesis of [Os(dmebpy)2Cl(ampy)]+
The synthesis of [Os(dmebpy)2Cl(ampy)]+ was based on previously reported protocols.30,31 However, in this work, we substituted the bipyridyl ligands for dmebpy to decrease the redox potential of the final complex, ensuring that it falls within a voltage window below the surface oxidation of gold (<0.3 V vs. Ag/AgCl).32 The synthesis of this complex involves two straightforward steps (Fig. S1, ESI†). First, we reacted 200 mg of (NH4)2OsCl6 with 180 mg dmebpy in 5 mL of DMF under reflux and N2 atmosphere for 1 hour. After cooling down the mixture for 30 min, the product was filtered to separate the NH4Cl precipitate, and 3 mL of MeOH was added to the mixture. Then, 200 mL of diethyl ether was slowly added under vigorous stirring, causing the precipitation of the intermediate complex (Os(dmebpy)2Cl)Cl. This complex was isolated by vacuum filtering and washed with diethyl ether. In the second step, 200 mg of the (Os(dmebpy)2Cl)Cl were reacted with 40 mg of ampy in 5 mL of ethylene glycol under reflux and N2 atmosphere for 3 hours. After cooling down the product to 25 °C for 1 hour, 5 mL of a saturated NH4PF6 aqueous solution was slowly added to the mixture while vigorously stirring, observing the precipitation of [Os(dmebpy)2Cl(ampy)]PF6 (dark solid). The final product was vacuum filtered, washed with deionized water, and air-dried overnight at 25 °C. For both steps, we obtained yields higher than 85%.Electrode cleaning and electrochemical measurements
Gold electrodes were polished using silicon carbide grinding paper and a cloth pad with alumina slurry, and then rinsed with water and sonicated for 1 min to remove polishing debris. The freshly polished electrodes were electrochemically activated by serially scanning via cyclic voltammetry, first from −0.3 to −1.6 V in 0.5 M NaOH, then from 0 to 1.6 V in 0.5 M H2SO4, a total of two hundred cycles each at a scan rate of 0.5 V s−1. After this process, the electrodes were ready for DNA functionalization. Unless otherwise stated, all square wave voltammograms in this work were measured using a square wave amplitude of 75 mV and a voltage step of 1 mV. The square wave frequency varied as indicated in figure panels and captions. Cyclic voltammograms were measured using a scan rate of 5 V s−1 and a voltage step of 1 mV. All voltammetric measurements are reported relative to the Ag/AgCl (1 M KCl) reference electrode.Redox reporter immobilization on electrode surfaces
For monolayer experiments, freshly polished and activated electrodes were incubated in 1 μM solutions of MUA prepared in ethanol for 2 hours at 25 °C. This dilute concentration prevented the effect of unreacted, free carboxylic groups on the sensors electrochemical response at different pH values. Then, the redox reporter, either NMB or the Os complex, was chemically bound to the surface via EDC chemistry (Fig. S2†). For this step, electrodes were incubated in a 30 mM EDC solution containing 5 mM of MgCl2, 150 mM of NaCl, and 100 μM of the redox reporter for 3 hours under constant stirring using a Thermomixer (1500 rpm). Controls were prepared incubating the electrodes in the same solution in the absence of EDC. After the coupling reaction, electrodes were rinsed with water and then with two separate washes for 30 min each under vigorous stirring using the Thermomixer. The first wash used acetonitrile containing 1 M of H2SO4. The second wash used 6 M guanidinium chloride solution prepared in ethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (v/v 4:1). These washes were critical to remove non-specifically bound redox reporter molecules from electrode surfaces, particularly in the case of the highly insoluble osmium complex (Fig. S3, ESI†). Last, electrodes were incubated overnight at 25 °C in 10 mM MCH.
:water (v/v 4:1). These washes were critical to remove non-specifically bound redox reporter molecules from electrode surfaces, particularly in the case of the highly insoluble osmium complex (Fig. S3, ESI†). Last, electrodes were incubated overnight at 25 °C in 10 mM MCH.
E-AB sensor fabrication
1 μL of either procaine or tobramycin aptamer solutions at a 100 μM concentration were treated with 2 μL of a 5 mM TCEP solution for 1 h. Then, freshly polished and activated electrodes were immersed in 2 μM solutions of the aptamer prepared in PBS for 4 h. After rinsing with water, electrodes were incubated overnight in EDC solutions containing the redox reporter, as indicated above. After the coupling reaction, electrodes were rinsed with water and washed in acetonitrile and guanidinium chloride solutions. Last, electrodes were incubated in 10 mM MCH solutions for 2 h under vigorous stirring. After rinsing with water, sensors were ready for use. In contrast to most E-AB sensors, the aptamer sequences provided by the manufacturer and used in this work had a COOH– modification at the 5′ end, which we used to attach the redox reporter. We evaluated if the position of MB and alkanethiol moieties influenced the performance of tobramycin aptamers, observing minimal differences (Fig. S4, ESI†). However, as reported by Chamorro and colleagues,33 other sequences including procaine-binding aptamers are affected by the surface orientation of aptamer molecules.10,12DNA hybridization assays
Two different hybridization assays were demonstrated in this work. For the first, we challenged [Os(dmebpy)2Cl(ampy)]+-modified sensors made using the tobramycin-binding aptamer with saturating concentrations (10 μM) of fully complementary DNA strands prepared in PBS, and measured the change in voltammetric peak currents as a function of time. The second assay consisted of the attachment of pre-hybridized duplex DNA to the gold electrodes. For this, we mixed 1 μL of a tobramycin aptamer solution prepared at a concentration of 100 μM with 2 μL of TCEP for 1 hour. Then, this solution was mixed with 2 μL of a 100 μM solution of the complementary strand, progressively increasing the temperature up to 85 °C and kept the solution there for 30 min. After this, the temperature was decreased to 25 °C. The total incubation time was 1 hour. Next, gold electrodes were functionalized with the duplex DNA in the same manner as described above. To minimize the melting of DNA duplexes during washing, we did not wash the sensors with guanidinium chloride in this case. Instead, we performed two washes with acetonitrile/sulfuric acid solutions. After these washes, the electrodes were ready for interrogation.Results and discussion
In this study we evaluated the suitability of the complex [Os(dmebpy)2Cl(ampy)]+ (Fig. 1D, Fig. S1, ESI†) to act as redox reporter in E-AB sensing. We first assessed the electrochemical performance of this reporter when covalently bound to self-assembled monolayers of MUA/MCH on gold electrodes via carbodiimide reaction with its free amine (Fig. 2A and B, Fig. S2 and S3, ESI†). To evaluate the performance of this complex relative to benchmark MB, we employed the commercially available analog new methylene blue (NMB),13 which has two amines available for coupling reactions.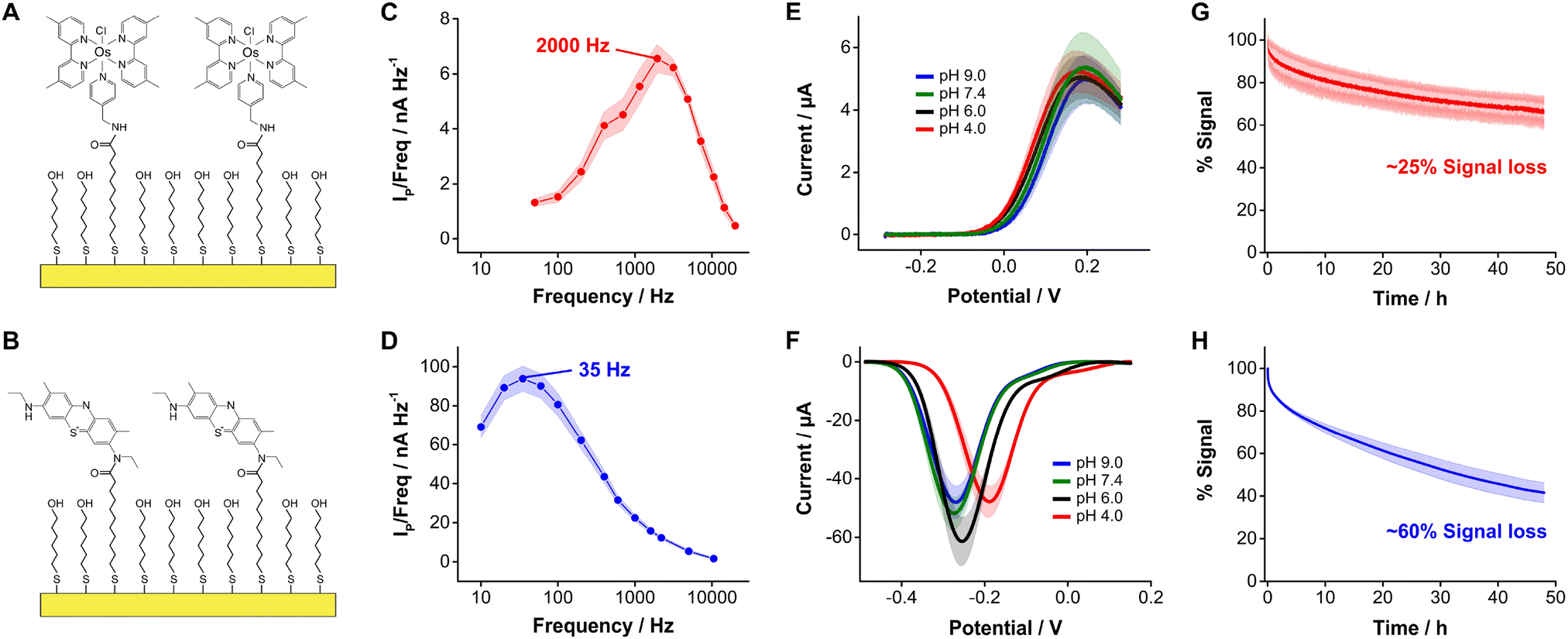 | ||
| Fig. 2 Evaluation of the electrochemical performance of monolayer bound [Os(dmebpy)2Cl(ampy)]+. (A) We formed mixed, self-assembled monolayers consisting of mercaptoundecanoic acid and mercaptohexanol at a ratio of ∼1:100. We then coupled the complex to these monolayers via amide bonding. (B) We used the same strategy with new methylene blue (NMB) to serve as a control. (C) Square wave frequency map corresponding to monolayer bound [Os(dmebpy)2Cl(ampy)]+. The maximum Ip/freq value indicates an electron transfer rate of ket ∼ 2000 s−1. (D) Frequency map for monolayer-bound NMB, indicating an electron transfer rate of ket ∼ 35 s−1. (E) Because the redox reaction of [Os(dmebpy)2Cl(ampy)]+ is not proton-dependent, square wave voltammograms display pH-independent peak currents and potentials. (F) In contrast, voltammograms for NMB show a strong pH dependence. The voltammograms in panels E and F were measured at a square wave frequency of 1000 Hz, which provided high signal to noise voltammetric measurements for both reporters based on panels C and D. (G) Sensorgram displaying peak currents over time from square wave voltammograms of monolayer-bound [Os(dmebpy)2Cl(ampy)]+ serially measured every 8 s for 48 h (∼20000 scans). (H) Equivalent sensorgram for monolayer bound NMB. Solid dots and lines in all panels represent the average of 4 sensors. Shaded areas indicate the standard deviation for each case. | ||
Surface-bound [Os(dmebpy)2Cl(ampy)]+ complexes undergo reversible electron transfer at rates that are fifty times faster than NMB. We illustrate this by showing frequency maps built from square wave voltammograms for each reporter (Fig. 2C and D). Briefly, we interrogated the modified electrodes via square wave voltammetry across a range of frequencies and calculated the ratio of maximum peak current from each voltammogram over its corresponding frequency, and plotted the data vs. the log of frequency.34 The maxima observed in the resulting frequency maps provide an assessment of electron transfer rates, obtaining values of ∼2000 s−1 and ∼35 s−1 for [Os(dmebpy)2Cl(ampy)]+ and NMB, respectively. In contrast to the slower electron transfer rate measured from NMB, which qualifies as a quasi-reversible process, the electron transfer rate of [Os(dmebpy)2Cl(ampy)]+ corresponds to that of a fully reversible electron transfer event. Such a high electron transfer rate could enable new future sensor applications. For example, the electrochemical interrogation at faster frequencies than benchmark sensors would allow the time-resolved monitoring of short-lived biological processes in vivo such as rapid neuronal firing in the brain. These events occur at time scales of milliseconds, much faster than the electron transfer from MB in benchmark E-AB sensors.35
Because electron transfer from [Os(dmebpy)2Cl(ampy)]+ does not involve the exchange of protons with the media (Fig. 1D), it is pH insensitive. To evaluate this effect, we measured voltammograms of the surface-bound reporter in PBS first adjusted to pH = 7.4, and then titrated either down to pH = 4.0 and 6.0, or up to pH = 9.0. Given the ability of the complex to undergo electron transfer regardless of proton concentration, we obtained voltammograms with peak currents and peak potentials within the standard deviation of the batch (shaded areas in Fig. 2E). In contrast, voltammograms measured from NMB-functionalized electrodes showed a strong pH-dependency with changes in formal redox potential of up to 100 mV and in peak currents of up to 15 μA (i.e., 25% to 40% signal change depending on pH, Fig. 2F). These results indicate that [Os(dmebpy)2Cl(ampy)]+ could be a promising alternative to MB as redox reporter in DNA-based sensors in general, and in E-AB sensors in particular, for molecular monitoring applications where pH cannot be tightly controlled by the user.
An additional benefit of [Os(dmebpy)2Cl(ampy)]+ is that the complex displays longer cycling stability under continuous voltammetric interrogation relative to NMB. We show this by serially interrogating complex-functionalized electrodes in PBS every 8 s for 48 h (20000 scans). To minimize the effect of voltage differences between the observed current decays, we used the same voltage window (0.3 V to −0.4 V vs. Ag/AgCl) and square wave frequency (700 Hz) in both cases. Doing so, we observed similar exponential decays in peak currents during the first 2 h of interrogation (∼15% of the initial currents), presumably due to voltage-induced reorganization of the respective monolayers. However, in the case of [Os(dmebpy)2Cl(ampy)]+-functionalized electrodes, such initial decay was followed by a linear decay with a rate of ∼0.45% h−1 (Fig. 2G). In contrast, NMB-modified electrodes showed a three-fold faster linear decay rate of ∼1.25% h−1 (Fig. 2H). We speculate the improved stability of [Os(dmebpy)2Cl(ampy)]+ may arise from two factors: first, the complex is highly hydrophobic, thereby minimizing passive desorption from the electrode surface under serial interrogation; and second, MB and NMB are known to be sensitive to reactive oxygen species released during electrochemical reduction of molecular oxygen at ∼-0.4 V vs. Ag/AgCl.36 This reaction occurs even in the presence of well-formed alkylthiol monolayers, albeit slowly, and increases the sensor decay rate over long periods of continuous cycling.
After characterizing the electrochemical performance of surface bound [Os(dmebpy)2Cl(ampy)]+, we evaluated if the complex could be used for E-AB sensing. To attach the complex to the aptamers used in E-AB sensors, we evaluated both 3′ and 5′ modifications on the aptamers targeting the aminoglycoside antibiotic tobramycin (see Fig. S4 for MB controls, ESI†). Because the two modifications achieved similar signal gains, we moved forward with the 5′ modification. The aptamers were also modified with a 3′ terminal alkylthiol group for monolayer self-assembly. After forming the monolayer, we conjugated [Os(dmebpy)2Cl(ampy)]+ to the aptamers via amide bonds using the carbodiimide reaction (Fig. 3A). As a control we again used NMB. The resulting [Os(dmebpy)2Cl(ampy)]+-modified E-AB sensors enabled the measurement of tobramycin concentrations in buffered solutions.
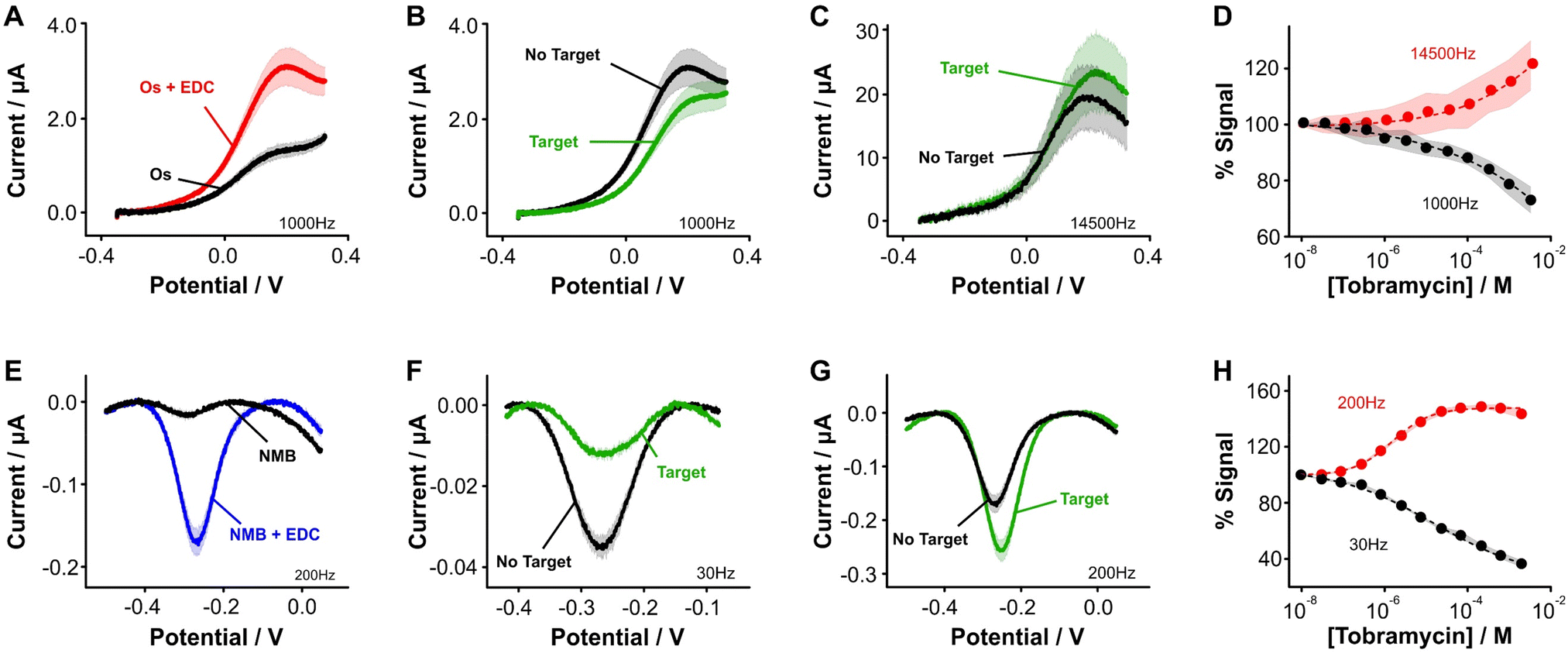 | ||
| Fig. 3 Aptamer-bound [Os(dmebpy)2Cl(ampy)]+ supports E-AB sensing. (A) Using the carboxylic group at the terminal end of immobilized tobramycin aptamers and the free amine group in the Os complex, we successfully attached the complex to the DNA strands via EDC chemistry. (B and C) [Os(dmebpy)2Cl(ampy)]+ sensors signal output to target additions at ∼1000 Hz and ∼14500 Hz, respectively. These square wave frequencies were determined from the frequency maps shown in Fig. S5.† (D) Such signaling is used to build calibration curves where the normalized response changes monotonically with target concentration. The signaling is reversible under continuous voltammetric interrogation (Fig. S7, ESI†). (E–G) Control NMB-modified sensors also display signal-OFF and signal-ON responses, albeit at lower frequencies: ∼30 Hz and ∼200 Hz, respectively. These square wave frequencies are the standard for interrogation of MB-based tobramycin sensors.5 (H) NMB-modified sensors achieve larger signal gain and display better affinity for the same target (tobramycin). Solid lines/dots and shaded areas represent the average and standard deviation of 4 sensors. | ||
Specifically, when we challenged the sensors with saturating concentrations of the drug (1 mM), we observed square wave frequency-dependent “signal-ON” and “signal-OFF” responses that are characteristic of E-AB sensors (Fig. 3B and C, Fig. S5, ESI†).5,37 We also observed this behavior in NMB-functionalized sensors (Fig. 3E–G); however, the frequencies required to observe the two responses in Os-based sensors were approximately 70-fold larger in magnitude than those required for NMB-modified sensors. This effect matches the difference in electron transfer kinetics we previously measured between the two reporters when directly bound to self-assembled monolayers (Fig. 2C and D).
Although both sets of sensors respond to the presence of target, the analytical performance that they exhibit in terms of sensitivity, dynamic range, and signal change is different (Fig. 3D and H). For example, while the dynamic range of the Os-based sensors begins at ∼1 × 10−6 M tobramycin and a plateau in the response is not observed, the dynamic range of NMB-based sensors begins at ∼5 × 10−7 M and achieves a plateau at ∼10−4 M. Moreover, at a concentration of 1 mM tobramycin, Os-based sensors achieve ∼30% worse signal gain (in both ON and OFF responses) than NMB-modified sensors. Such a difference in performance between the two reporters may arise from three different sources. First, the surface modification protocol we followed to couple both reporters with the aptamer was less efficient than when directly attaching the complex to the monolayer. This point is shown by worse baseline currents in the resulting square wave voltammograms when the aptamer is present (Fig. 3A and Evs. Fig. 2E) and by lower surface coverages obtained for both types of sensors (ΓOs = 0.6 ± 0.1 pmol cm−2, ΓNMB = 0.4 ± 0.1 pmol cm−2, Fig. S6, ESI†) relative to those obtained from benchmark E-AB sensors fabricated with alkylthiol and methylene blue-modified aptamers (ΓMB–SH = 1–2 pmol cm−2).12 This lower efficiency could be due to the short linker length used for the amide coupling reaction, which contains only one carbon prior to the amine we use for coupling (Fig. 2A and B). The amine may be strongly affected by steric or coulombic effects from the Os center. In addition, the complex presents strong non-specific binding to the DNA and has proven difficult to remove via solvent washes. To overcome these challenges, future efforts in our laboratory will focus on testing different linker lengths to increase the efficiency of the coupling reaction, as well as bottom-up synthesis of the complex-modified aptamer via solid phase oligo synthesis.
A second consideration to explain the worse signaling performance of [Os(dmebpy)2Cl(ampy)]+ is that its electron transfer rate may be too fast to measure target binding to the aptamer under conditions of thermodynamic equilibrium. Specifically, the conformation dynamics of nucleic acids typically fluctuate with time constants of a few nanoseconds.38 Such time constants are expected to slow down for surface-bound oligos because of changes in persistence length and folding degrees of freedom to slower time constants, potentially to periods of microseconds. At the optimal square wave frequency for signal-ON interrogation of the [Os(dmebpy)2Cl(ampy)]+-modified aptamer (14500 Hz in Fig. 3C), current is being sampled every 70 μs, a time constant that may be approaching the folding rates of surface bound oligos. Although this is speculation, similar effects (lower gain) have been observed with ferrocene-modified aptamers.37 Ferrocene is another redox reporter that undergoes electron transfer at rates much faster than NMB (and MB). More research beyond the scope of this work is needed to gain further insight into this question.
Two final considerations for the difference in sensing behavior are the fact that [Os(dmebpy)2Cl(ampy)]+ is highly hydrophobic and has a larger size (M.W. = 702.29 g mol−1) compared to NMB (M.W. = 484.22 g mol−1). The hydrophobicity of the complex may induce strong interactions with the hydrophobic alkylthiol backbone of the monolayer, thus affecting the conformational dynamics of the aptamer. The complex's large size may present steric hindrance that also affects folding dynamics of the aptamer, leading to the lower signal gain observed. This last effect may be dependent on aptamer secondary structure. To illustrate this point, we evaluated the signaling of a procaine-binding sensor that folds into a triple foil secondary structure and is known to generate larger signal gains relative to the tobramycin aptamer,10 which folds into a stem loop (Fig. S8†). When we functionalized procaine-binding sensors (Fig. 4A and B, Fig. S9, ESI†), we observed that the analytical performance of sensors fabricated with both reporters is closer than in the case of the tobramycin aptamer. This observation may indicate that the binding-induced conformational change is less affected by the chemical nature of the osmium complex (Fig. 4C and D). The effect that different redox reporters have on the conformational change of surface bound aptamer molecules, and thus on the analytical performance of the E-AB sensors, remains an open question to be addressed in future work.
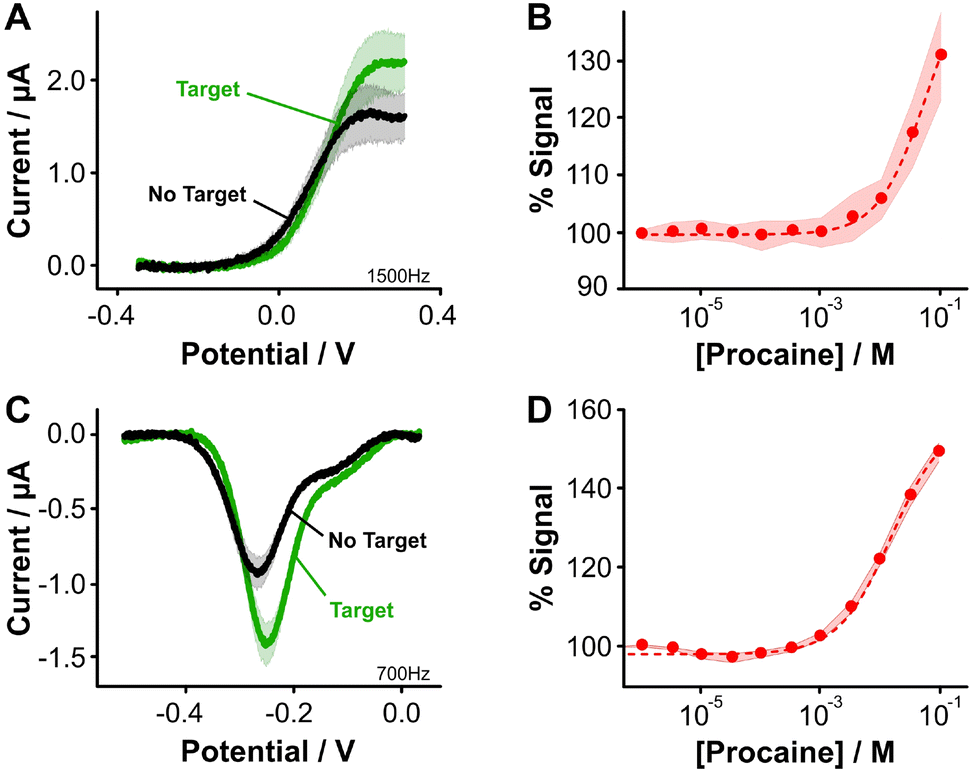 | ||
| Fig. 4 Aptamer-bound [Os(dmebpy)2Cl(ampy)]+ and NMB enable the measurement of procaine. Following the modification protocol used for tobramycin E-AB sensors, we successfully attached each reporter to the terminal end of procaine-binding aptamers using their free carboxylic group via EDC chemistry. Sensors fabricated with (A and B) [Os(dmebpy)2Cl(ampy)]+ or (C and D) NMB respond to saturating concentrations of procaine (100 mM, panels A and C), observing in both cases an increase in the voltammetric response when procaine was added to the solution. Titration curves (panels B and D) compared favorably in signal gain and binding affinity between reporters. The square wave frequencies used in these measurements were selected from sensor frequency maps based on best peak current signal-to-noise ratios. Solid lines and dots, and shaded areas represent the average and standard deviation of 4 sensors, respectively. | ||
Seeking to highlight the versatility of [Os(dmebpy)2Cl(ampy)]+ as a reporter for other DNA-based platforms beyond E-AB sensors, we evaluated its use for DNA hybridization assays. Upon hybridization, the redox reporter moves away from the electrode surface, which decreases its freedom of movement and, therefore, its electron transfer rate.4 For such measurements, we challenged electrode-bound tobramycin aptamers with saturating concentrations of fully complementary strands. Hybridization of the strands repositions the reporter further away from the electrode surface, leading to a decrease in voltammetric peak currents of ∼20% after 90 min of incubation in complement strands (Fig. 5A). This effect was not observed when challenging the same sensors with control, non-complimentary strands (Fig. S10, ESI†). We observed larger signal differences when we prepared pre-hybridized DNA duplexes of the tobramycin aptamer plus its complement in solution, and then formed sensor monolayers using the duplexes. In this case, duplex-functionalized sensors presented a decrease of ∼50% in voltammetric currents relative to unhybridized E-AB sensors (Fig. 5B). The lower signal gain observed between the real-time measurement (Fig. 5A) and the pre-hybridization experiment (Fig. 5B) could be explained by the slow hybridization kinetics induced by the larger size and hydrophobic nature of [Os(dmebpy)2Cl(ampy)]+. Nevertheless, these results indicate that the complex can support signaling across different DNA-based sensing platforms, and that future optimization of aptamer–reporter coupling strategies may significantly improve the signaling output of [Os(dmebpy)2Cl(ampy)]+-modified sensors.
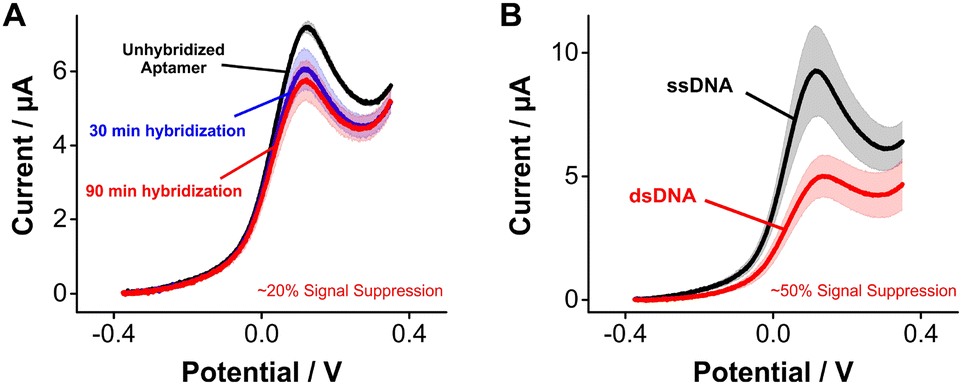 | ||
| Fig. 5 [Os(dmebpy)2Cl(ampy)]+ supports sensing of nucleic acid hybridization events. (A) Voltammograms from a 26 nucleotide-long, electrode-bound tobramycin aptamer functionalized with the osmium-based reporter at t = 0, t = 30 min, and t = 90 min after exposure to a 10 μM solution of fully complementary strands. (B) Control voltammograms for non-hybridized sensors (black), and sensors that were fabricated after pre-hybridizing the aptamer with its complement for 1 h. Voltammograms were measured using a square wave frequency of 1800 Hz, as determined from frequency maps based on best peak current signal-to-noise ratios. Lines and dots represent the average of 4 sensors. Shaded areas represent their standard deviation. | ||
Conclusions
We report the first use of [Os(dmebpy)2Cl(ampy)]+ as a redox reporter for E-DNA sensors. This complex can be efficiently coupled to mixed self-assembled monolayers of mercaptoundecanoic acid in mercaptohexanol via EDC chemistry and undergoes reversible and pH-insensitive electron transfer at rates that are fifty times faster than the benchmark methylene blue reporter used in E-DNA sensors. Additionally, Os-modified sensor monolayers are 35% more stable under continuous voltammetric interrogation than methylene blue-modified analogs. In a similar way, this Os complex can also be coupled to surface-bound nucleic acid aptamers to enable aptamer-based electrochemical sensing, also enabling the electrochemical monitoring of hybridization events. However, the hydrophobic nature of [Os(dmebpy)2Cl(ampy)]+ poses the challenge of non-specific adsorption on sensor surfaces when attached to surface-bound aptamers post monolayer formation. Future efforts from our group will focus on developing phosphoramidite-coupled [Os(dmebpy)2Cl(ampy)]+ for the bottom-up incorporation of the complex to oligonucleotide strands during solid phase synthesis. This approach should eliminate non-specific binding of bulk [Os(dmebpy)2Cl(ampy)]+ molecules to sensor surfaces while simultaneously allowing for targeted and highly efficient coupling to aptamers. Finally, the relevance of ultrafast electron transfer to E-DNA sensing is still to be demonstrated; however, the ability to probe sensor surfaces at the sub millisecond scales enabled by [Os(dmebpy)2Cl(ampy)]+ may open the door for the real-time study of ultra-fast biological processes in vivo, such as rapid neuronal firing in the brain.Author contributions
M. A. P. and N. A. C. contributed equally to research work conceptualization, data curation, formal analysis, methodology and manuscript writing. M. A. P. performed the investigation, data analysis and image processing. N. A. C. acquired the funding, supervised the work, and administered the project.Conflicts of interest
The authors report no conflicts of interest.Acknowledgements
This study was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM140143. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.References
- A. Victorious, Z. Zhang, D. Chang, R. Maclachlan, R. Pandey, J. Xia, J. Gu, T. Hoare, L. Soleymani and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202204252 CAS.
- J. Das, S. Gomis, J. B. Chen, H. Yousefi, S. Ahmed, A. Mahmud, W. Zhou, E. H. Sargent and S. O. Kelley, Nat. Chem., 2021, 13, 428–434 CrossRef CAS PubMed.
- Y. Xiao, A. A. Lubin, A. J. Heeger and K. W. Plaxco, Angew. Chem., Int. Ed., 2005, 44, 5456–5459 CrossRef CAS PubMed.
- M. A. Pellitero, A. Shaver and N. Arroyo-Currás, J. Electrochem. Soc., 2019, 167, 037529 CrossRef.
- N. Arroyo-Currás, J. Somerson, P. A. Vieira, K. L. Ploense, T. E. Kippin and K. W. Plaxco, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 645–650 CrossRef PubMed.
- N. Arroyo-Currás, P. Dauphin-Ducharme, K. Scida and J. L. Chávez, Anal. Methods, 2020, 12, 1288–1310 RSC.
- A. Shaver and N. Arroyo-Currás, Curr. Opin. Electrochem., 2022, 32, 100902 CrossRef CAS PubMed.
- A. Shaver, S. D. Curtis and N. Arroyo-Currás, ACS Appl. Mater. Interfaces, 2020, 12, 11214–11223 CrossRef CAS PubMed.
- Z. Watkins, A. Karajić, T. Young, R. White and J. Heikenfeld, ChemRxiv, 2022, This content is a preprint and has not been peer-reviewed.
- A. Shaver, N. Kundu, B. E. Young, P. A. Vieira, J. T. Sczepanski and N. Arroyo-Currás, Langmuir, 2021, 37, 5213–5221 CrossRef CAS PubMed.
- N. Arroyo-Currás, P. Dauphin-Ducharme, G. Ortega, K. L. Ploense, T. E. Kippin and K. W. Plaxco, ACS Sens., 2018, 3, 360–366 CrossRef PubMed.
- M. A. Pellitero, S. D. Curtis and N. Arroyo-Currás, ACS Sens., 2021, 6, 1199–1207 CrossRef CAS PubMed.
- M. A. Pellitero and N. Arroyo-Currás, Anal. Bioanal. Chem., 2022, 414, 5627–5641 CrossRef CAS PubMed.
- V. Clark, K. Waters, B. Orsburn, N. N. Bumpus, N. Kundu, J. T. Sczepanski, P. Ray and N. Arroyo-Currás, Angew. Chem., Int. Ed., 2022, e202211292 CAS.
- S. Lin, X. Cheng, J. Zhu, B. Wang, D. Jelinek, Y. Zhao, T.-Y. Wu, A. Horrillo, J. Tan, J. Yeung, W. Yan, S. Forman, H. A. Coller, C. Milla and S. Emaminejad, Sci. Adv., 2022, 8, eabq4539 CrossRef CAS PubMed.
- D. Kang, R. J. White, F. Xia, X. Zuo, A. Vallée-Bélisle and K. W. Plaxco, NPG Asia Mater., 2012, 4, e1–e1 CrossRef.
- Y. Liu, Y. Liu, Z. Matharu, A. Rahimian and A. Revzin, Biosens. Bioelectron., 2015, 64, 43–50 CrossRef CAS PubMed.
- J. C. Fornaciari, L.-C. Weng, S. M. Alia, C. Zhan, T. A. Pham, A. T. Bell, T. Ogitsu, N. Danilovic and A. Z. Weber, Electrochim. Acta, 2022, 405, 139810 CrossRef CAS.
- D. Kang, F. Ricci, R. J. White and K. W. Plaxco, Anal. Chem., 2016, 88, 10452–10458 CrossRef CAS PubMed.
- A. Singh, D. R. Chowdhury and A. Paul, Analyst, 2014, 139, 5747–5754 RSC.
- S. Li, A. Ferrer-Ruiz, J. Dai, J. Ramos-Soriano, X. Du, M. Zhu, W. Zhang, Y. Wang, M. Á. Herranz, L. Jing, Z. Zhang, H. Li, F. Xia and N. Martín, Chem. Sci., 2022, 13, 8813–8820 RSC.
- K. K. Leung, A. M. Downs, G. Ortega, M. Kurnik and K. W. Plaxco, ACS Sens., 2021, 6, 3340–3347 CrossRef CAS PubMed.
- T. J. Ohara, R. Rajagopalan and A. Heller, Anal. Chem., 1993, 65, 3512–3517 CrossRef CAS PubMed.
- R. Kumar and D. Leech, J. Electrochem. Soc., 2014, 161, H3005 CrossRef.
- E. Tremey, C. Stines-Chaumeil, S. Gounel and N. Mano, ChemElectroChem, 2017, 4, 2520–2526 CrossRef CAS.
- M. Marquitan, A. Ruff, M. Bramini, S. Herlitze, M. D. Mark and W. Schuhmann, Bioelectrochemistry, 2020, 133, 107487 CrossRef CAS PubMed.
- G. McGarraugh, Diabetes Technol. Ther., 2009, 11, S-17–S-24 CrossRef CAS PubMed.
- E. Paleček and M. A. Hung, Anal. Biochem., 1983, 132, 236–242 CrossRef PubMed.
- M. Fojta, P. Kostečka, M. Trefulka, L. Havran and E. Paleček, Anal. Chem., 2007, 79, 1022–1029 CrossRef CAS PubMed.
- F. P. Dwyer, H. A. Goodwin and E. C. Gyarfas, Aust. J. Chem., 1963, 16, 42–50 CrossRef CAS.
- A. M. Ricci, E. J. Calvo, S. Martin and R. J. Nichols, J. Am. Chem. Soc., 2010, 132, 2494–2495 CrossRef CAS PubMed.
- A. Ricci, C. Rolli, S. Rothacher, L. Baraldo, C. Bonazzola, E. J. Calvo, N. Tognalli and A. Fainstein, J. Solid State Electrochem., 2007, 11, 1511–1520 CrossRef CAS.
- A. Chamorro-Garcia, G. Ortega, D. Mariottini, J. Green, F. Ricci and K. W. Plaxco, Chem. Commun., 2021, 57, 11693–11696 RSC.
- V. Mirceski, R. Gulaboski, M. Lovric, I. Bogeski, R. Kappl and M. Hoth, Electroanalysis, 2013, 25, 2411–2422 CrossRef CAS.
- B. Wang, W. Ke, J. Guang, G. Chen, L. Yin, S. Deng, Q. He, Y. Liu, T. He, R. Zheng, Y. Jiang, X. Zhang, T. Li, G. Luan, H. D. Lu, M. Zhang, X. Zhang and Y. Shu, Front. Cell. Neurosci., 2016, 10, 239 Search PubMed.
- A. Y. Satoh, J. E. Trosko and S. J. Masten, Environ. Sci. Technol., 2007, 41, 2881–2887 CrossRef CAS PubMed.
- P. Dauphin-Ducharme and K. W. Plaxco, Anal. Chem., 2016, 88, 11654–11662 CrossRef CAS PubMed.
- H. Ma, C. Wan, A. Wu and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 712–716 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2an01901a |
| This journal is © The Royal Society of Chemistry 2023 |