
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBottom-up/cross-linking mass spectrometry via simplified sample processing on anion-exchange solid-phase extraction spin column†
Ayako
Takemori
 a,
Yusuke
Kawashima
b and
Nobuaki
Takemori
*a
a,
Yusuke
Kawashima
b and
Nobuaki
Takemori
*a
aDivision of Analytical Bio-Medicine, Advanced Research Support Center, Ehime University, Toon, Ehime, Japan. E-mail: takemori@m.ehime-u.ac.jp
bDepartment of Applied Genomics, Kazusa DNA Research Institute, Kisarazu, Chiba, Japan
First published on 12th December 2021
Abstract
We introduce a simple single-column protein digestion method for low-microgram-level samples containing sodium dodecyl sulfate and Coomassie dye that can be completed within a few hours.
Comprehensive multimolecular characterization within cellular components is essential for a systematic understanding of biological processes at the molecular level. Proteomic analysis that attempts to capture the dynamics of intracellular protein components on a large scale is currently driven by mass spectrometry (MS). The bottom-up MS approach, which is employed in many proteomic analyses, is initiated by digesting protein components extracted from cells.1 The resulting mixture of digested peptides is separated by reversed-phase liquid chromatography (RPLC) and finally ionized in an MS instrument connected to the RPLC. The ionized peptides are subjected to tandem MS (MS/MS) analysis in a data-dependent acquisition (DDA) mode, and the sequences are identified based on the MS/MS spectral information.2,3 Data-independent acquisition (DIA) analysis can be performed using the acquired tandem MS information to comprehensively quantify the ionized peptides, and quantitative information for several thousand proteins can be obtained in a single DIA analysis.4–9 The use of MS is not limited to the identification and quantification of proteins, however, and has recently also been the focus as a means of analyzing the higher-order structure of proteins.10 The establishment of highly sensitive structural analysis methods for protein complexes is a technological frontier in the field of structural biology, and interest in cross-linking MS (XL-MS), which combines chemical protein XL with bottom-up MS, is rapidly growing.11–13
In bottom-up MS workflows, sample pretreatment is an essential step that determines the sensitivity and reproducibility of the analysis. This process includes protein extraction from the biological sample, digestion of the extracted protein using proteases with high substrate specificity such as trypsin and/or Lys-C, and purification of the resulting digested peptide. General sample pretreatment protocols unfortunately require many steps, are sensitive to the skill of the operator, and often result in loss of trace amounts of sample. Thus, there has recently been a movement to develop sample preparation methods that can be completed in a single device, minimize the number of steps, and be used universally for any type of sample.14–17
In addition to solubilized protein samples, sample pretreatment often includes in-gel proteins that have been separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as well. High-resolution proteome fractionation by SDS-PAGE is effective for improving protein identification in DDA analysis18,19 and is often used to separate cross-linked protein complexes in XL-MS analysis.20 Since it is difficult to recover PAGE-separated proteins into solution, they are generally enzymatically digested in gels.21 However, the low-recovery rate of in-gel digested peptides has always been a problem.22 We recently reported a workflow for efficient and rapid recovery of proteins from SDS-PAGE gels called PEPPI-MS, or passively eluting proteins from polyacrylamide gels as intact species for MS, and demonstrated that proteins of a wide range of molecular weights can be recovered in less than 10 minutes by using Coomassie Brilliant Blue (CBB) dye and SDS as extraction enhancers.23 A constraint of the original PEPPI-MS workflow is CBB and SDS in the recovered solution that need to be removed before sample preparation, which can be done by methanol–chloroform–water precipitation. However, such precipitation is often difficult to apply for trace amounts of protein.
As a simple sample preparation method for bottom-up MS analysis, Mann's group reported an in-StageTip method16 in 2014 that achieves protein extraction and peptide purification in a single pipette tip filled with a C18 RP solid-phase extraction (SPE) disc called stop-and-go-extraction tip (StageTip).24 The in-StageTip method allows the whole sample pretreatment process from reductive alkylation to peptide purification to be completed in a single StageTip, thus minimizing sample loss even for small amounts of sample. In addition, the use of a unique StageTip fitted with two different SPE discs, an anion exchange (AX) disc and a C18 RP disc, allows detailed peptide fractionation followed by enzymatic digestion, which can be used for in-depth proteome analysis.25 However, this conventional in-StageTip method is not suitable for use with samples containing strong anionic surfactants such as SDS and CBB because of the use of C18 or combined AX/C18 StageTips. Heretofore, the use of AX SPE disc alone has not been studied. In this study, we focused on the use of a single AX StageTip fitted with only an AX SPE disc and developed a sample pretreatment method by performing the entire process from enzymatic digestion to peptide purification in the AX StageTip.
We began by investigating the conditions for effective protein digestion using the AX StageTips as a reactor. First, we examined the localization of the proteins entrapped in the closely packed AX SPE discs using the fluorescent protein β-phycoerythrin. A 200 μL AX StageTip was used to examine the conditions (Fig. S1a, ESI†). For comparison, the localization was also examined in the C18 RP StageTip using the in-StageTip method. In the C18 RP StageTips, β-phycoerythrin was entrapped inside the disc and showed a wide distribution (Fig. S1b, ESI†). In contrast, in the AX StageTip, β-phycoerythrin was localized on the disc surface in a layered manner and did not penetrate into the disc. These properties of the AX-SPE disc are effective in the enrichment of dilute protein samples, and the accumulation of proteins on the disc surface facilitates access for proteases.
We then evaluated the enzymatic digestion performance on the AX SPE disc for 10 μg of human cellular protein extracts (HCPE) dissolved in 10 μL of 0.05% (w/v) SDS/100 mM ABC. The protein extracts were first loaded onto the discs, washed with 100 mM ammonium bicarbonate (ABC), and then covered with a thin layer of Trypsin/Lys-C mixture solution. The proteases were adsorbed onto the discs by centrifugation at low rpm to initiate the digestion reaction, which was performed at 25 °C. During the reaction, 10 μL of ABC solution were stacked on the discs to prevent drying of the sample. The results of monitoring the digestion reaction against time by SDS-PAGE are shown in Fig. 1a. Typically, a substrate enzyme ratio of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 100:1 is selected for Trypsin/Lys-C digestion. We first observed rapid digestion performance at a substrate enzyme ratio of 20:1. At this lower ratio, most of the substrate disappeared within 30 minutes and complete digestion was achieved in 1–4 hours. In contrast, when the substrate enzyme ratio was set to 100:1, digestion was not complete even after 4 hours (Fig. S2, ESI†).
:1 to 100:1 is selected for Trypsin/Lys-C digestion. We first observed rapid digestion performance at a substrate enzyme ratio of 20:1. At this lower ratio, most of the substrate disappeared within 30 minutes and complete digestion was achieved in 1–4 hours. In contrast, when the substrate enzyme ratio was set to 100:1, digestion was not complete even after 4 hours (Fig. S2, ESI†).
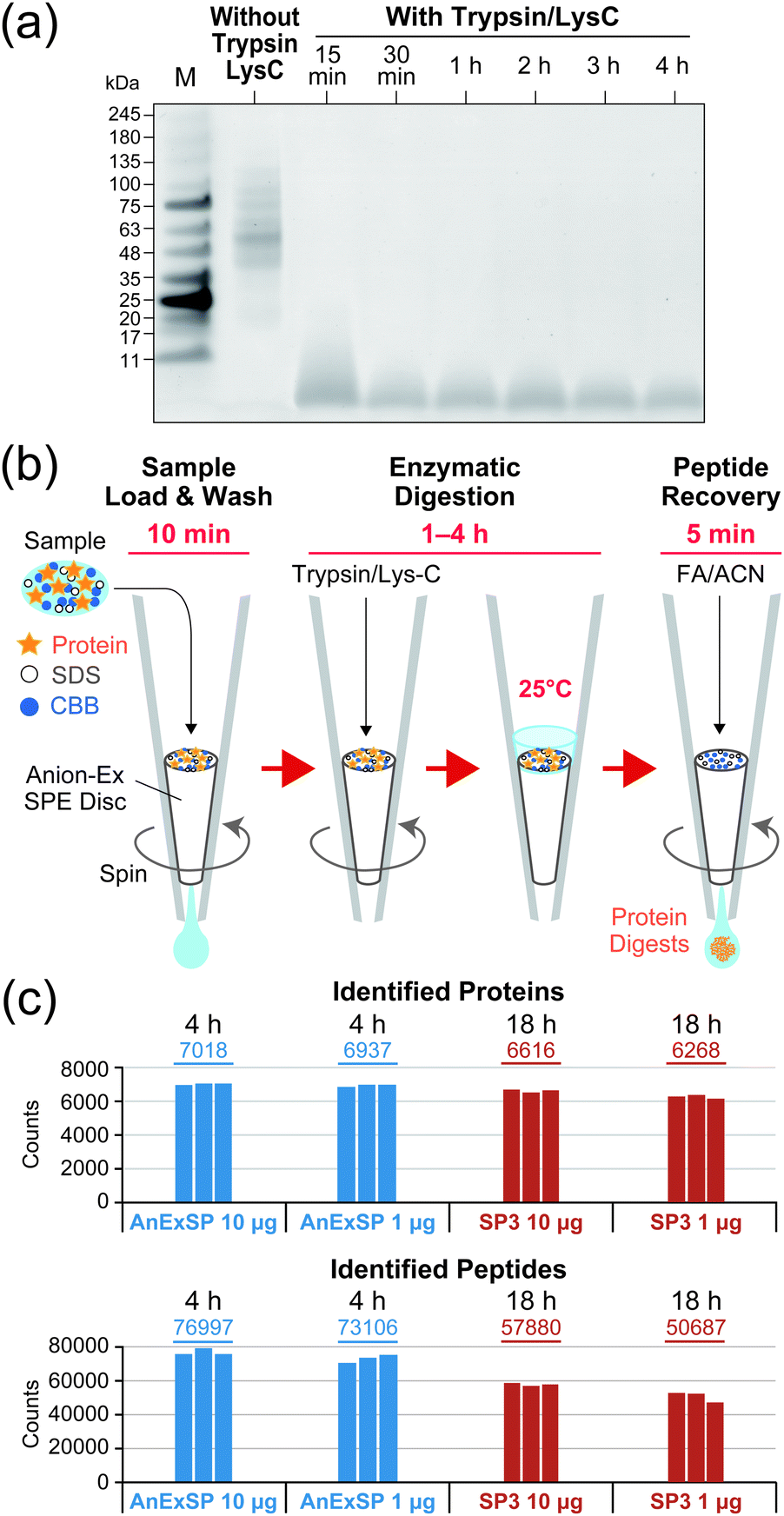 | ||
| Fig. 1 Sample pretreatment for bottom-up MS using AnExSP. (a) SDS-PAGE of digested HCPE recovered from AX SPE discs. After digestion of HCPE with Trypsin/Lys-C, digestion products recovered from AX SPE discs with 10% SDS were separated by SDS-PAGE and visualized with CBB. The substrate enzyme ratio was set to 20:1. M: molecular weight marker. (b) Schematic workflow of AnExSP. FA: formic acid. ACN: acetonitrile (c) DIA analysis of digestion products. | ||
When SDS is contained in the sample, it is trapped on the disc along with the protein. However, there is a limit to the SDS concentration that AnExSP can handle, and if the sample is contaminated with a high concentration of SDS, the protein will penetrate into the disc (Fig. S3, ESI†), making digestion on the disc difficult. While an SDS concentration of 0.05% (w/v) is optimal for a 50 μL sample, it can be as high as 0.1% (w/v). When eluting peptides after digestion, it is necessary to collect only the peptides while retaining the SDS on the disc. The elution can be accomplished with a formic acid solution containing as much as 50% (v/v) acetonitrile, but higher concentrations of acetonitrile cause SDS to be eluted as well. Our study demonstrated that acetonitrile concentration can be reduced to as low as 30% (v/v) without affecting peptide recovery (Fig. S4, ESI†). If the 50 μL HCPE sample contains less than 0.1% (w/v) SDS, elution of SDS can be inhibited with the use of 0.5% (v/v) formic acid/30% (v/v) acetonitrile.
Based on the results described above, we established a sequential sample preparation method inside a single AX StageTip, which we have named AnExSP (anion-exchange disc-assisted sequential sample preparation). The experimental workflow of AnExSP is shown in Fig. 1b. AnExSP can be applied to cell/tissue extracts and PEPPI-MS fractions containing SDS and CBB, which have been difficult to handle via previous in-StageTip methods. When processing a protein sample after reductive alkylation, the whole process from enzymatic digestion to digested peptide purification can be completed in as little as 1.5 hours, at most 4.5 hours. Fig. 1c shows the results of DIA analysis of Trypsin/Lys-C digestion products of HCPE samples recovered from the AX SPE discs. Digestion of 10 μg of HCPE at 25 °C for 4 hours identified approximately 7000 proteins. Overnight digestion (18 hours) was also conducted, but no difference in protein identification was observed (Fig. S5, ESI†). These results indicate that on the AX SPE discs, even a quick treatment at room temperature is sufficient to achieve excellent digestion performance.
Single-pot solid-phase-enhanced sample preparations (SP3) and filter-aided sample preparation (FASP) are popular as sample pretreatment methods that can be used for samples containing SDS.14,15 SP3 has been reported to provide better results than FASP in processing low-microgram samples because it concentrates and digests protein samples on a magnetic bead.26 We consequently compared AnExSP and SP3 in sample preparation for 1 μg HCPE (Fig. 1c). DIA analysis of the digested product of 1 μg HCPE showed that more than 6000 proteins were identified by both digestion methods. The digestion performance of AnExSP did not change significantly when the sample amount was reduced from 10 μg to 1 μg, and AnExSP outperformed SP3 in the number of proteins and peptides detected. In AnExSP, the entire process from digestion to purification is completed in a single tip, and the digestion reaction is performed in a short time (4 hours) on a very small disc. These features contribute to the reduction of sample loss. In contrast, the cleaning of the magnetic beads and the long digestion reaction time in SP3 (18 hours) lead to sample loss, resulting in a lower performance than AnExSP.
In addition to SDS, compatibility with CBB is required when processing PEPPI-MS fractions in AnExSP. The effect of the presence of CBB on the enzymatic digestion in AnExSP was examined by using HCPE samples with and without CBB. When samples were loaded onto AX SPE discs, those containing CBB clearly showed that it was retained stably at the top of the disc (Fig. 2a). When the digested peptides were recovered, CBB remained on the disc and did not elute into the peptide solution. DIA analysis of the recovered peptides showed no significant difference in protein identification between samples with and without CBB (Fig. S6, ESI†), indicating that amounts of CBB normally used for gel-staining do not hinder results in AnExSP.
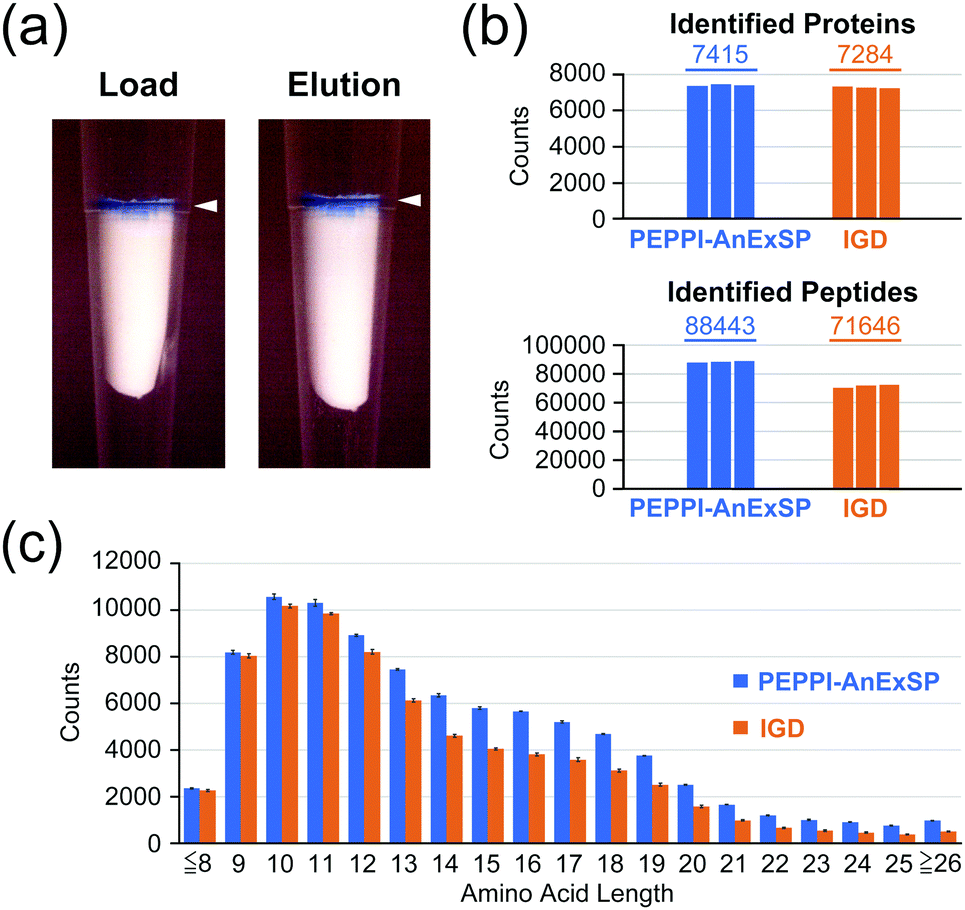 | ||
| Fig. 2 AnExSP for polyacrylamide gel-extracted proteins containing CBB. (a) AX SPE disc: after loading a CBB-containing HCPE (Load) and after peptide elution (Elution). White arrows: Trapped CBB. (b and c) Comparison between PEPPI-AnExSP and IGD: identified proteins/peptides (b) and amino acid lengths of identified peptides (c). | ||
We next established a sample preparation strategy for PAGE-separated proteins (PEPPI-AnExSP) combining PEPPI-MS and AnExSP, and validated its performance compared to IGD-based sample preparation. HCPE samples (8 μg) separated by SDS-PAGE and stained with CBB were used for validation (Fig. S7a, ESI†). In the case of PEPPI-AnExSP, the proteins in the gel were collected by PEPPI-MS using 0.05% SDS/100 mM ammonium bicarbonate and subjected to trypsin digestion by AnExSP at 25 °C for 4 hours. For IGD, the standard protocol of trypsin digestion performed at 37 °C overnight (18 hours) was followed. In DIA analysis, PEPPI-AnExSP showed comparable protein identification to IGD in spite of the significantly reduced digestion time (Fig. 2b). Moreover, the number of peptides detected was 1.2 times higher in PEPPI than in IGD. The reproducibility between PEPPI-AnExSP analyses was high (Fig. S7b, ESI†) and better than that of IGD. In particular, PEPPI was superior to IGD in the detection of long peptides longer than 14 amino acids, with 1.4–2 times more peptides detected than IGD (Fig. 2c and Fig. S7c, ESI†). These analyses demonstrate that PEPPI-AnExSP has excellent reproducibility and can detect more peptides in a shorter digestion time than IGD. In particular, the excellent performance of PEPPI-AnExSP in detecting long-chain peptides will be useful for middle-down MS analysis and XL-MS analysis where long-chain peptides are the target of analysis.
In order to validate the efficacy of PEPPI-AnExSP in XL-MS, sample pretreatment of cross-linked hemoglobin (Hb) complexes with PEPPI-AnExSP was performed. Disuccinimidyl dibutyric urea (DSBU), which can be cleaved by MS,27 was used to cross-link Hb in solution, and the resulting cross-linked products were separated by SDS-PAGE (Fig. 3a). The gel bands of Hb tetramer and dimer visualized by CBB were cut out and treated with PEPPI-AnExSP or IGD. DDA analyses identified more cross-linked peptides in samples treated with PEPPI-AnExSP than IGD for both Hb tetramer and Hb dimer (Fig. 3b). The cross-linked peptides detected only in PEPPI-AnExSP were all large in size (>2000 Da) and likely retained in the gel during IGD (Table S1, ESI†). The PEPPI-AnExSP treatment successfully avoided this loss of cross-linked peptides by recovering cross-linked Hbs from the gel and digesting them on the disc, demonstrating that PEPPI-AnExSP is a more effective sample preparation method than IGD for XL-MS analysis.
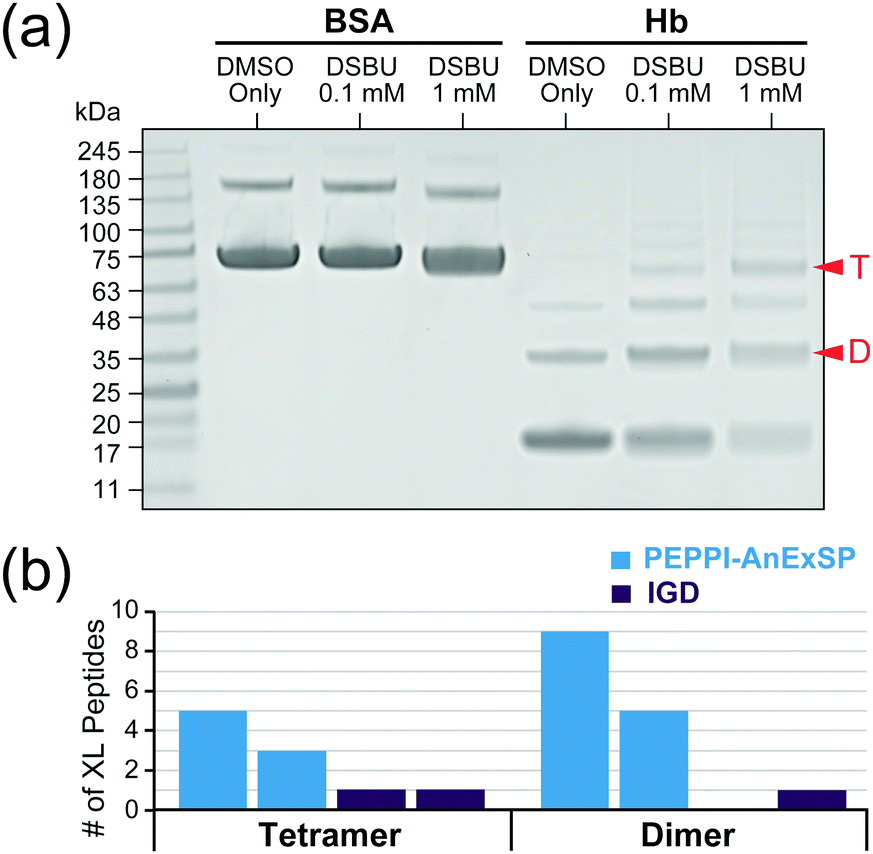 | ||
| Fig. 3 PEPPI-AnExSP workflow for XL-MS. (a) SDS-PAGE of Hb and BSA cross-linked with DSBU. Red arrows: The bands of Hb dimer (D) and tetramer (T) subjected to sample pretreatment. (b) The number of cross-linked Hb peptides observed by DDA. The experiments were performed twice independently. | ||
In summary, we have developed AnExSP, a sample preparation method for bottom-up MS analysis within a single AX StageTip. In this method, protein enrichment on a single AX SPE disc enables enzymatic digestion and peptide purification with minimal loss, and we attained an outstanding streamlined sample processing for low-microgram-level samples. AnExSP has fewer steps and reduced digestion times with better digestion performance than SP3, which will accelerate the speed of sample preparation for bottom-up MS. Moreover, the combination of AnExSP with PEPPI-MS enables rapid sample pretreatment of PAGE-separated proteins, which is difficult with conventional IGD. Our study revealed that AnExSP has a 1.4- to 2-fold advantage over IGD in the recovery of long-chain HCPE peptides, but the advantage of AnExSP over IGD is even more remarkable in the recovery of cross-linked long-chain peptides in XL-MS analysis of PAGE-separated Hb. Its application is certainly not limited to purified protein complexes but has the potential to be extended to XL-MS analysis targeting protein complexes in crude biological samples as well.
This work was supported by JSPS KAKENHI (19K05526 and 20H04713 to N. T.). The authors thank Professor Emeritus Robert J Beynon (University of Liverpool) for valuable discussion on AnExSP development. The authors also thank Erika Teraoka for editing and reviewing this manuscript for use of the English language and helpful discussion.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. K. Eng, A. L. McCormack and J. R. Yates, J. Am. Soc. Mass Spectrom., 1994, 5, 976–989 CrossRef CAS PubMed.
- A. L. McCormack, D. M. Schieltz, B. Goode, S. Yang, G. Barnes, D. Drubin and J. R. Yates 3rd, Anal. Chem., 1997, 69, 767–776 CrossRef CAS PubMed.
- A. J. Link, J. Eng, D. M. Schieltz, E. Carmack, G. J. Mize, D. R. Morris, B. M. Garvik and J. R. Yates 3rd, Nat. Biotechnol., 1999, 17, 676–682 CrossRef CAS PubMed.
- S. Purvine, J. T. Eppel, E. C. Yi and D. R. Goodlett, Proteomics, 2003, 3, 847–850 CrossRef CAS PubMed.
- J. D. Venable, M. Q. Dong, J. Wohlschlegel, A. Dillin and J. R. Yates, Nat. Methods, 2004, 1, 39–45 CrossRef CAS PubMed.
- R. S. Plumb, K. A. Johnson, P. Rainville, B. W. Smith, I. D. Wilson, J. M. Castro-Perez and J. K. Nicholson, Rapid Commun. Mass Spectrom., 2006, 20, 1989–1994 CrossRef CAS PubMed.
- A. Panchaud, A. Scherl, S. A. Shaffer, P. D. von Haller, H. D. Kulasekara, S. I. Miller and D. R. Goodlett, Anal. Chem., 2009, 81, 6481–6488 CrossRef CAS PubMed.
- T. Geiger, J. Cox and M. Mann, Mol. Cell. Proteomics, 2010, 9, 2252–2261 CrossRef CAS PubMed.
- L. C. Gillet, P. Navarro, S. Tate, H. Rost, N. Selevsek, L. Reiter, R. Bonner and R. Aebersold, Mol. Cell. Proteomics, 2012, 11, O111 016717 CrossRef PubMed.
- P. Lossl, M. van de Waterbeemd and A. J. Heck, EMBO J., 2016, 35, 2634–2657 CrossRef PubMed.
- A. Sinz, J. Mass Spectrom., 2003, 38, 1225–1237 CrossRef CAS PubMed.
- A. Leitner, T. Walzthoeni, A. Kahraman, F. Herzog, O. Rinner, M. Beck and R. Aebersold, Mol. Cell. Proteomics, 2010, 9, 1634–1649 CrossRef CAS PubMed.
- A. Sinz, Angew. Chem., Int. Ed., 2018, 57, 6390–6396 CrossRef CAS PubMed.
- J. R. Wisniewski, A. Zougman, N. Nagaraj and M. Mann, Nat. Methods, 2009, 6, 359–362 CrossRef CAS PubMed.
- C. S. Hughes, S. Foehr, D. A. Garfield, E. E. Furlong, L. M. Steinmetz and J. Krijgsveld, Mol. Syst. Biol., 2014, 10, 757 CrossRef PubMed.
- N. A. Kulak, G. Pichler, I. Paron, N. Nagaraj and M. Mann, Nat. Methods, 2014, 11, 319–324 CrossRef CAS PubMed.
- A. Zougman, P. J. Selby and R. E. Banks., Proteomics, 2014, 14, 1006–1010 CrossRef CAS PubMed.
- D. Pflieger, J. P. Le Caer, C. Lemaire, B. A. Bernard, G. Dujardin and J. Rossier, Anal. Chem., 2002, 74, 2400–2406 CrossRef CAS PubMed.
- M. Schirle, M. A. Heurtier and B. Kuster, Mol. Cell. Proteomics, 2003, 2, 1297–1305 CrossRef CAS PubMed.
- C. Iacobucci, M. Gotze, C. H. Ihling, C. Piotrowski, C. Arlt, M. Schafer, C. Hage, R. Schmidt and A. Sinz, Nat. Protoc., 2018, 13, 2864–2889 CrossRef CAS PubMed.
- A. Shevchenko, M. Wilm, O. Vorm and M. Mann, Anal. Chem., 1996, 68, 850–858 CrossRef CAS PubMed.
- A. Takemori, J. Ishizaki, K. Nakashima, T. Shibata, H. Kato, Y. Kodera, T. Suzuki, H. Hasegawa and N. Takemori, J. Proteome Res., 2021, 20, 1535–1543 CrossRef CAS PubMed.
- A. Takemori, D. S. Butcher, V. M. Harman, P. Brownridge, K. Shima, D. Higo, J. Ishizaki, H. Hasegawa, J. Suzuki, M. Yamashita, J. A. Loo, R. R. O. Loo, R. J. Beynon, L. C. Anderson and N. Takemori, J. Proteome Res., 2020, 19, 3779–3791 CrossRef CAS PubMed.
- J. Rappsilber, Y. Ishihama and M. Mann, Anal. Chem., 2003, 75, 663–670 CrossRef CAS PubMed.
- W. Chen, S. Adhikari, L. Chen, L. Lin, H. Li, S. Luo, P. Yang and R. Tian, J. Chromatogr. A, 2017, 1498, 207–214 CrossRef CAS PubMed.
- M. Sielaff, J. Kuharev, T. Bohn, J. Hahlbrock, T. Bopp, S. Tenzer and U. Distler, J. Proteome Res., 2017, 16, 4060–4072 CrossRef CAS PubMed.
- M. Q. Muller, F. Dreiocker, C. H. Ihling, M. Schafer and A. Sinz, Anal. Chem., 2010, 82, 6958–6968 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc05529a |
| This journal is © The Royal Society of Chemistry 2022 |