
Hydrogel nanoparticle degradation influences the activation and survival of primary macrophages†
Bader M.
Jarai
 ,
Zachary
Stillman
and
Catherine A.
Fromen
*
,
Zachary
Stillman
and
Catherine A.
Fromen
*
Department of Chemical and Biomolecular Engineering, University of Delaware, 150 Academy St., Newark, DE 19716, USA. E-mail: cfromen@udel.edu; Tel: +1 302-831-3649
First published on 28th June 2021
Abstract
The effect of nanoparticle (NP) internalization on cell fate has emerged as an important consideration for nanomedicine design, as macrophages and other phagocytes are the primary clearance mechanisms of administered NP formulations. Pro-survival signaling is thought to be concurrent with phagocytosis and recent work has shown increased macrophage survival following lysosomal processing of internalized NPs. These observations have opened the door to explorations of NP physiochemical properties aimed at tuning the NP-driven macrophage survival at the lysosomal synapse. Here, we report that NP-induced macrophage survival and activation is strongly dependent on NP degradation rate using a series of thiol-containing poly(ethylene glycol) diacrylate-based NPs of equivalent size and zeta potential. Rapidly degrading, high thiol-containing NPs allowed for dramatic enhancement of cell longevity that was concurrent with macrophage stimulation after 2 weeks in ex vivo culture. While equivalent NP internalization resulted in suppressed caspase activity across the NP series, macrophage activation was correlated with increasing thiol content, leading to increased lysosomal activity and a robust pro-survival phenotype. Our results provide insight on tuning NP physiochemical properties as design handles for maximizing ex vivo macrophage longevity, which has implications for improving macrophage-based immune assays, biomanufacturing, and cell therapies.
Introduction
Over the past few decades, interactions of synthetic biomaterials with macrophages, a class of innate phagocytic immune cell, have offered new opportunities to both study cell responses and to modulate cell phenotype with the overall goal of directing host immune response.1 Certainly, synthetic microenvironments have emerged as powerful tools to study macrophage migration and phenotype progression in the context of disease-altered tissue properties,2 with biomaterials-based nano- and microparticle platforms also providing increased understanding of how various internalized physiochemical stimuli drive macrophage function and activation.3–5 With ever growing advances in synthetic approaches, particulates ranging from lipid-6 to metal-7 to polymer-based materials8,9 have demonstrated the significance of particle size,10 shape,11 modulus,12 surface charge,13 and degradability14 on biological effects of cellular uptake, trafficking, and cargo release. In addition to delivery of known stimuli as therapeutic cargos, particulate platforms afford a unique opportunity to modulate cell phenotype through the cell internalization process, i.e. phagocytosis. Innate immune cells, including macrophages, dendritic cells, and neutrophils, are inherently phagocytic, allowing them to engulf foreign materials, and are equipped with high sensitivity at the phagocytic synapse, where everything from surface charge to particle shape can influence subsequent downstream signaling.15,16 Increasing investigation into the role of various physiochemical properties of particulate carriers that alter the phagocytic synapse and downstream signaling is warranted to both improve understanding of the overall process of phagocytosis in these critical innate immune cells and leverage this increased understanding for therapeutic benefit.One such physiochemical property deserving of further investigation is particle degradation rate and its effect in the regulation of intracellular signaling following phagocytosis. Our recent work has demonstrated that macrophage lifespan is intimately linked to phagocytic events that can dramatically increase the cell longevity through enhanced lysosomal signaling, even in the absence of cell activation. We previously demonstrated that treatment of inert17 poly(ethylene glycol) (PEG) diacrylate (PEGDA)-based nanoparticles (NPs) drives pro-survival signaling following NP internalization in a range of ex vivo and in vivo macrophages.18 Combined with supporting studies of pro-survival signaling centered in the lysosome,19,20 this prior work highlights an untapped opportunity for intelligently-designed NP platforms to further modulate this response. Upon phagocytosis, the phagosome undergoes compartment acidification and fusion with the lysosome to form the phagolysosome, a strongly acidic and hydrolytic environment enriched with a wide range of enzymes and signaling molecules that are responsible for breakdown of the internalized material and the triggering of subsequent cell activation signaling.21,22 Intracellular NP degradation following phagocytosis has been shown to impact lysosomal signaling and compartment acidification.23,24 Thus, variations in NP intracellular degradation rates may directly correspond to macrophage viability.
While the particle surface charge and size are known to impact tissue distribution,25 cellular internalization,26 and cellular response,27,28 the role of NP degradability has largely been studied in terms of cargo delivery and sustained released and likely represents an important opportunity for regulation of phagocytotic and subsequent lysosomal signaling. Sustained release of antigens and immune-modifying cargoes have been advantageous in NP vaccination and therapeutic strategies that target phagocytic cells,29–31 while the renowned stimulatory efficacy of alum, a commonly used vaccine adjuvant, has been attributed in part to its slow degradation profile.32 Despite the many tangential observations that slow-degrading NPs can provide distinct immune stimulation on the cellular level, studies of intracellular degradation of such NP platforms in the absence of therapeutic cargos are less frequently pursued for sustained phagocyte modulation. Depending on the desired effect (stimulation, suppression, or avoidance), application, or rate of degradation, different NP systems may offer distinct advantages to phagocyte stimulation through controlled degradation,33 with biodegradable aliphatic polyesters such as polylactic acid (PLA), polyglycolic acid (PGA), poly(lactic-co-glycolic acid) [PLGA], and poly-ε-caprolactone (PCL) representing the most widely studied platforms of tunable breakdown. PLGA NPs in particular can have varied rates of degradation based on the ratio of PLA to PGA34,35 and degrade into acidic lactic and glycolic acid-related products that can stimulate the immune system similar to an adjuvant,27,36 with potential for deleterious side effects in some cases.37,38 PCL-based NPs provide slower degradation and gradual cargo release, with no acidic byproducts and thus no autocatalytic degradation,39 providing slow-release profiles with the downside of potential long-term accumulation in the body.40 Given the multitude of other NP platforms used for various immune engineering applications, consideration of the degradation rate and associated byproducts of intracellular degradation is likely critical to tuning temporal regulation of phagocytic phenotype and individual lifespan following NP-based cues.
Given the role of lysosomal involvement in pro-survival signaling19,20 and the importance of NP design in tuning degradation occurring in the lysosome, we sought to directly investigate the role of tunable particle degradation rates on phagocyte lifespan. In this study, we modulate the degradability of PEGDA-based hydrogel NPs through the inclusion of varying amounts of thiol–PEG–thiol (HS–PEG–SH) in the NP formulation, increasing the acid-sensitivity of the NP and providing more degradable points for intracellular breakdown. The resulting degradable formulations are investigated to tune the NP-induced survival of primary macrophages. We report that macrophage survival is enhanced following treatment with rapidly degradable NPs relative to their slowly degrading counterparts. This effect is coupled with the upregulation of immunostimulatory molecules likely caused by acidic degradation products, as well as increased lysosomal activity and signaling in rapidly degrading NPs. The results provide a platform to tune the ex vivo survival of macrophages for a range of applications including biomanufacturing, in vitro drug screening assays, vaccine development, and autologous cell therapies.
Experimental
Nanoparticle synthesis & characterization
Hydrogel NPs were generated as described previously,41 but with modifications to pre-particle compositions. Briefly, to generate 0% HS–PEG–SH, 10% HS–PEG–SH, 20% HS–PEG–SH PEGDA NPs (referred to as 0%, 10%, and 20% NPs, respectively hereafter), pre-particle mol% compositions according to Table 1 were formulated by combining varying amounts of poly(ethylene glycol) diacrylate (PEGDA) Mn = 700 (Millipore Sigma), thiol–PEG–thiol (HS–PEG–SH) Mn = 600 (Creative PEGWorks), 1,6-hexanediol dimethacrylate (HDDMA) (Millipore Sigma), and 2-carboxyethyl acrylate (CEA) (Millipore Sigma). HDDMA and a higher amount of CEA were included to improve the resulting hydrogel NP modulus and surface charge, which are notable differences to our previously used formulations in studying NP-macrophage interactions.18 1 mg of photoinitiator diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (PI) (Millipore Sigma) and 0.05 mg fluorescent label cyanine 5 (Cy5) maleimide (AAT Bioquest) were added and the formulations were diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by mass in methanol (Fisher Scientific) to arrive at 50 wt% mixtures. 100 μL of the mixture was emulsified in 1 mL of silicone oil AP1000 (Millipore Sigma) by vortex mixing for 1 minute followed by sonicating for 30 seconds. The emulsion was then irradiated with UV light (APM LED UV Cube, wavelength of 365 nm at a distance of ∼28 cm from the light source, ∼5–10 mW cm−2) for 44, 50, and 52 seconds for 0%, 10%, and 20% NP formulations, respectively. The polymerized emulsions were washed with 1 mL of n-hexanes, followed by two more washes with 1 mL of ethanol.
:1 by mass in methanol (Fisher Scientific) to arrive at 50 wt% mixtures. 100 μL of the mixture was emulsified in 1 mL of silicone oil AP1000 (Millipore Sigma) by vortex mixing for 1 minute followed by sonicating for 30 seconds. The emulsion was then irradiated with UV light (APM LED UV Cube, wavelength of 365 nm at a distance of ∼28 cm from the light source, ∼5–10 mW cm−2) for 44, 50, and 52 seconds for 0%, 10%, and 20% NP formulations, respectively. The polymerized emulsions were washed with 1 mL of n-hexanes, followed by two more washes with 1 mL of ethanol.
| NP | Formulation (mol%) | PEGDA | HS–PEG–SH | HDDMA | CEA |
|---|---|---|---|---|---|
| 0% | 0% HS–PEG–SH | 75 | 0 | 5 | 20 |
| 10% | 10% HS–PEG–SH | 65 | 10 | 5 | 20 |
| 20% | 20% HS–PEG–SH | 55 | 20 | 5 | 20 |
Nanoparticle degradation analysis via thermogravimetric analysis (TGA)
In preparation for degradation studies, the synthesized NPs (0%, 10%, and 20%, respectively) were isolated from ethanol via centrifugation at 18200 RCF for 5 minutes, the ethanol removed, and NPs dispersed into water via vortex mixing for 20 seconds followed by sonication for 30 seconds. This procedure was repeated a second time to ensure removal of ethanol. Following concentration determination via thermogravimetric analysis (TGA) in water using a TA instruments TGA 550, requisite volumes of the three respective NP types (0%, 10%, and 20%) were added to microcentrifuge tubes to achieve concentrations of 3 mg mL−1 of NPs in 1 mL of the medium of choice (either artificial lysosomal fluid or ALF,42 phosphate buffered saline or PBS, ALF with 10 mM glutathione or ALF + GSH, or PBS with 10 mM glutathione or PBS + GSH). The NPs were isolated from water via centrifugation 18200 RCF for 5 minutes. Following isolation, the water supernatant was removed and 1 mL of the medium of choice (either ALF, PBS, ALF + GSH, or PBS + GSH) was added to the microcentrifuge tube. For each NP type (0%, 10%, and 20%), there were 12 total samples (NPs dispersed in each medium with N = 3). The NPs were then dispersed via vortex mixing for 20 seconds followed by sonication for 30 seconds and then incubated in a shaker kept at 37 °C and 1000 rpm. At designated time points, a 50 μL aliquot was analyzed via thermogravimetric analysis (TGA) to determine the mass of non-degraded NPs remaining; select samples were also analyzed via scanning electron microscopy (see sections below).
Dynamic light scattering (DLS) and zeta potential
DLS of the NPs was performed using a Malvern Zetasizer Nano ZS. NP samples were prepared for DLS measurement by diluting samples in water to ∼0.1 mg mL−1. Hydrodynamic diameters (Dh) and polydispersity indices (PDIs) were measured from two independently synthesized samples. NP samples were prepared for zeta potential measurement by diluting samples in water to ∼0.5 mg mL−1 in 10 mM NaCl or in PBS. Zeta potentials were measured from two independently synthesized samples.Cryogenic scanning electron microscopy (cryo-SEM)
As-synthesized 0%, 10%, and 20% NP samples 10 μL in volume were added to a sample holder for cryo-SEM and flash frozen with liquid nitrogen. Samples were prepared at 3 mg mL−1 for imaging. The samples were sputter-coated for 60 seconds with a platinum coating and then imaged using an Apreo VolumeScope Scanning Electron Microscope at 2 kV from 5000× to 40000× magnifications under high vacuum.
X-Ray energy dispersive spectroscopy (XEDS)
2 μL of PEG-SH NP samples were dropped onto a glass slide and allowed to dry overnight. The samples were then sputter-coated for 65 seconds with gold–palladium coating (thickness of ∼5 nm) using a Denton Desk IV Sputter Coater and imaged using a JSM-7400F Scanning Electron Microscope at 3 kV from 1000× to 40000× magnifications under high vacuum. XEDS was performed using the JSM-7400F that is equipped with an OXFORD INCAx-sight energy-dispersive XEDS detector. Samples were analyzed for 100 seconds and elemental data collected using the INCA software for elemental analysis.
Liquid chromatography mass spectrometry (LC-MS)
Similar to degradation studies, the synthesized NPs (0%, 10%, and 20%, respectively) were isolated from ethanol via centrifugation at 18200 RCF for 5 minutes, the ethanol removed, and NPs dispersed into water via vortex mixing for 20 seconds followed by sonication for 30 seconds. This procedure was repeated a second time and a third time to ensure removal of ethanol. Following concentration determination via thermogravimetric analysis (TGA) in water using a TA instruments TGA 550, requisite volumes of the three respective NP types (0%, 10%, and 20%) were added to microcentrifuge tubes to achieve concentrations of 3 mg mL−1 of NPs in 1 mL of water, chosen to prevent ion interference with mass spectrometry. At 1 day, 2 days, 7 days, and 14 days time points, the NPs were isolated from water via centrifugation 18200 RCF for 5 minutes. Following isolation, the water supernatant was removed for analysis via LC-MS. The particle degradation products were then analyzed using a Q-Exactive Orbitrap coupled with an HPLC. Analysis was then performed in the Xcalubur software and species identified by the authors.
Animals
Animals were housed in a pathogen-free facility at the University of Delaware. Studies involving animals were performed according to the National Institutes of Health (NIH) guidelines for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Delaware. Female C57BL/6J (Jackson Laboratories) six to twelve weeks of age were used to isolate primary BMMs.Primary cell isolation and culture
Bone marrow-derived macrophages (BMMs) were generated according to standard protocols as previously described.43 Briefly, bone marrow cells from femurs and tibias of mice were plated in BMM differentiation media composed of DMEM/F-12 media (Corning) with 20% fetal bovine serum, 30% L929 cell conditioned media, and 1% penicillin–streptomycin. An equal volume of BMM differentiation media was added on day 3 and cells were used on day six for experiments in DMEM/F-12 media containing 10% fetal bovine serum.Assessment of cell viability
BMMs were seeded in 96-well plates (1 × 105 cells per well) and allowed to adhere for at least 4 h prior to NP treatment. BioTek Cytation 5 Multimode Imager was utilized to continuously determine cell counts. Caspase-Glo® 3/7 Assay System (Promega) was used according to manufacturer's guidelines to determine the levels of caspase 3 and caspase 7 in BMMs and luminescence was measured using BioTek Cytation 5 Multimode Imager.NP internalization and trafficking
BMMs were plated in 24-well plates (2 × 105 cells per well) and allowed to adhere overnight prior to NP treatment. BMMs were then dosed with 50 μg mL−1 Cy5-labelled NPs. Cells were detached using Accutase® (Innovative Cell Technologies, Inc.) at 0, 4, 16, 24, 48, and 72 hours (h) and analyzed for % Cy5+ cells using ACEA NovoCyte Flow Cytometer to determine kinetic NP uptake. For lysosomal imaging, BMMs were cultured in glass bottom 96-well plates (1 × 105 cells per well) and Cell Navigator™ Lysosome Staining Kit (AAT Bioquest) was used according to manufacturer's guidelines. Cells were imaged using BioTek Cytation 5 Multimode Imager.Macrophage polarization studies
BMMs were plated in 6-well plates (1.5 × 106 cells per well) and allowed to adhere overnight prior to NP treatment. BMMs were then dosed with 100 μg mL−1 Cy5-labelled NPs. At 24 h and 72 h timepoints, cells were detached using Accutase® (Innovative Cell Technologies, Inc.) and washed twice with PBS supplemented with 2% FBS. Cells were then incubated with anti-CD16/32 (Fc block, Biolegend) for 10 minutes and then stained with CD80-Pacific Blue, CD86-AlexaFluor700, and I-A/I-E-Brilliant Violet 785™ antibodies (All from Biolegend) for 30 minutes in the dark at 4 °C. Cells were then fixed with 4% paraformaldehyde in PBS (Alfa Aesar) for 15 minutes at room temperature and then permeabilized by washing twice with Intracellular Staining Permeabilization Wash Buffer (Biolegend) and stained with CD206-PE-Cy7 antibodies (Biolegend) and analyzed using ACEA NovoCyte Flow Cytometer.Cytokine analysis
Enzyme-Linked Immunosorbent Assay (ELISA) kits for Interleukin-6 (IL-6), Interleukin-10 (IL-10), and Tumor Necrosis Factor-α (TNF-α) (all from BD Biosciences) were used to determine cytokine concentrations in culture supernatants according to manufacturer's guidelines.Statistical analysis
GraphPad Prism 9 (GraphPad Software Inc.) was used to perform all the statistical analyses. All quantitative data are represented as mean ± standard deviation (SD) or standard error of the mean (SEM). Tukey's multiple-comparisons tests were used to generate p-values in ANOVA multiple comparisons, unless stated otherwise.Results & discussion
Nanoparticle synthesis and characterization
To form a set of NPs that could aid in the determination of the cause and extent of primary cell longevity, we synthesized a set of PEGDA and HS–PEG–SH-based NPs, which had varying amounts of HS–PEG–SH (as described in the Experimental section). Given the varied thiol content, we hypothesized that these formulations would lead to variable intracellular degradation rates and thus variable longevity of primary cells. The main scheme of NP synthesis is shown in Fig. 1, which shows the polymerization of PEGDA with HS–PEG–SH being capped primarily by CEA groups at its surface. As shown in Fig. 1, the polymer NP will primarily be comprised of PEGDA and HS–PEG–SH, with each NP type having a variable amount of HS–PEG–SH (either 0%, 10%, or 20% of PEGDA replaced with HS–PEG–SH by mole). The assumption of the reaction scheme is that carbons 1 and 4 react with other with CEA and HS–PEG–SH (PEGDA for the 0% NPs), respectively, leaving carbons 2 and 3 to react with other molecules such as PEGDA, HS–PEG–SH (not for the 0% NPs), methanol (hydrogen abstraction), CEA, or Cy5-maleimide (which are collectively represented as R groups). In reality, there will likely be many varieties of reactions between the molecules present to form the polymer NPs such that carbons 1–4 can react with many combinations of the aforementioned molecules, though reactions of PEGDA with itself or with HS–PEG–SH (for the 10% and 20% NPs), or with the solvent will be much more probable than reactions with CEA or Cy5-maleimide because of the larger relative number of moles of PEGDA and HS–PEG–SH since the reaction rates for vinyl carbons in photopolymerization are similar, though they may have slight effects from steric hindrance in the case of the Cy-5 maleimide.44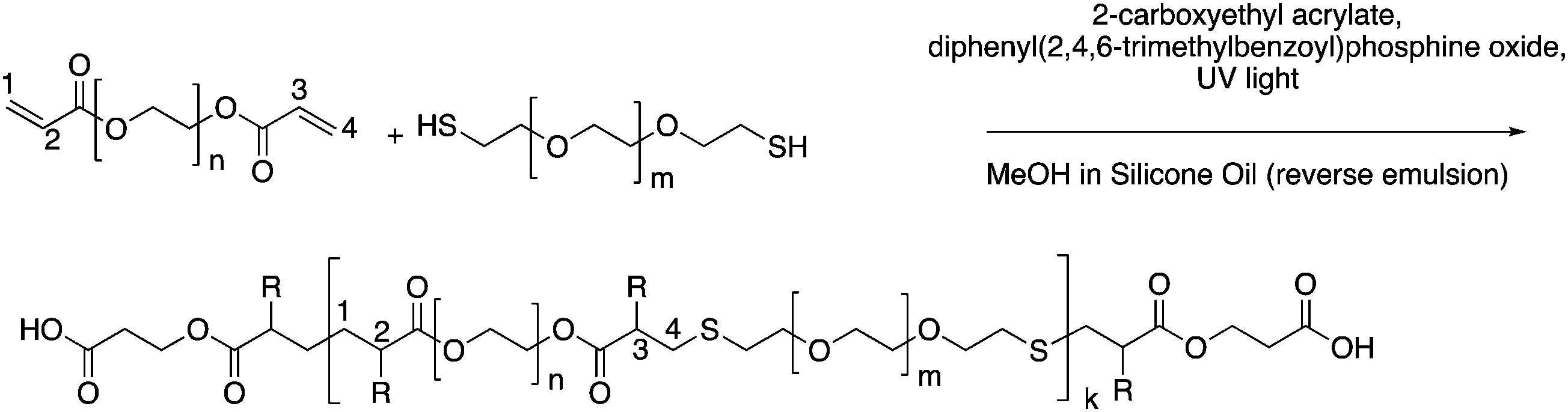 | ||
| Fig. 1 Representative reaction scheme of PEGDA monomer with HS–PEG–SH to form polymer NPs in a reverse emulsion where R groups could be PEGDA chains, HS–PEG–SH chains, hydrogen, CEA, or Cy5-maleimide. Bonds shown can also be via carbons 2 and 3 instead of 1 and 4, as well. | ||
Following the synthesis of the 0%, 10%, and 20% NPs, we characterized the NPs via DLS, cryo-SEM, zeta potential, and EDS to obtain NP sizes (DLS, cryo-SEM), overall surface charge (zeta potential), and relative sulfur content (XEDS). Similar to our previous syntheses of PEGDA-based NPs,18,41 the synthesized NPs were typically ∼500 nm in diameter, as measured via DLS (Fig. 2A–C) and confirmed via cryo-SEM (examples shown in Fig. 2D–F and in the ESI,† in Fig. S1). The z-average diameters of the three sets of NPs were 524.9 ± 121.6 nm, 467.8 ± 14.3 nm, and 584.3 ± 14.1 nm, respectively, and, as can be seen from the NP size distributions, the sizes and size ranges for the three NPs are similar, indicating that size will not significantly affect interactions with cells, nor their internalization. The NP sizes for the three formulations are also within the desired size range for macrophage phagocytosis (0.1–10 μm),45 which is critical for determination of the effect of variable degradation rates on primary cell longevity and can also affect immune response.45,46
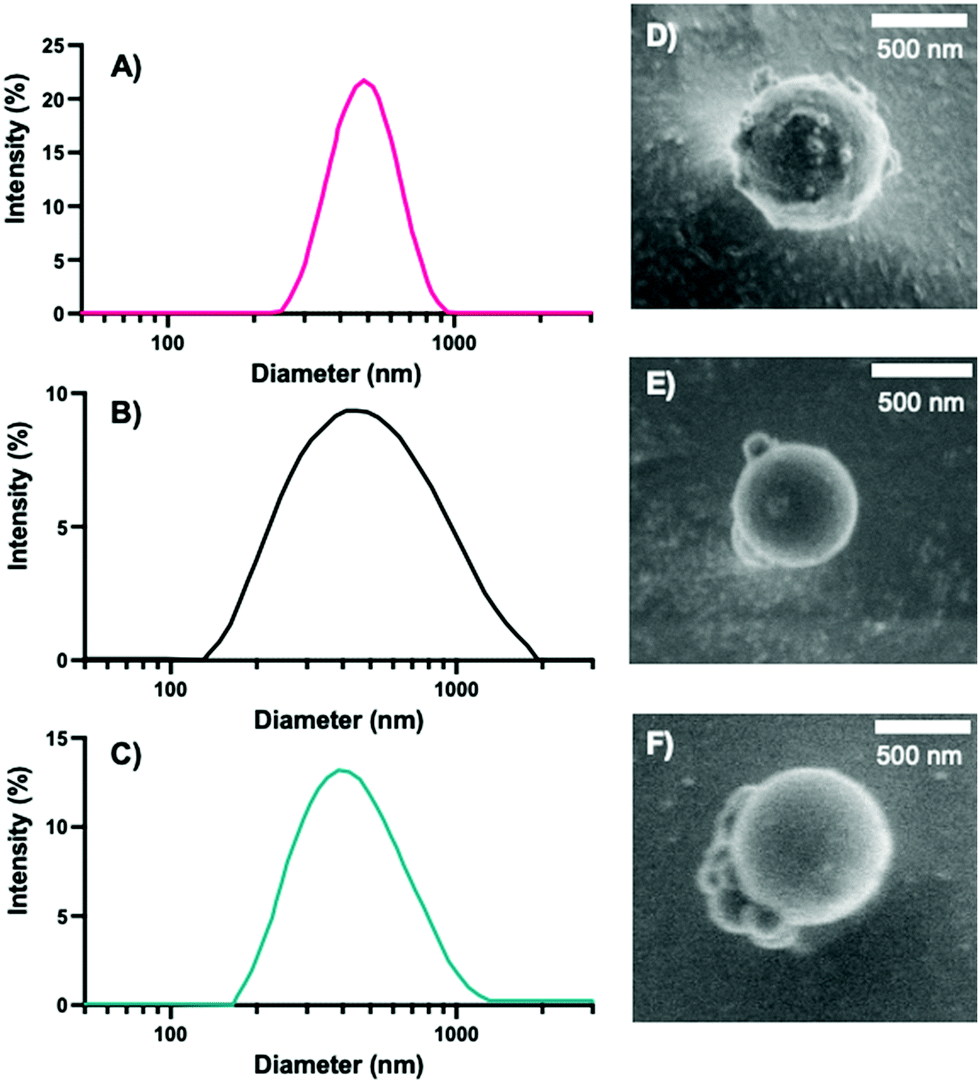 | ||
| Fig. 2 Intensity distribution of (A) 0%, (B) 10%, and (C) 20% NPs acquired from DLS. Representative cryo-SEM images of (D) 0%, (E) 10%, and (F) 20% NPs. | ||
The zeta potentials of the NPs, −11.1 ± 0.4, −10.4 ± 0.6, and −10.3 ± 0.4 mV, for the 0%, 10%, and 20% NPs, respectively (Fig. 3A), are all negative, as expected of NPs with CEA incorporated into their formulations. The slight differences in zeta potential can likely be attributed to increases in the relative number of thiol groups on the surface of the NPs in the 10% and 20% formulations, which will make them less negative overall, though the values are not statistically significant as determined via Tukey's multiple comparisons as part of a one-way ANOVA. The zeta potentials were also determined in PBS (Fig. S2, ESI†), were slightly negative, and not statistically significantly different, as was the case in NaCl. Regardless of solvent, the zeta potentials are all slightly negative and not different enough in magnitude to cause significant differences in uptake.26 Other advantages of their negative surface charge are the reduction of NP aggregation,47 increased NP uptake by phagocytic cells relative to neutral or positively charged NPs,13,48 and lower relative inflammatory potential relative to positively charged NPs.13,26,49
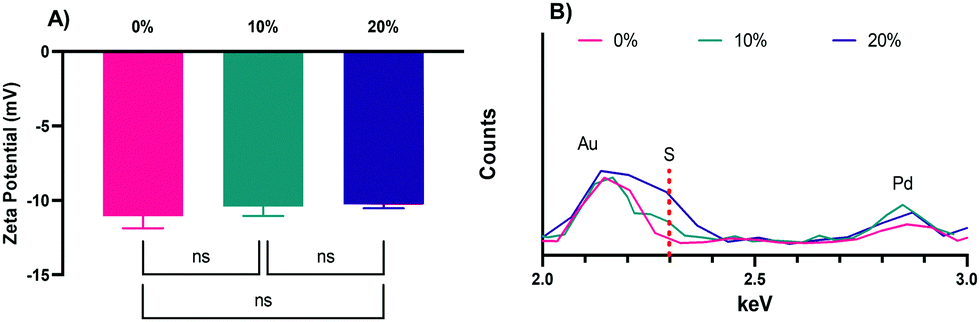 | ||
| Fig. 3 (A) Zeta potential measurements for the 0%, 10%, and 20% SH NPs. The graph shows the mean and SD from two independently synthesized samples measured 3 times each in 10 mM NaCl solution (6 total measurements, N = 2). (B) Overlapping XEDS spectra of the 0%, 10%, and 20% SH NPs to highlight differences in the detection of the key sulfur peak between the three NP formulations. Dashed red indicates main S peak at 2.307 keV. | ||
To confirm the incorporation of HS–PEG–SH into the 10% and 20% NPs and to confirm the absence of sulfur in the 0% NPs, XEDS was performed during SEM with results shown for the 0%, 10%, and 20% NPs, respectively, in Fig. 3B (the full spectra can be found in in Fig. S3, ESI†). As Fig. 3B shows, the peak for sulfur for the 20% NPs was the largest relative to the peaks for other elements present, though still relatively small because of the small amount of sulfur present in the NPs. The peak height of sulfur for the 10% NPs was between that of the 20% NPs and the 0% NPs, the latter of which was at baseline, indicating no discernable amount of sulfur present. This result confirmed that there was variable incorporation of HS–PEG–SH into the 10% and 20% NPs, as desired.
Nanoparticle degradation
Following the synthesis of the 0%, 10%, and 20% SH NPs, NPs were introduced to variable pH and glutathione (GSH) environments to determine their in vitro degradation rates. GSH is a reducing agent that functions to neutralize reactive oxygen species (examples shown in the ESI† in Scheme S1A) and can also function as a nucleophile (examples shown in Scheme S1B, ESI†). The pH buffers were chosen to mimic extracellular pH (∼7.0–7.4)50 and intracellular pH in a phagolysosome (∼4.5–5),51 which the NPs would encounter upon internalization by a cell such as a macrophage. The two pH environments are mimicked by PBS (mimicking extracellular, pH 7.4) and artificial lysosomal fluid (ALF, mimicking intracellular, pH 4.5). GSH is commonly found in the phagolysosome52 and thus was added to attempt to better mimic the lysosomal environment in the case of ALF (ALF + GSH medium) or as a point of comparison in the case of PBS (PBS + GSH medium). The results of the degradation of the three NP types in the four media (ALF, PBS, ALF + GSH, PBS + GSH) can be found in Fig. 4A (ALF), Fig. 4B (PBS), Fig. 4C (ALF + GSH), and Fig. 4D (PBS + GSH) (comparisons between conditions for a single NP type can be found in the ESI,† in Fig. S4–S6).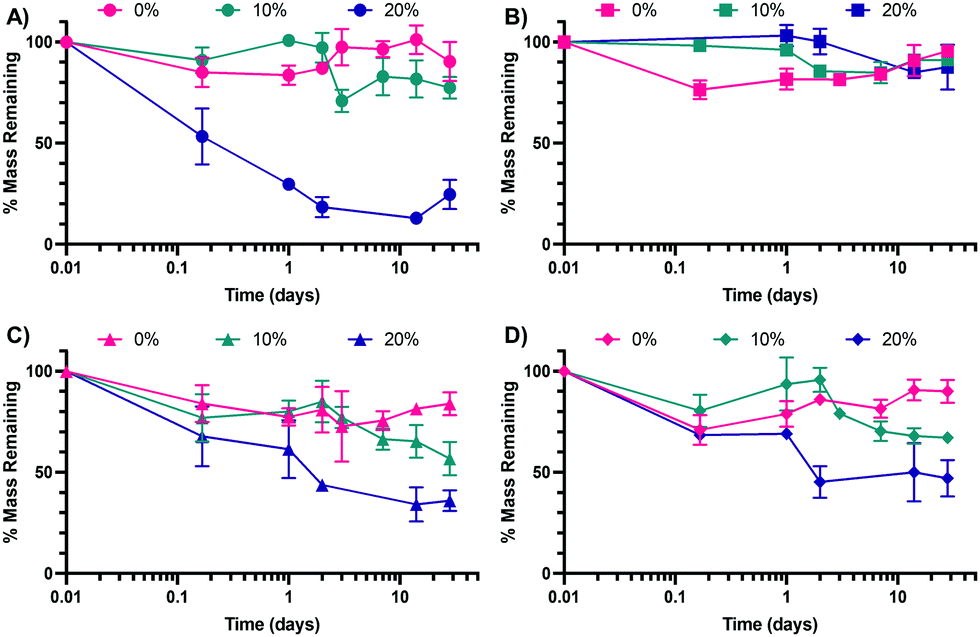 | ||
| Fig. 4 Degradation by mass of 0%, 10%, and 20% NPs in (A) ALF, (B) PBS, (C) ALF + GSH, and (D) PBS + GSH. Data points represent the mean and error bars represent the SEM (N = 3). Comparisons were made via a two-way ANOVA with Tukey's multiple comparisons. | ||
As can be seen from Fig. 4B, none of the NPs experience significant degradation in PBS, which mimics the extracellular environment, up to 28 days. This is consistent with our prior work studying the degradation of similar formulations of PEGDA NPs.41 The 0% NPs experience some initial degradation from 0 h to 4 h, though this could be the result of partial degradation of NPs prior to dispersion into PBS. Outside of the 4 h time point, the percentage of mass remaining is not statistically significantly different between the three NP types as determined via multiple comparisons as part of a two-way ANOVA. In contrast, for the ALF condition, the 20% NPs are statistically significantly different from the 10% NPs and the 0% NPs at all time points beyond 4 h. This would indicate that the 20% NPs are the most sensitive to acidic degradation, which, based on the variable chemistries, may indicate that its larger relative percentage of S–C bonds makes it more susceptible to acid-catalyzed degradation. Interestingly, the trends are less clear in the cases of the ALF–GSH and PBS–GSH conditions, for which the 20% NP degradation is not statistically significantly different from the degradation of the 0% or 10% NPs until the 2 days time point. Furthermore, the 10% NP degradation is not statistically significantly different from the degradation of the 0% NPs until the 28 days time point for the PBS–GSH condition or the 14 days time point for the ALF–GSH condition. The extent of degradation for the 20% NPs is less in the ALF–GSH condition and the PBS–GSH condition than in the ALF condition. We hypothesize that this may be the case because, despite the presence of the nucleophilic GSH (particularly in its deprotonated form, GS−), the greater concentration of protons in the ALF relative to PBS causes greater protonation of the GSH to keep it in its less nucleophilic, protonated form, which both utilizes the protons in the ALF solution and reduces the ability of the GSH to perform nucleophilic attack to degrade the NPs. In the case of the PBS + GSH condition, the concentration of protons is significantly lower and thus the proportion of GSH in its deprotonated form will commensurately be much greater than in the ALF + GSH condition. Accordingly, the GSH will more readily be able to perform nucleophilic attack than in the ALF–GSH case. The rates of degradation of the NPs are approximately equal for all three NPs in the ALF–GSH condition and the PBS–GSH condition, which may indicate that the greater activity of the GSH in the PBS–GSH condition counterbalances the relative lack of free protons, which seem to aid in the degradation of the 20% NPs in particular. Overall, our results suggest that variable thiol incorporation does result in variable degradation under relevant intracellular conditions. We expect the degradation rates of the 20% NPs to be the greatest when internalized by cells into low-pH (∼4.5–5) phagolysosomes, as our intracellular-mimicking degradation confirms that NP breakdown is highest in all of the conditions studied for the 20% NPs. This is expected since it can not only undergo acid-catalyzed ester hydrolysis but can also undergo nucleophilic attack at the sulfide (–S–C–) bonds, both of which are expected to occur in the phagolysosome.53,54
In addition to exploring the degradation rates of the three nanoparticle formulations, we also explored the possible mechanisms and products that could be formed from the degradation of the PEGDA- and HS–PEG–SH-based NPs. From the results of the degradation (Fig. S7–S20, ESI†), we were able to identify many products from the degradation of the 0%, 10%, and 20% NPs (Table S1, ESI†), which may influence the longevity of primary cells. Most of the products were PEGDA, HS–PEG–SH, and CEA or combinations therein and were indicative of hydrolysis being the primary breakdown mechanism. Ester hydrolysis was observed from the PEG-based products with losses of 54 MW relative to a base PEGDA or HS–PEG–SH molecule. This corresponds to the mass of the acrylate group (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CO, which would also have an –OH group or other nucleophile on the ketone) that has undergone nucleophilic attack and left the remainder of the molecule as the leaving group (which is subsequently protonated). There was also evidence of nucleophilic attack at the more ether-like carbons toward the ends of the HS–PEG–SH as evidenced by mass losses of 68 MW. This decrease corresponds to losses of HS–CH2–CH2– groups from either end of the HS–PEG–SH followed by protonation of the product PEG. The relative lower abundance of these patterns indicates that hydrolysis is likely the primary mechanism, but the availability of both mechanisms as well as sulfur-based leaving groups allows for more rapid degradation of the 10% and 20% NPs relative to the 0% particles in non-PBS (only) environments. Over the 14 days study, the degradation products of the 10% NPs were very similar to those from the 20% NPs, but generally were generated at a later time point (Fig. S20, ESI†). Unsurprisingly, the resultant spectra of 0% NPs, unlike the 10% and 20% NPs, did not contain peaks corresponding to the HS–PEG–SH or its derivatives and thus lacked peaks at m/z of 320, 521, 389, and more.
CH–CO, which would also have an –OH group or other nucleophile on the ketone) that has undergone nucleophilic attack and left the remainder of the molecule as the leaving group (which is subsequently protonated). There was also evidence of nucleophilic attack at the more ether-like carbons toward the ends of the HS–PEG–SH as evidenced by mass losses of 68 MW. This decrease corresponds to losses of HS–CH2–CH2– groups from either end of the HS–PEG–SH followed by protonation of the product PEG. The relative lower abundance of these patterns indicates that hydrolysis is likely the primary mechanism, but the availability of both mechanisms as well as sulfur-based leaving groups allows for more rapid degradation of the 10% and 20% NPs relative to the 0% particles in non-PBS (only) environments. Over the 14 days study, the degradation products of the 10% NPs were very similar to those from the 20% NPs, but generally were generated at a later time point (Fig. S20, ESI†). Unsurprisingly, the resultant spectra of 0% NPs, unlike the 10% and 20% NPs, did not contain peaks corresponding to the HS–PEG–SH or its derivatives and thus lacked peaks at m/z of 320, 521, 389, and more.
Ex vivo primary macrophage longevity is dependent on NP degradation rate
To test our hypotheses regarding whether NP degradation rate impacts the ex vivo survival of primary macrophages, BMMs were dosed with 100 μg mL−1, 50 μg mL−1, and 10 μg mL−1 of the 0%, 10%, and 20% NPs and cell counts were continuously monitored following treatment with the different NP formulations (Fig. 5A and Fig. S5, ESI†). In all of the tested formulations, NP treatment enhanced the survival of ex vivo BMMs in a concentration-dependent manner. This trend agrees with results from our previous study with other PEGDA-based NPs,18 even with the notably different additions to the PEGDA NP compositions. Treatment of NPs to BMMs at a concentration of 100 μg mL−1 resulted in statistically significantly higher % viability than the untreated (UT) cells for the three tested NP formulations as early as 72 h following treatment (p < 0.05 using Tukey's multiple comparisons tests as part of a two-way ANOVA). The 0%, 10%, and 20% NP formulations resulted in differences in BMM survival profiles. Overall, rapidly degrading 20% NPs resulted in the highest survival levels over two weeks when compared to the other formulations at the same dosage conditions, which was the case for 100 μg mL−1, 50 μg mL−1, and 10 μg mL−1 dosage concentrations. The general pattern of the BMM survival shows greater longevity associated with treatment with 20% NPs followed by treatment with the 10% and 0% NPs, respectively. This suggests that NP degradation rate plays a major role in regulating the survival of the phagocytosing cell. This behavior was evident from treatment with 100 μg mL−1 and 50 μg mL−1 of NPs, but not for the 10 μg mL−1 dosage (Fig. S21, ESI†), which indicates that there is likely a critical threshold NP dosage required before any effects of internalization and degradation of NPs on BMM longevity are observed.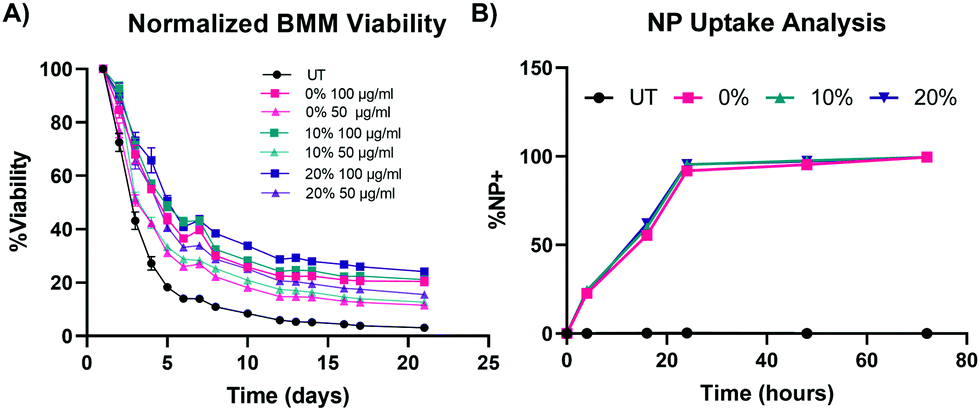 | ||
| Fig. 5 Effect of NP degradation rate on macrophage survival. (A) Normalized cell counts over time of BMMs treated with 100 μg mL−1 and 50 μg mL−1 of 0%, 10%, and 20% NPs (N = 8). (B) Kinetic profiles of BMM uptake of 0%, 10%, and 20% NP formulations at a concentration of 50 μg mL−1 (N = 3). Data points represent the mean and error bars represent the SEM; error bars are too small to be visible for some data points. | ||
The results of concentration-dependent cell viability point to the strong effect of the amount of internalized NPs on macrophage survival. Therefore, we investigated whether the enhanced survival following treatment with rapidly degrading 20% NPs relative to its slower degrading counterparts stems from differential uptake across the three NP formulations, as opposed to degradation rate. NP uptake was kinetically quantified via flow cytometric analysis of % Cy5+ populations (representative flow cytometry gating analysis in Fig. S22, ESI†). BMM uptake of 0%, 10%, and 20% NPs was identical, which was expected since the particles have effectively the same size and charge; after 24 h, more than 90% of the cells were determined to be NP+ following treatment with 0%, 10%, and 20% NPs (Fig. 5B). More than 95% and 99% of BMMs in all the NP groups were NP+ at 48 h and 72 h after NP dosage, respectively. The rapid and homogenous levels of NP internalization for all of the tested formulations indicate that the differential macrophage survival is unlikely to be occurring due to variations in NP uptake between the three NP types. Therefore, other NP–cell interactions are possibly responsible for the enhanced macrophage survival caused by the internalization of rapidly degrading NPs.
After determining that the observed effects of longevity are likely not a result of differences in NP uptake, we sought to further explore the effects of the degradable NPs on the BMMs. We began this exploration by investigating the effect of NP degradation rate on pro-apoptotic effectors. We have previously demonstrated that NP internalization by BMMs enhances survival through the upregulation of anti-apoptotic Bcl-2 family genes and proteins,18 which have been shown to suppress caspase-dependent apoptotic pathways.55 Caspase-3/7 activity was measured in BMMs treated with 100 μg mL−1, 50 μg mL−1, and 10 μg mL−1 of 0%, 10%, and 20% NPs and caspase-3/7 activity was normalized to the corresponding cell count in each group (Fig. 6). Unsurprisingly, untreated BMMs exhibited the highest levels of active caspases-3/7, which indicates the strong apoptotic potential of ex vivo macrophages.56 Active caspase-3/7 levels were statistically significantly reduced following dosage with 0%, 10%, and 20% NP formulations at all of the tested concentrations (p < 0.0001 for all the NP groups compared to untreated BMMs using Tukey's multiple comparisons tests as part of a two-way ANOVA). Suppression of pro-apoptotic caspase-3/7 expression following NP treatment occurred in a concentration-dependent manner, where treatment with 100 μg mL−1 of 0%, 10%, and 20% NPs resulted in the greatest reduction of active caspase-3/7 levels while the treatments at 10 μg mL−1 concentrations resulted in the least. Surprisingly, active caspase-3/7 levels did not statistically significantly differ among the 0%, 10%, and 20% NP formulations at this 72 h timepoint when dosed at the same concentration (p > 0.05 using Tukey's multiple comparisons tests as part of a two-way ANOVA). This result contrasts with the cell viability data (Fig. 5A), which showed significant differences in % viability between the 0%, 10%, and 20% NP groups, where the NPs with the highest rates of degradation resulted in the greatest cell survival. The disagreement between cell viability data and suppression of pro-apoptotic signaling for the 0%, 10%, and 20% NP groups indicates the possible involvement of alternate pathways that promote cell survival independent of those relying on caspase-3/7 suppression, which may possibly include cell activation markers.
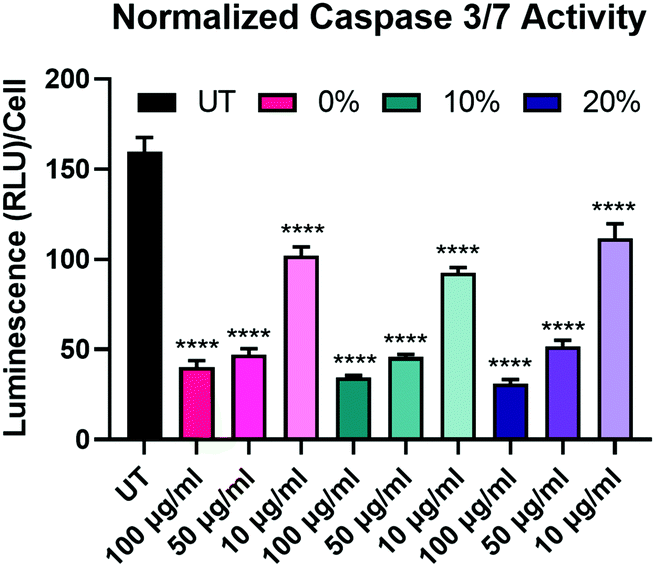 | ||
| Fig. 6 Cell count-normalized caspase-3/7 activity in BMMs treated with 100 μg mL−1, 50 μg mL−1, and 10 μg mL−1 of 0%, 10%, and 20% NPs 72 h following NP treatment. ****p < 0.0001 comparison to UT using Tukey's multiple comparisons tests as part of a one-way ANOVA (N = 3). Error bars represent SEM. | ||
We next assayed BMMs 2 weeks following NP treatment with 0%, 10%, and 20% NPs to investigate the effect of NP dosing on macrophage stimuli responsiveness. The effects of 24 h pulsing with 25 ng mL−1 LPS of untreated and NP-treated BMMs on IL-6 and TNF-α inflammatory cytokine secretion was monitored via ELISA (Fig. 7A and B). Treatment with 0%, 10%, and 20% NPs, especially at the 100 μg mL−1 concentration, resulted in notably higher IL-6 and TNF-α, though only TNF-α secretions for 0% NPs were statistically significantly higher than those of untreated BMMs. Nonetheless, all of the tested conditions showed responsiveness to LPS stimulation as evident by the detectable levels of inflammatory cytokines compared to undetectable secretions in the unstimulated counterparts. This indicated that surviving BMMs at two weeks were still stimuli responsive and presented with functional phenotypes that were enhanced over the UT controls.
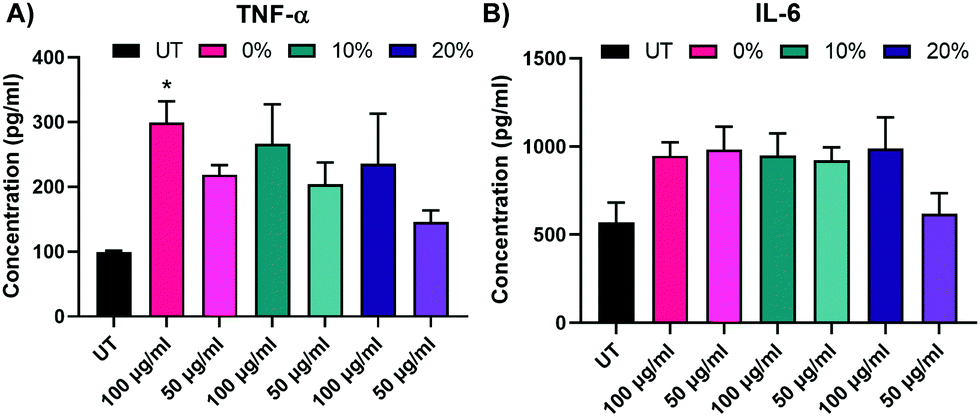 | ||
| Fig. 7 TNF-α and IL-6 concentrations of BMM supernatants two weeks following treatment with 0%, 10%, and 20% NP formulations after a 24 h LPS challenge. *p < 0.05 comparison to UT using Tukey's multiple comparisons tests as part of a one-way ANOVA (N = 3). Error bars represent SEM. | ||
NP degradation rate promotes the activation of BMMs into an M1-like state
We next probed the effect of degradable NPs on cellular response by investigating whether NP degradation rate plays a role in the activation of macrophages. BMMs were dosed with 100 μg mL−1 of the 0%, 10%, and 20% NP formulations and flow cytometric analysis of macrophage activation markers of the M1 and M2 paradigm was executed on BMMs 24 h and 72 h following treatment. Median fluorescence intensity (MFI) as a measure of activation marker expression was recorded (Representative flow cytometry gating analysis in Fig. S22, ESI†). Relative to untreated BMMs, all three NP formulations sharply increased the expression of CD86 costimulatory molecule (p < 0.0001 using Tukey's multiple comparisons tests as part of a one-way ANOVA) at both the 24 h and the 72 h timepoints (Fig. 8A and B), indicating potent activation of BMMs following treatment with 0%, 10%, and 20% NP formulations. The slowly degrading 0% NPs resulted in the smallest increase in CD86 expression, while the 10% and 20% NPs with faster degradation resulted in higher expression at the 24 h and 72 h timepoints. The NP-induced upregulation of CD86 is accompanied by a statistically significant increase in the expression major histocompatibility complex class II (MHCII) as early as 24 h, which is even further augmented at 72 h following NP treatment (Fig. 8C and D). Similar to CD86 expression, the upregulation of MHCII was observed to be dependent on NP degradation rate, where 10% and 20% NPs were superior to 0% NPs. Overall, the 72 h results showed dramatic increases in the two M1 activation markers, indicating the strong kinetic effects of degradable NPs on macrophage activation, which correspond to notable breakdown from in vitro degradation studies. It is noteworthy to mention that significant stimulatory effects with 0% NPs contrast with our previous studies of macrophage phenotypical changes in response to internalization of NPs formulated with PEGDA- and CEA-only. This is likely due to changes in NP formulations, namely the inclusion of HDDMA co-monomer and increase in the amount of CEA used, which are hypothesized to account for the differences between the two formulations. Interestingly, CD80 expression was mostly unchanged 24 h following NP treatment and was suppressed at 72 h (Fig. S23, ESI†). This could be in part due to the naturally lower abundance and the sluggish response of CD80 relative to CD86.57 CD86, along with other activation markers, has been shown to be stimulated in dendritic cells upon interactions with polymeric particles of varying extents of degradation;58 however, it is unclear whether the degraded particles affect the survival of the primary dendritic cells.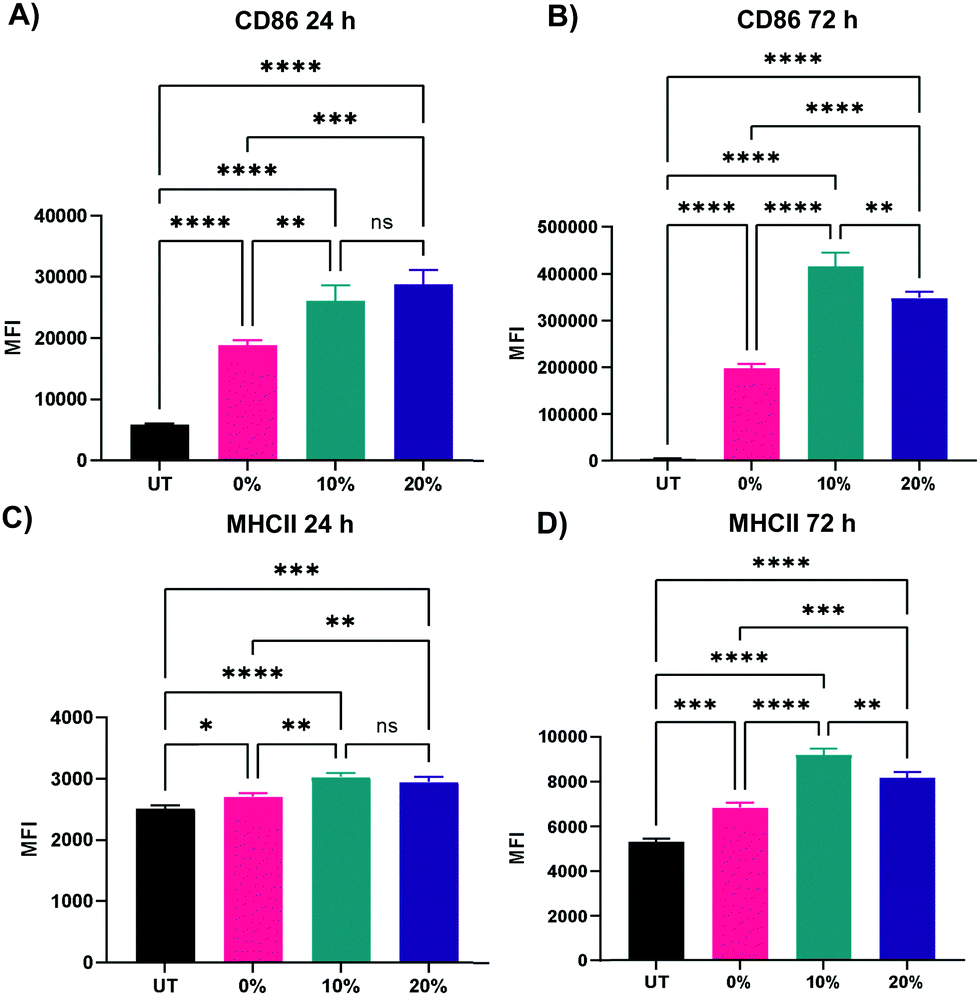 | ||
| Fig. 8 Expression of representative M1 activation markers of BMMs treated with 100 μg mL−1 of 0%, 10%, and 20% NPs (A) 24 h CD86 expression (B) 72 h CD86 expression (C) 24 h MHCII expression (D) 72 h MHCII expression. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns = not significant using Tukey's multiple comparisons tests as part of a one-way ANOVA (N = 3). Error bars represent SEM. | ||
In addition to the upregulation of M1 activation markers in BMMs following the treatment with the three NP formulations, an M2 marker, CD206, was significantly downregulated at both 24 and 72 h following treatment with 0%, 10%, and 20% NPs (Fig. 9A and B), indicating a potent activation towards an M1 phenotypical state. Similar to patterns observed with CD86 and MHCII markers, NP degradation rate played a crucial role in the downregulation of CD206. At the 24 h analysis timepoint, rapidly degrading 20% NPs resulted in the greatest suppression of CD206 expression relative to untreated BMMs (p < 0.0001 using Tukey's multiple comparisons tests as part of a one-way ANOVA). The suppression of CD206 expression by 10% NPs was the second highest followed by that of the 0% NPs (p < 0.001 and p < 0.01, respectively using Tukey's multiple comparisons tests as part of a one-way ANOVA). These results were less pronounced at the 72 h timepoint, with the 20% NPs holding the pattern of the sharpest decrease in CD206 expression relative to untreated BMMs (p < 0.01), while 0% and 10% NPs were statistically insignificant relative to untreated BMMs (p > 0.05), indicating that rapidly degrading NPs play a major role in controlling the macrophage phenotype. This also potentially explains the enhanced primary macrophage survival following internalization of rapidly degrading NPs.
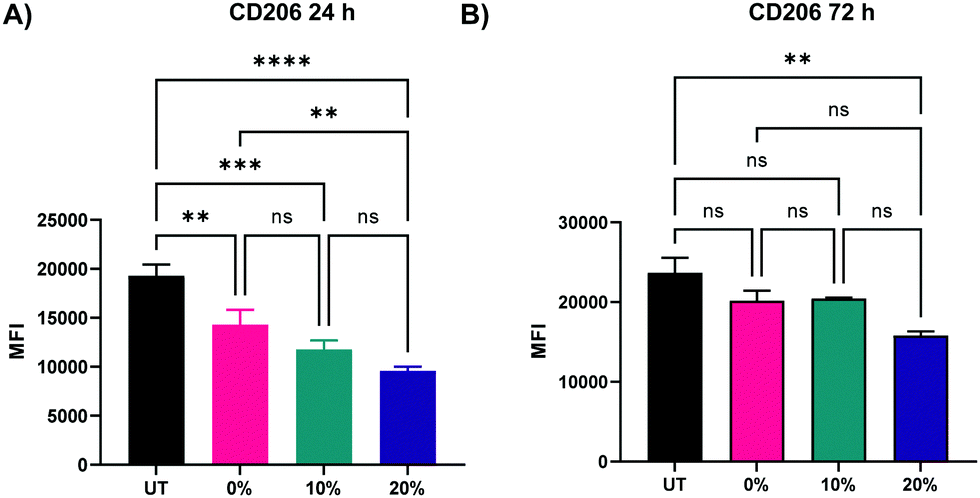 | ||
| Fig. 9 Expression of CD206 M2 activation marker of BMMs treated with 100 μg mL−1 of 0%, 10%, and 20% NPs (A) 24 h (B) 72 h following treatment. **p < 0.01, ***p < 0.001, ****p < 0.0001, ns = not significant using Tukey's multiple comparisons tests as part of a one-way ANOVA (N = 3). Error bars represent SEM. | ||
Interestingly, IL-6 and TNF-α inflammatory cytokines were not present in supernatants of untreated and NP-treated BMMs within 72 h of NP dosing, with cytokine concentrations below the detectable limit via ELISA analysis (data not shown). While the absence of secretions may be surprising given the potent stimulation of CD86 and MHCII markers, the lack of potent toll-like receptor (TLR) agonists and pathogen-associated molecular patterns (PAMPs) in the NP formulations, which are often required for a robust secretory response,59 may explain the undetectable cytokine levels. In addition, while detectable, IL-10 levels in supernatants of untreated and NP-treated BMMs were statistically indistinguishable 72 h following NP treatment (Fig. S24, ESI†) (p > 0.05 using Tukey's multiple comparisons tests as part of a one-way ANOVA), indicating the inability of 0%, 10%, and 20% NPs to stimulate either pro- or anti-inflammatory cytokine secretions. These results are in agreement with low inflammatory cytokine secretion profiles of macrophages upon interactions with PEG-based materials.17
The results of immunostimulatory behavior stemming from NP degradation rate present a contrast to other PLGA-based degradable particles, which caused the downregulation of M1 markers including both CD86 and MHCII and was attributed to immunomodulatory acidic degradation products, namely lactic acid.60 On the other hand, degradable poly(beta-amino-ester) (PBAE) particles provide supporting evidence of M1-like stimulation in dendritic cells, but does not point to any survival effects as a result of the degradable particle-induced stimulation.58 These reports of enhanced immune stimulation may also be because of intracellular processing of specific degradation products of the particles. Therefore, the degradation products of the 0%, 10%, and 20% NPs could play an instrumental intracellular role in causing the activation of macrophages as seen by the potent upregulation of CD86 and MHCII M1 markers and the downregulation of CD206 M2 marker. Further investigations are required to understand the direct impact, if any, of NP degradation products on inherent adjuvanticity of these platforms in driving the activation state of macrophages and its link to cell survival. The direct effect of NP degradation on cell survival is contrary to our initial hypothesis that slowly degrading NPs will enable sustained effects relative to rapidly degrading NPs, especially when compared to slowly degrading NPs for antigen delivery and cargo release applications;30 this likely is attributed to the stimulation of the intracellular degradation products driving an M1 phenotype.
Lysosomal activity is enhanced with degradable NPs
Lysosomal tracking was utilized to gain insight on the intracellular trafficking of degradable NPs and to determine whether NP degradation rate affects intracellular NP processing. Imaging analysis revealed high intensity LysoBrite™ Green activity in all NP-treated BMMs as compared to their untreated counterparts, which is indicative of NP trafficking in late lysosomal compartments (Fig. 10), especially given the strong overlap between LysoBrite™ Green and NP fluorescence signals. From LysoBrite™ Green fluorescence, lysosomal activity was strongest in BMMs treated with 20% NPs followed by activity in the 10%, 0%, and untreated conditions, respectively, as evident by the bright green fluorescence. As a result, imaging showed drastically increased lysosomal activity in BMMs dosed with rapidly degradable NPs as compared to those dosed with slowly degrading NPs or untreated conditions. This observation is expected, as degradation of phagocytosed materials occurs following the fusion of the phagosome with the lysosome.61 We have previously shown that NP internalization stimulates the expression of late endosomal/lysosomal adaptor, MAPK and mTOR activator (LAMTOR) genes and proteins,18 which have been linked to survival.62,63 The enhanced lysosomal activity may potentially trigger increased expression of lysosomal signaling proteins, which have been reported to contribute to cell survival. Administration of biodegradable NPs with acidic byproducts have been shown to restore lysosomal acidity and degradative capacity,23,24 which may further contribute to cell stimulation. Potent activation of lysosomal signaling by degradable NPs may explain the resulting enhanced survival and could provide insight to possible links to macrophage activation evident by the upregulation of CD86 and MHCII and the subsequent enhancement of antigen presentation and interface with adaptive immune cells, which has been shown in dendritic cells64 and could extend to macrophage behavior upon phagocytosis and processing of NPs.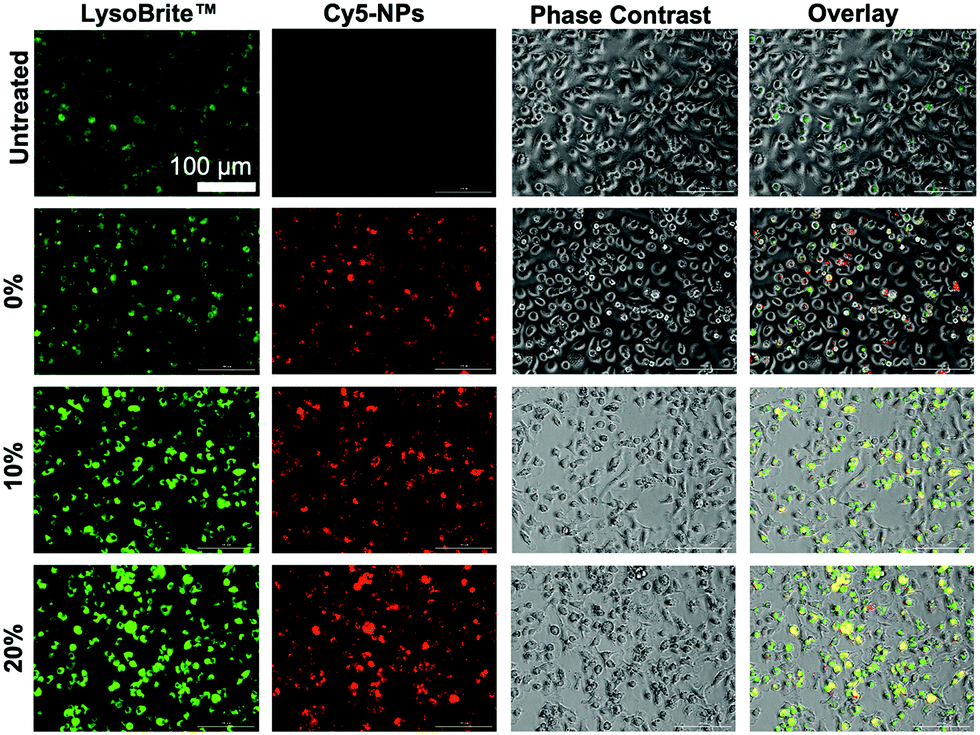 | ||
| Fig. 10 Lysosomal tracking with LysoBrite™ Green and imaging at 20× magnification of BMMs treated with 100 μg mL−1 of 0%, 10%, and 20% NPs 72 h NP treatment. Images are representative of two experiments. | ||
Overall, based on mass-based degradation profiles of 0%, 10%, and 20% NPs, the 20% formulation experienced the greatest levels of degradation in acidic and reducing environments that simulate lysosomal fluids, with drastic mass loss occurring as early as 24 h following incubation. These results correspond to improved cell survival and enhanced expression of activation markers, likely as a result of increased lysosomal stimulation. The initial enhancement from rapidly degrading 20% NPs result in cell survival beyond 2 weeks, whereas the 0% and 10% NPs of slower rates of degradation are associated with lower lysosomal involvement and enhancement of activation signaling. Therefore, while pro-survival cues may be present from all treatment conditions, including those of slowly degrading NPs, they may not be sufficient to overcome the initial boost from the rapidly degrading 20% NPs. Extended phagocyte viability following phagocytosis has often been observed following internalization of bacteria, where cells become highly activated and M1-polarized through TLR signalling65 and potent Nuclear Factor (NF)-κB activation,66 which results in the production of inflammatory cytokines and soluble factors contributing to polarization. Autophagy signaling may also be responsible for prolonging phagocyte survival, which was the case for internalization of apoptotic cells and survival resulting from interactions with mitogen-activated protein kinases (MAPK) pathways.67 Phagocyte survival associated with autophagy or TLR signaling is potentially initiated at the phagocytic synapse; likely the M1 activation stemming from the acidic degradation components serves to synergize with pro-survival signaling to enhance viability.68 Thus, the polarization observed following PEGDA NP internalization and resultant M1 polarization is expected to enhance pro-survival signaling to directly influence cell fate.
To gain deeper insight on these NP degradation-induced macrophage longevity profiles, potentially immunomodulatory NP degradation products, such as those listed in Table S1 (ESI†) must be investigated in future studies. Immunostimulatory HS–PEG–SH-based degradable formulations are in contrast to immunosuppressive properties of other particle chemistries such as PLGA, where lactic acid degradation products are hypothesized to suppress M1 phenotypical changes,60 but are in agreement with M1-like polarization as a result of degradable PBAE particles.58 Therefore, NP chemistry and the specific nature of NP depredation products may play a critical role in macrophage activation and the resultant pro-survival mechanisms. Our work draws attention to this important influence of NP-induced phagocyte longevity enhancement and the link to various physiochemical properties that requires future evaluations.
Conclusion
In this study, we report that degradation rate and resultant degradation products of PEG-based NPs are critical parameters for tuning the survival of ex vivo primary macrophages. NPs with higher degradation rates show dramatic effects in stimulating M1-like macrophage activation markers in the absence of inflammatory cytokine secretions, corresponding to in vitro evaluations of mass-based NP degradation. Lysosomal stimulation is dramatically enhanced in the presence of rapidly degrading NPs relative to their slowly degrading counterparts. These phenomena are hypothesized to be caused by the increased presence of degradation products in rapidly degrading NP groups, which have been recently shown in other works to drive phenotypical changes in innate immune cells. Further studies are needed to characterize the independent effects of PEG-based degradation products on primary macrophage longevity and activation state. In addition, different degradable chemistries must be compared to better understand the impact of downstream intracellular NP processing events on cell survival. This work opens the door to future investigations of physiochemical properties of NP-based strategies aimed at tuning the survival and function of macrophages and phagocytes for therapeutic applications and models.Funding information
Research reported in this publication was supported by the National Institutes of Health and the State of Delaware under Award Number P20GM103446 and P20GM104316, as well as a Research Starter Grant in Pharmaceutics from the PhRMA Foundation, and a University of Delaware Research Foundation Award. Z. S. S. was supported by a T32GM008550 training grant. Thermo Scientific™ Apreo VS SEM microscope equipment was acquired with a shared instrumentation grant (S10 OD025165) and access was supported by the NIH-NIGMS (P20 GM103446), the NSF (IIA-1301765) and the State of Delaware. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
TOC figure was created using biorender.com. LC-MS was performed by Dr P. Asare-Okai. Cryo-SEM was performed with the assistance of D. Powell.References
- R. Sridharan, A. R. Cameron, D. J. Kelly, C. J. Kearney and F. J. O’Brien, Mater. Today, 2015, 18, 313–325 CrossRef.
- C. E. Witherel, K. Sao, B. K. Brisson, B. Han, S. W. Volk, R. J. Petrie, L. Han and K. L. Spiller, Biomaterials, 2021, 269, 120667 CrossRef PubMed.
- K. L. Wofford, D. K. Cullen and K. L. Spiller, J. Biomed. Mater. Res., Part A, 2019, 107, 1213–1224 CrossRef PubMed.
- B. M. Jarai, Z. Stillman, K. Bomb, A. M. Kloxin and C. A. Fromen, ACS Biomater. Sci. Eng., 2021, 7(5), 1742–1764 CrossRef PubMed.
- E. C. Wayne, C. Long, M. J. Haney, E. V. Batrakova, T. M. Leisner, L. V. Parise and A. V. Kabanov, Adv. Sci., 2019, 6, 1900582 CrossRef.
- N. Shobaki, Y. Sato, Y. Suzuki, N. Okabe and H. Harashima, J. Controlled Release, 2020, 325, 235–248 CrossRef.
- B. M. Jarai, Z. Stillman, L. Attia, G. E. Decker, E. D. Bloch and C. A. Fromen, ACS Appl. Mater. Interfaces, 2020, 12, 38989–39004 CrossRef.
- S. Soares, J. Sousa, A. Pais and C. Vitorino, Front. Chem., 2018, 6, 360 CrossRef PubMed.
- B. M. Jarai, E. L. Kolewe, Z. S. Stillman, N. Raman and C. A. Fromen, in Nanoparticles for Biomedical Applications, ed. E. J. Chung, L. Leon and C. Rinaldi, Elsevier, 2020, pp. 303–324 DOI:10.1016/B978-0-12-816662-8.00018-7.
- J. W. Hickey, J. L. Santos, J.-M. Williford and H.-Q. Mao, J. Controlled Release, 2015, 219, 536–547 CrossRef PubMed.
- J.-M. Williford, J. L. Santos, R. Shyam and H.-Q. Mao, Biomater. Sci., 2015, 3, 894–907 RSC.
- D. Guo, G. Xie and J. Luo, J. Phys. D: Appl. Phys., 2013, 47, 013001 CrossRef.
- E. Fröhlich, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef PubMed.
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602–2663 CrossRef.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef.
- J. A. Champion and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4930 CrossRef PubMed.
- R. A. Roberts, T. Shen, I. C. Allen, W. Hasan, J. M. DeSimone and J. P. Y. Ting, PLoS One, 2013, 8, e62115 CrossRef.
- B. M. Jarai and C. A. Fromen, bioRxiv, 2021 DOI:10.1101/2021.04.22.440822.
- P. Zhou, X.-L. Yang, X.-G. Wang, B. Hu, L. Zhang, W. Zhang, H.-R. Si, Y. Zhu, B. Li, C.-L. Huang, H.-D. Chen, J. Chen, Y. Luo, H. Guo, R.-D. Jiang, M.-Q. Liu, Y. Chen, X.-R. Shen, X. Wang, X.-S. Zheng, K. Zhao, Q.-J. Chen, F. Deng, L.-L. Liu, B. Yan, F.-X. Zhan, Y.-Y. Wang, G.-F. Xiao and Z.-L. Shi, Nature, 2020, 579, 270–273 CrossRef.
- A. Parihar, T. D. Eubank and A. I. Doseff, J. Innate Immun., 2010, 2, 204–215 CrossRef PubMed.
- L. M. Stuart and R. A. B. Ezekowitz, Immunity, 2005, 22, 539–550 CrossRef.
- W. K. E. Ip, A. Sokolovska, G. M. Charriere, L. Boyer, S. Dejardin, M. P. Cappillino, L. M. Yantosca, K. Takahashi, K. J. Moore, A. Lacy-Hulbert and L. M. Stuart, J. Immunol., 2010, 184, 7071–7081 CrossRef PubMed.
- G. C. Baltazar, S. Guha, W. Lu, J. Lim, K. Boesze-Battaglia, A. M. Laties, P. Tyagi, U. B. Kompella and C. H. Mitchell, PLoS One, 2012, 7, e49635 CrossRef CAS PubMed.
- J. Zeng, A. Martin, X. Han, O. S. Shirihai and M. W. Grinstaff, Ind. Eng. Chem. Res., 2019, 58, 13910–13917 CrossRef CAS.
- C. A. Fromen, T. B. Rahhal, G. R. Robbins, M. P. Kai, T. W. Shen, J. C. Luft and J. M. DeSimone, Nanomedicine, 2016, 12, 677–687 CrossRef CAS.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657–3666 CrossRef CAS.
- R. Nicolete, D. F. d. Santos and L. H. Faccioli, Int. Immunopharmacol., 2011, 11, 1557–1563 CrossRef CAS PubMed.
- C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. W. Chan, J. Am. Chem. Soc., 2012, 134, 2139–2147 CrossRef CAS.
- S. L. Demento, W. Cui, J. M. Criscione, E. Stern, J. Tulipan, S. M. Kaech and T. M. Fahmy, Biomaterials, 2012, 33, 4957–4964 CrossRef CAS.
- N. Chen, M. M. Johnson, M. A. Collier, M. D. Gallovic, E. M. Bachelder and K. M. Ainslie, J. Controlled Release, 2018, 273, 147–159 CrossRef CAS PubMed.
- X. Xie, J. Wang, L. Zhang, S. Zeng, X. Su and Q. Chen, Bioconjugate Chem., 2021, 32(5), 1008–1016 CrossRef CAS.
- J. A. Hamilton, J. Leukocyte Biol., 2003, 73, 702–712 CrossRef CAS PubMed.
- B. S. Zolnik, A. González-Fernández, N. Sadrieh and M. A. Dobrovolskaia, Endocrinology, 2010, 151, 458–465 CrossRef CAS PubMed.
- S. Su and P. M. Kang, Nanomaterials, 2020, 10(4), 656 CrossRef CAS PubMed.
- S. Rezvantalab, N. I. Drude, M. K. Moraveji, N. Güvener, E. K. Koons, Y. Shi, T. Lammers and F. Kiessling, Front. Pharmacol., 2018, 9, 1260 CrossRef CAS.
- T. M. Fahmy, S. L. Demento, M. J. Caplan, I. Mellman and W. M. Saltzman, Nanomedicine, 2008, 3, 343–355 CrossRef CAS.
- J. Da Silva, S. Jesus, N. Bernardi, M. Colaço and O. Borges, Front. Bioeng. Biotechnol., 2019, 7, 137 CrossRef PubMed.
- B. K. Lee, Y. Yun and K. Park, Adv. Drug Delivery Rev., 2016, 107, 176–191 CrossRef CAS.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS PubMed.
- E. Malikmammadov, T. E. Tanir, A. Kiziltay, V. Hasirci and N. Hasirci, J. Biomater. Sci., Polym. Ed., 2018, 29, 863–893 CrossRef CAS.
- Z. Stillman, B. M. Jarai, N. Raman, P. Patel and C. A. Fromen, Polym. Chem., 2020, 11, 568–580 RSC.
- A. Pelfrêne, M. R. Cave, J. Wragg and F. Douay, Int. J. Environ. Res. Public Health, 2017, 14, 112 CrossRef PubMed.
- X. Zhang, R. Goncalves and D. M. Mosser, Curr. Protoc. Immunol., 2008, ch. 14, p. 14.11 Search PubMed.
- D. L. Kurdikar and N. A. Peppas, Polymer, 1994, 35, 1004–1011 CrossRef CAS.
- H. Chikaura, Y. Nakashima, Y. Fujiwara, Y. Komohara, M. Takeya and Y. Nakanishi, Biosurf. Biotribol., 2016, 2, 18–25 CrossRef.
- M. O. Oyewumi, A. Kumar and Z. Cui, Expert Rev. Vaccines, 2010, 9, 1095–1107 CrossRef CAS PubMed.
- E. Joseph and G. Singhvi, in Nanomaterials for Drug Delivery and Therapy, ed. A. M. Grumezescu, William Andrew Publishing, 2019, pp. 91–116 DOI:10.1016/B978-0-12-816505-8.00007-2.
- O. Lunov, T. Syrovets, C. Loos, J. Beil, M. Delacher, K. Tron, G. U. Nienhaus, A. Musyanovych, V. Mailänder, K. Landfester and T. Simmet, ACS Nano, 2011, 5, 1657–1669 CrossRef CAS.
- M. A. Dobrovolskaia and S. E. McNeil, Nat. Nanotechnol., 2007, 2, 469–478 CrossRef CAS PubMed.
- J. R. Griffiths, Br. J. Cancer, 1991, 64, 425–427 CrossRef CAS PubMed.
- Y. Ishida, S. Nayak, J. A. Mindell and M. Grabe, J. Gen. Physiol., 2013, 141, 705–720 CrossRef CAS.
- H. B. Fleit and M. B. Furie, in Pathobiology of Human Disease, ed. L. M. McManus and R. N. Mitchell, Academic Press, San Diego, 2014, pp. 289–299 DOI:10.1016/B978-0-12-386456-7.01807-4.
- A. Tang, Y. Wang, H. Ye, C. Zhou, C. Yang, X. Li, H. Peng, F. Zhang, Y. Hou and F. Teng, Nanotechnology, 2013, 24, 355602 CrossRef.
- F. Hoelscher, P. B. Cardoso, G. Candiotto, C. Guindani, P. Feuser, P. H. H. Araújo and C. Sayer, J. Polym. Environ., 2021 DOI:10.1007/s10924-021-02139-w.
- R. J. Clem, E. H. Y. Cheng, C. L. Karp, D. G. Kirsch, K. Ueno, A. Takahashi, M. B. Kastan, D. E. Griffin, W. C. Earnshaw, M. A. Veliuona and J. M. Hardwick, Proc. Natl. Acad. Sci. U. S. A., 1998, 95(2), 554–559 CrossRef PubMed.
- H. Lin, C. Chen and B. D. Chen, Biochem. J., 2001, 353, 299–306 CrossRef.
- D. M. Sansom, Immunology, 2000, 101, 169–177 CrossRef PubMed.
- J. I. Andorko, K. L. Hess, K. G. Pineault and C. M. Jewell, Acta Biomater., 2016, 32, 24–34 CrossRef.
- D. A. Hume, D. M. Underhill, M. J. Sweet, A. O. Ozinsky, F. Y. Liew and A. Aderem, BMC Immunol., 2001, 2, 11 CrossRef PubMed.
- R. P. Allen, A. Bolandparvaz, J. A. Ma, V. A. Manickam and J. S. Lewis, ACS Biomater. Sci. Eng., 2018, 4, 900–918 CrossRef PubMed.
- H. H. Gustafson, D. Holt-Casper, D. W. Grainger and H. Ghandehari, Nano Today, 2015, 10(4), 487–510 CrossRef PubMed.
- F. Sparber, J. M. Scheffler, N. Amberg, C. H. Tripp, V. Heib, M. Hermann, S. P. Zahner, B. E. Clausen, B. Reizis, L. A. Huber, P. Stoitzner and N. Romani, Blood, 2014, 123(2), 217–227 CrossRef PubMed.
- S.-y. Zhu, R.-q. Yao, Y.-x. Li, P.-y. Zhao, C. Ren, X.-h. Du and Y.-m. Yao, Cell Death Dis., 2020, 11, 817 CrossRef PubMed.
- E. S. Trombetta, M. Ebersold, W. Garrett, M. Pypaert and I. Mellman, Science, 2003, 299, 1400 CrossRef PubMed.
- J. Baran, K. Guzik, W. Hryniewicz, M. Ernst, H. D. Flad and J. Pryjma, Infect. Immun., 1996, 64, 4242–4248 CrossRef PubMed.
- E. Lombardo, A. Alvarez-Barrientos, B. Maroto, L. Boscá and U. G. Knaus, J. Immunol., 2007, 178, 3731 CrossRef PubMed.
- S. M. Reddy, K. H. K. Hsiao, V. E. Abernethy, H. Fan, A. Longacre, W. Lieberthal, J. Rauch, J. S. Koh and J. S. Levine, J. Immunol., 2002, 169, 702 CrossRef PubMed.
- M.-Y. Wu and J.-H. Lu, Cells, 2019, 9, 70 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb00982f |
| This journal is © The Royal Society of Chemistry 2021 |