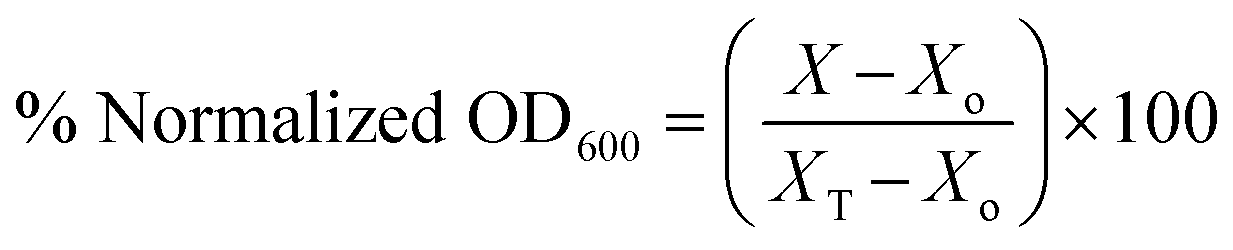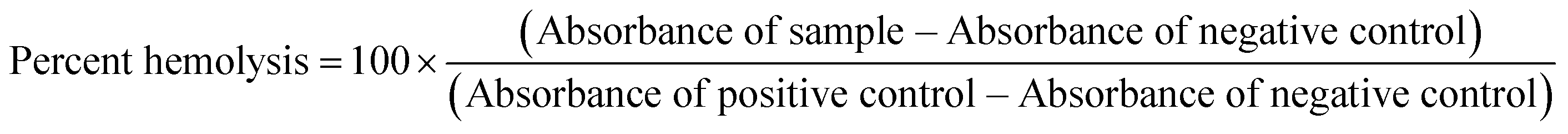DOI:
10.1039/D1MD00211B
(Research Article)
RSC Med. Chem., 2021,
12, 1879-1893
SF5- and SCF3-substituted tetrahydroquinoline compounds as potent bactericidal agents against multidrug-resistant persister Gram-positive bacteria†
Received
20th June 2021
, Accepted 9th August 2021
First published on 10th August 2021
Abstract
Bacteria persister cells are immune to most antibiotics and hence compounds that are active against persister bacteria are needed. We screened a chemical library of SF5- and SCF3-substituted tetrahydroquinoline compounds, synthesized via the Povarov reaction, for antibacterial activity and identified active compounds that displayed good activities against many Gram-positive bacteria, including persisters. The most potent of these compounds, HSD1835, inhibited the growth of drug-resistant Gram-positive bacterial pathogens (including clinical strains) at concentrations ranging from 1 μg mL−1 to 4 μg mL−1. Several of the SCF3- and SF5-containing compounds were active against methicillin-resistant Staphylococcus aureus (MRSA) and against the two most fatal strains of vancomycin-resistant Enterococcus (VRE), VRE faecalis and VRE faecium. The compounds showed bactericidal activity against stationary phase persister MRSA in time-kill assays. Mechanistic studies showed that HSD1835 acts by disrupting bacterial membranes. Scanning electron microscopy (SEM) was used to confirm bacterial membrane disruption. Interestingly, in a 30 day serial exposure experiment, MRSA remained susceptible to low-dose HSD1835 whilst resistance to ciprofloxacin and mupirocin emerged by day 10. Analogs of HSD1835, which did not bear the SF5 or SCF3 moieties, were inactive against bacteria. Recent reports (G. A. Naclerio, N. S. Abutaleb, K. I. Onyedibe, M. N. Seleem and H. O. Sintim, RSC Med. Chem. 2020, 11, 102–110 and G. A. Naclerio, N. S. Abutaleb, D. Li, M. N. Seleem and H. O. Sintim, J. Med. Chem. 2020, 63(20), 11934–11944) also demonstrated that adding the SF5 or SCF3 groups to a different scaffold (oxadiazoles) enhanced the antibacterial properties of the compounds, so it appears that these groups are privileged moieties that enhance the antimicrobial activities of compounds.
1.0 Introduction
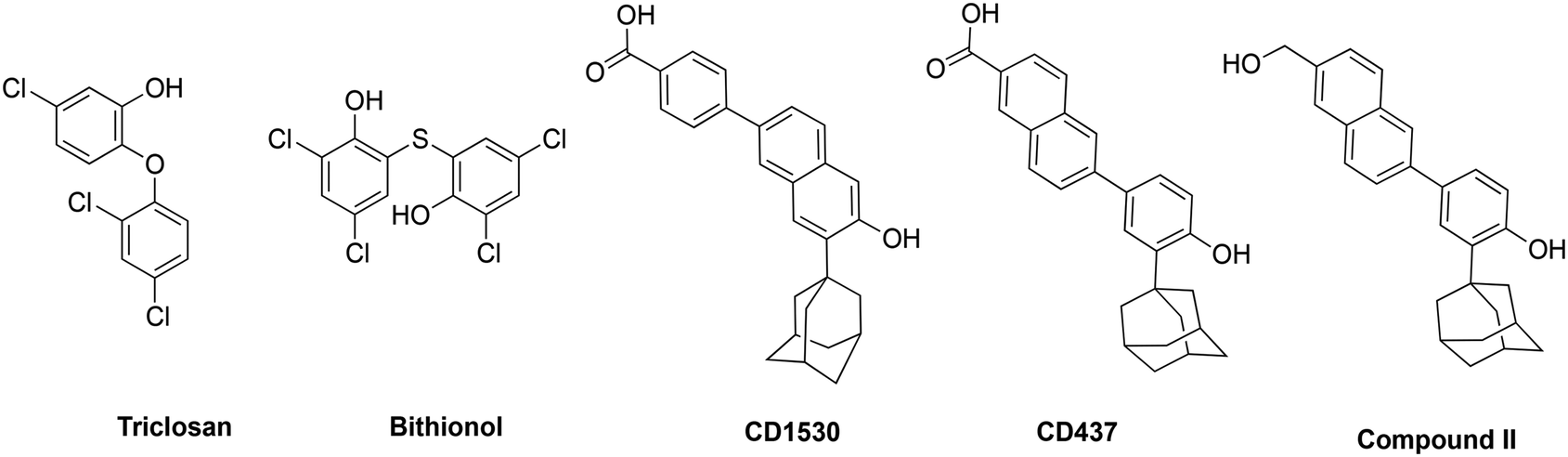
Gladys Hobby and her colleagues in 1942 first pointed out the presence of persister bacteria in Staphylococcus while observing the mechanism of action of the then newly found ‘magic bullet’, penicillin.1 This was swiftly followed by another independent and more widely known report in the Lancet by Joseph Bigger, who first used the term ‘persisters’ in 1944.2 Shortly afterwards, many more bacterial species showed the capacity for developing persister variants.3 Persister bacteria are growth-arrested bacteria with significantly reduced metabolism and are able to grow and multiply once the stressor is removed.3 Because most antibiotics target metabolism in bacteria, in the normally quiescent state of persisters, these conventional antibiotics are ineffective.4,5 Thus, even in the presence of most antibiotics, many bacterial strains that can form persisters persist in the host and stay unharmed for extended periods, leading to chronic infections,3,6 which could be fatal.7,8 For instance, Staphylococcus aureus bacteremia (SAB), caused by persister S. aureus variants, has a high mortality rate of 30%.6–10 Currently, antibiotic resistance poses a substantial threat to public health and is rapidly increasing globally. The Centers for Disease Control and Prevention (CDC) categorized vancomycin-resistant Enterococci (VRE) and methicillin-resistant S. aureus (MRSA) as serious threats to global health.11 VRE and MRSA are responsible for considerable medical and economic burden to patients, their families and health care systems globally.11,12 A recent Swedish study reported that the incidence of MRSA infections more than doubled from 17.6 per 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 inhabitants in 2008 to 37.3 per 100000 inhabitants in 2016.13 In the same study, besides intrinsic resistance to all β-lactams, MRSA also showed resistance rates as high as 69% to fluoroquinolones and an overwhelming 86% to erythromycin.13 Comparable levels of multidrug resistance of MRSA and VRE have also been reported in the United States, Africa, Asia and other parts of Europe.11,12,14 Additionally, there is a 50% higher chance of mortality in intensive care unit (ICU) patients with MRSA infection compared to methicillin-susceptible S. aureus (MSSA) infection.15 Drugs such as triclosan, bithionol and other experimental compounds (Fig. 1) can kill persister bacteria. Bithionol was banned by the FDA (CFR code 700.11) due to photosensitizing effects and there is some concern that triclosan could be carcinogenic.16–18 Increased use of triclosan also leads to bacterial resistance to other antibiotics.16–18 Recently, Mylonakis and co-workers reported that retinoids CD1530, CD437 and compound II (Fig. 1) could kill persister bacteria with a minimum inhibitory concentration (MIC) of ∼8 μg mL−1.19 Herein, we show that SF5- and SCF3-containing compounds, facilely synthesized via the Povarov reaction, are capable of killing persister multidrug-resistant Gram-positive bacteria.
000 inhabitants in 2008 to 37.3 per 100000 inhabitants in 2016.13 In the same study, besides intrinsic resistance to all β-lactams, MRSA also showed resistance rates as high as 69% to fluoroquinolones and an overwhelming 86% to erythromycin.13 Comparable levels of multidrug resistance of MRSA and VRE have also been reported in the United States, Africa, Asia and other parts of Europe.11,12,14 Additionally, there is a 50% higher chance of mortality in intensive care unit (ICU) patients with MRSA infection compared to methicillin-susceptible S. aureus (MSSA) infection.15 Drugs such as triclosan, bithionol and other experimental compounds (Fig. 1) can kill persister bacteria. Bithionol was banned by the FDA (CFR code 700.11) due to photosensitizing effects and there is some concern that triclosan could be carcinogenic.16–18 Increased use of triclosan also leads to bacterial resistance to other antibiotics.16–18 Recently, Mylonakis and co-workers reported that retinoids CD1530, CD437 and compound II (Fig. 1) could kill persister bacteria with a minimum inhibitory concentration (MIC) of ∼8 μg mL−1.19 Herein, we show that SF5- and SCF3-containing compounds, facilely synthesized via the Povarov reaction, are capable of killing persister multidrug-resistant Gram-positive bacteria.
 |
| | Fig. 1 Structures of membrane-acting compounds which are active against persister bacteria. | |
2.0 Results and discussion
2.1 Identification and screening of SF5 and SCF3 compounds for antibacterial activity
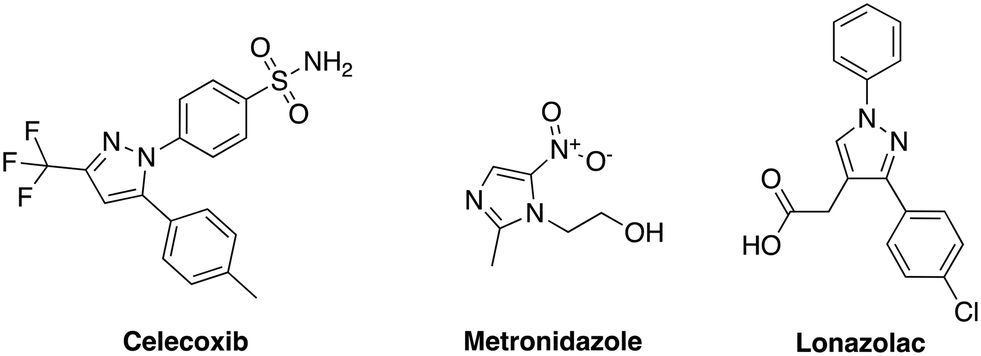
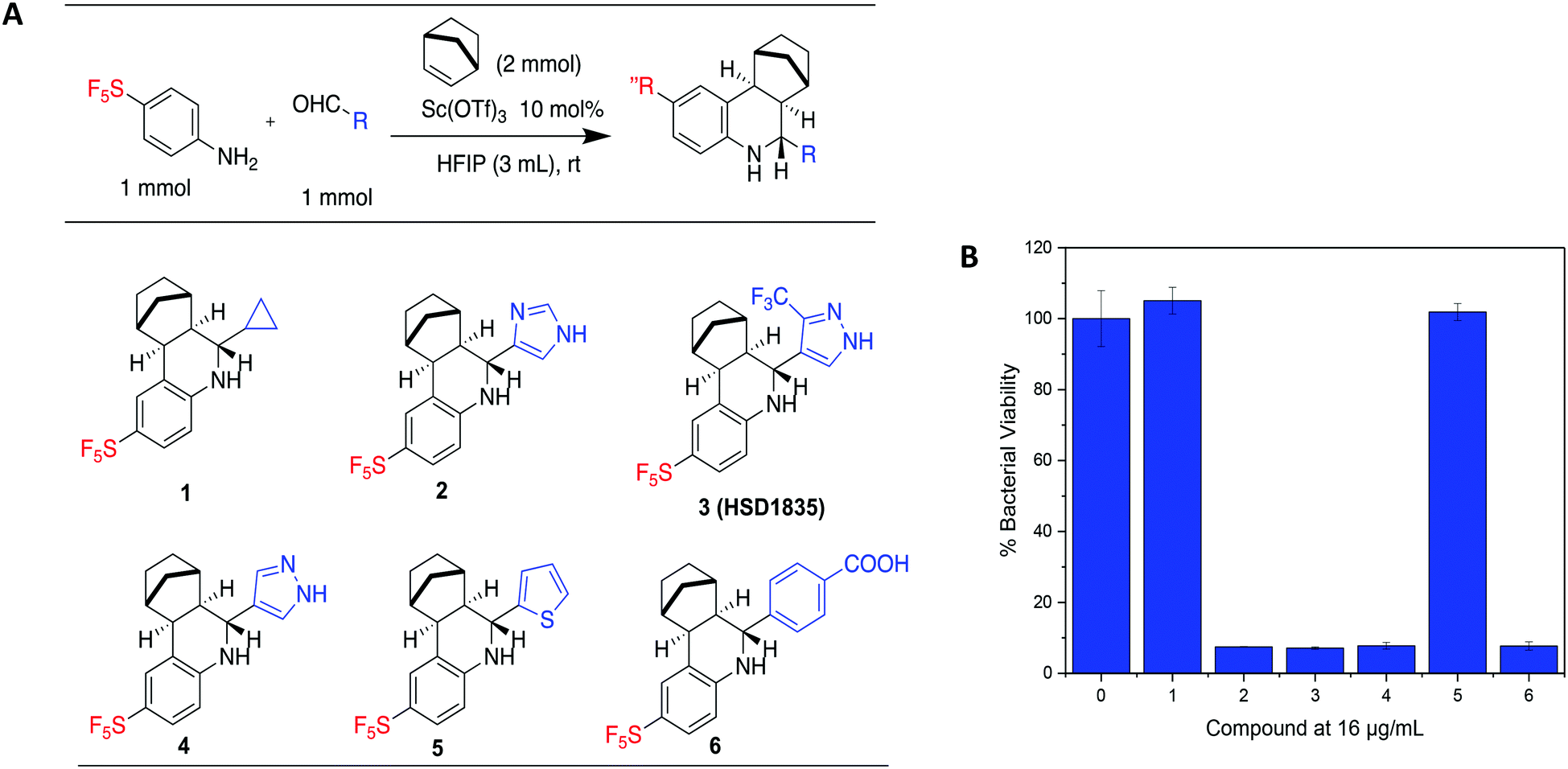
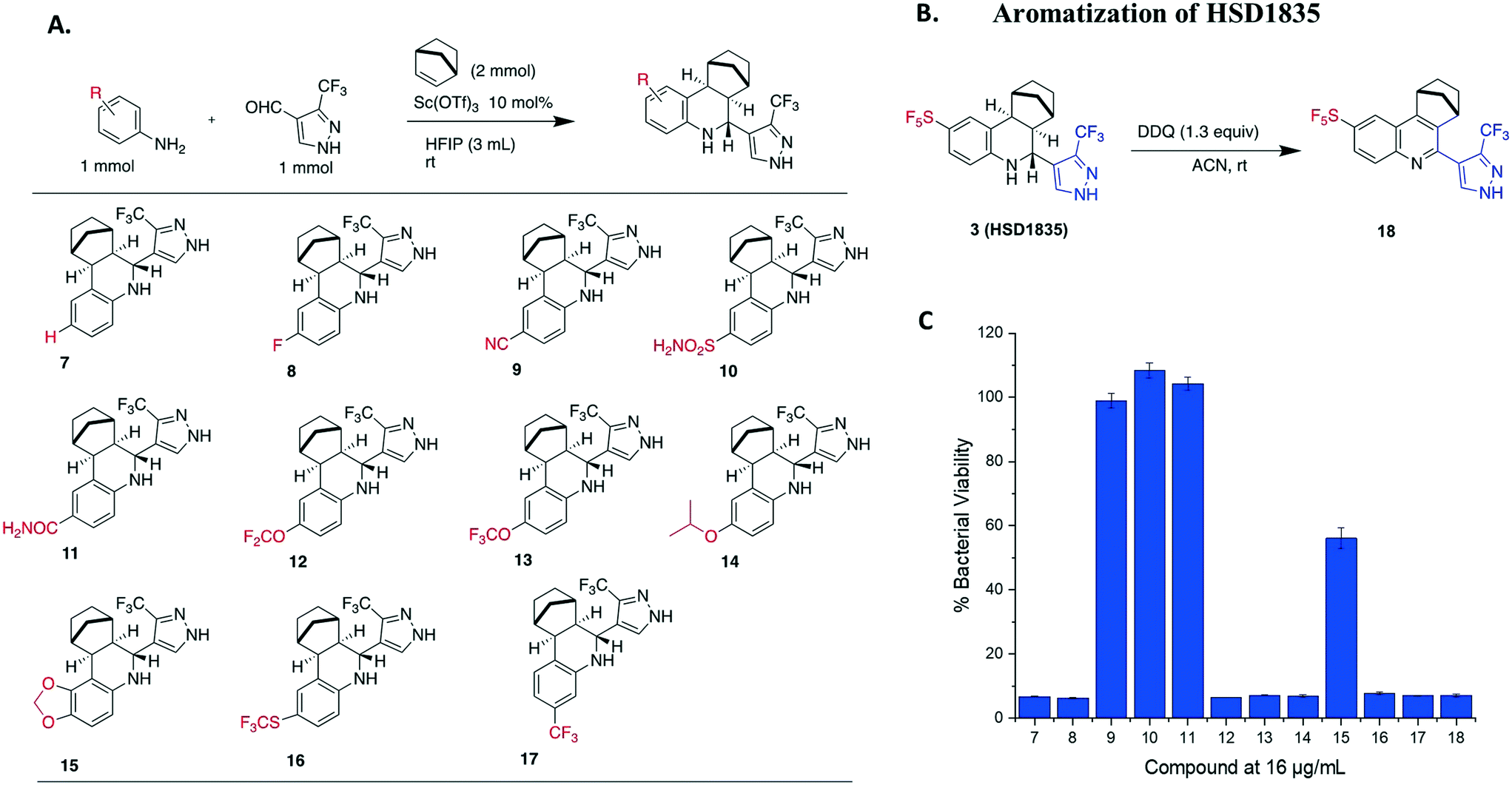
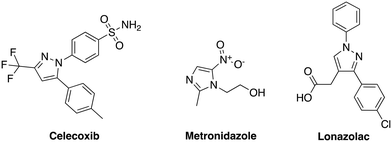
Our group has generated a diverse collection of compounds, synthesized via multi-component reactions (such as Povarov/Doebner), and screened such library against bacteria and cancer.20,21 Screening the library, which was generated via the Povarov/Doebner reaction, against persister bacteria however did not lead to any hit. In another project, which utilized oxadiazoles, we observed that adding the SF5 moiety to oxadiazoles enhanced the antibacterial activity.22 We therefore generated a new small library, which contained SF5 and pyrazole or imidazole moieties (commonly found in drugs, for examples, see Fig. 2), via the Povarov reaction and tested the compounds for antibacterial activities. The six initial synthesized compounds (Fig. 3) were screened against S. aureus ATCC 25923 at a concentration of 16 μg mL−1.
 |
| | Fig. 2 Bioactive compounds/drugs containing pyrazole or imidazole groups. | |
 |
| | Fig. 3 (A) Scheme and structures of SF5-containing Povarov compounds. (B) Antibacterial activities of compounds 1 to 6 at 16 μg mL−1 against S. aureus ATCC 23592. Compound 0 is DMSO control. HFIP = hexafluoroisopropanol. | |
2.2 Library synthesis
We prepared our Povarov library by utilizing an established method.23 Briefly, we used monocyclic aromatic amine, substituted aldehydes and norbornene as the alkene source in the presence of 10 mol% Lewis acid catalyst, scandium triflate, in hexafluoroisopropanol (HFIP) at room temperature. In agreement with a previous report,23 the exo–exo diastereomers were formed. The reader is referred to ref. 23, which explains the favored exo-facial cyclization and also provides the X-ray crystal structure of similar compounds.23
2.3 Evaluation of HSD1835 analogs
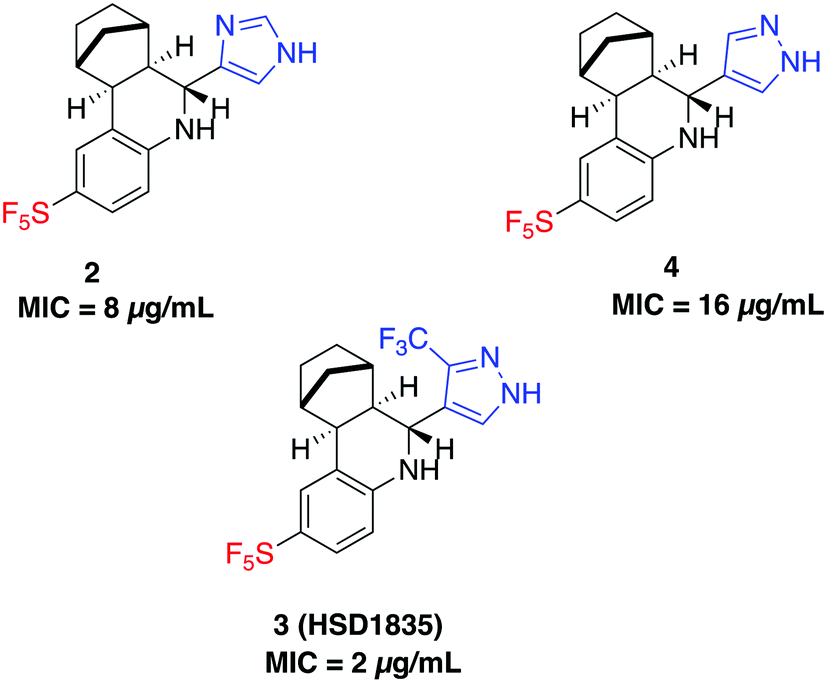
S. aureus and VRE infections are very difficult to treat and remain a major cause of death in the clinical setting despite the availability of several Gram-positive active antibiotics.24,25 Four of the compounds screened (compounds 2, 3, 4 and 6) were active against S. aureus ATCC 23592 (Fig. 3B). Thus, we evaluated the minimum inhibitory concentrations (MICs) of the active compounds synthesized by the Povarov reaction and found that the 3-(trifluoromethyl)-1H-pyrazol-containing compound 3 or HSD1835 had the best antibacterial activity against S. aureus ATCC 25923 with concentrations as low as 2–4 μg mL−1 for HSD1835 (Fig. 4). To investigate the importance of the SF5 group for antibacterial activity, we replaced the SF5 group with other moieties (see Fig. 5) and tested the antibacterial activities against S. aureus ATCC 25923. Compound 7, which did not contain the SF5 group, and compounds 8, 12–14 and 16–18 (substituted with one of F, OCF3, OiPr, SCF3 and CF3) also inhibited bacterial growth at 16 μg mL−1 (see Fig. 5). However, the MIC values for the aforementioned compounds were 4 μg mL−1 or higher (Table 1); thus HSD1835, which contained the SF5 group, remained the most potent antibacterial compound with MIC of 2 μg mL−1 in some cases (Table 1). None of the compounds screened had antibacterial activity against E. coli ATCC 25922 (data not shown).
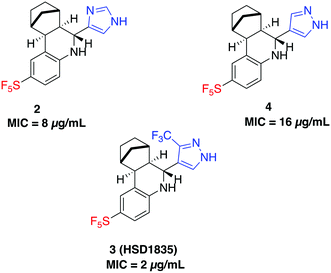 |
| | Fig. 4 Structures of SF5-containing active compounds and their minimum inhibitory concentrations (MICs) against S. aureus ATCC 25923. | |
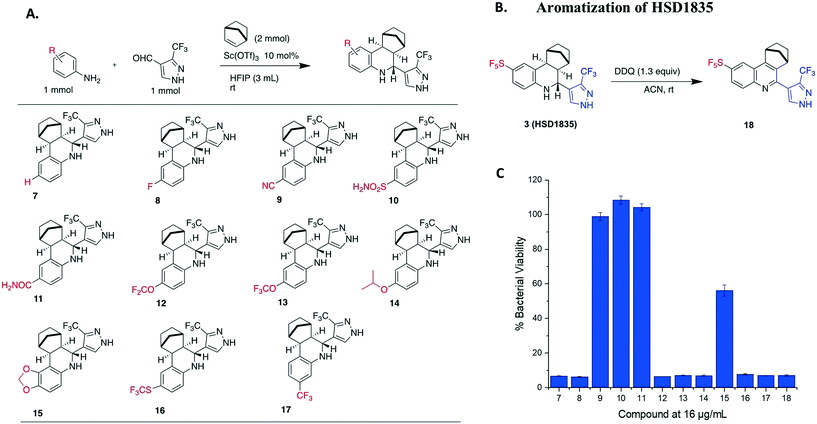 |
| | Fig. 5 (A) Synthesis and structures of 3-(trifluoromethyl)-1H-pyrazole-containing analogs. (B) Aromatization of HSD1835 to determine the importance of the tetrahydroquinoline core. (C) Antibacterial activity of compounds 7 to 18 at 16 μg mL−1 tested against S. aureus ATCC 23592. HFIP = hexafluoroisopropanol. | |
Table 1 Minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) in μg mL−1 of most active compounds against Gram-positive bacterial strains. NT = Not Tested
| Bacterial strain |
HSD1835
|
6
|
16
|
18
|
13
|
17
|
Vancomycin |
Linezolid |
| MIC |
MBC |
MIC |
MIC |
MIC |
MIC |
MIC |
MIC |
MBC |
MIC |
MBC |
|
S. aureus ATCC 25923 |
2–4 |
8 |
4 |
4 |
4 |
4 |
4 |
1 |
2 |
2 |
64 |
| MRSA USA300 |
4 |
8 |
16 |
4 |
8 |
8 |
8 |
2 |
2 |
2 |
64 |
| MRSA ATCC 33592 |
2–4 |
4 |
4 |
2 |
8 |
4 |
8 |
2 |
2 |
2 |
64 |
|
E. faecalis ATCC 29212 |
4 |
8 |
4 |
8 |
8 |
8 |
8 |
2 |
2 |
1 |
64 |
| VRE (E. faecalis) ATCC 51575 |
4 |
8 |
4 |
8 |
8 |
8 |
8 |
>128 |
>128 |
1 |
>128 |
| VRE (E. faecium) ATCC 700221 |
2 |
4 |
4 |
4 |
8 |
4 |
8 |
>128 |
>128 |
1 |
>128 |
|
L. monocytogenes ATCC 19115 |
2 |
4 |
4 |
4 |
8 |
4 |
8 |
1 |
NT |
2 |
NT |
Since the compounds showed remarkable antibacterial activity against S. aureus, we then tested the compounds against MRSA strains (MRSA USA300 and MRSA ATCC 33592) and VRE strains (VRE faecalis and VRE faecium) (Table 1). HSD1835 had an excellent MIC of 2–4 μg mL−1 against all tested strains, including VRE faecalis and VRE faecium. We further investigated to see if HSD1835 would be bactericidal to these MDR strains. Interestingly, HSD1835 was also bactericidal to all the MDR strains (MRSA and VRE) with a minimum bactericidal concentration (MBC) of 4–8 μg mL−1. Moreover, vancomycin and linezolid were totally ineffective against VRE faecalis and VRE faecium, whereas HSD1835 achieved total eradication of both strains at 4–8 μg mL−1.
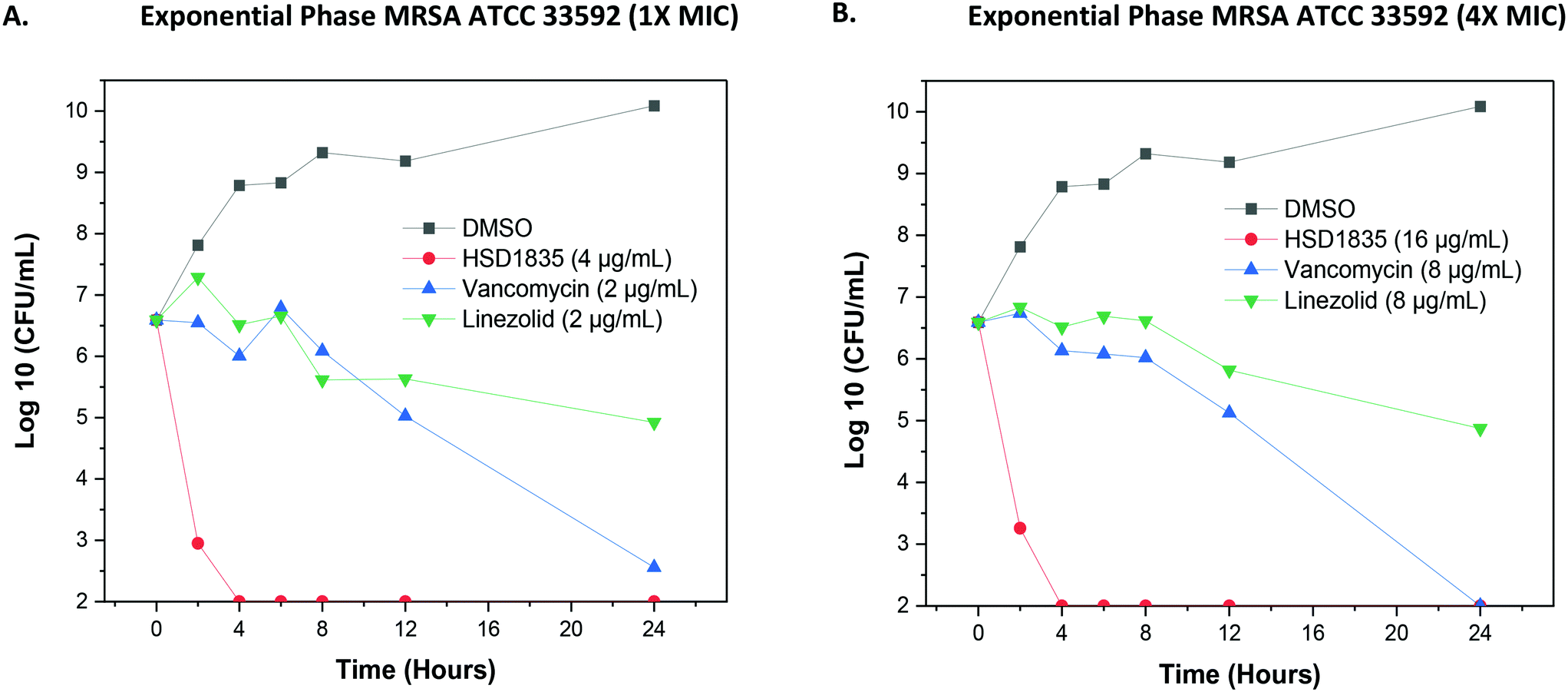
2.4 Bactericidal activity of HSD1835 is fast acting
Since the compounds showed good MBC against MDR strains, we proceeded to confirm the bactericidal activity and to determine the time to kill against MRSA strains. HSD1835 was highly active against log-phase MRSA ATCC 33592 (Fig. 6A and B). Specifically, at MIC and MBC concentrations, HSD1835 achieved total killing of MRSA in 4 h. In contrast, currently used antibiotics such as vancomycin and linezolid at 4× MIC were unable to achieve total bacteria eradication even after 24 h of incubation.
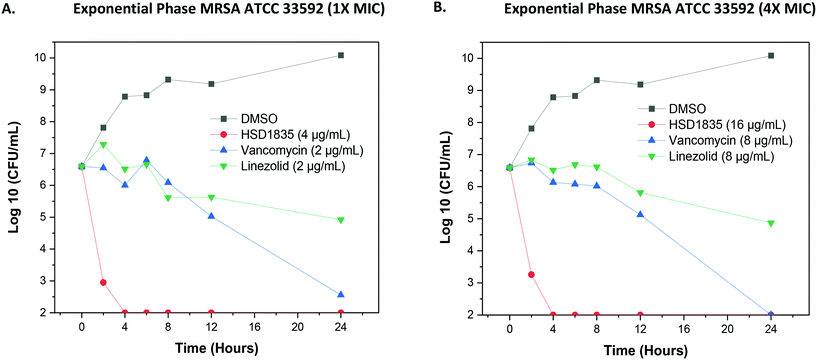 |
| | Fig. 6 Time-kill analysis of HSD1835 against MRSA ATCC 33592. (A) At 1× MIC and (B) 4× MIC of HSD1835, MRSA ATCC 33592 was completely eradicated in 4 h. Graphs plotted using Origin (Pro), Version 2020. OriginLab Corporation, Northampton, MA, USA. | |
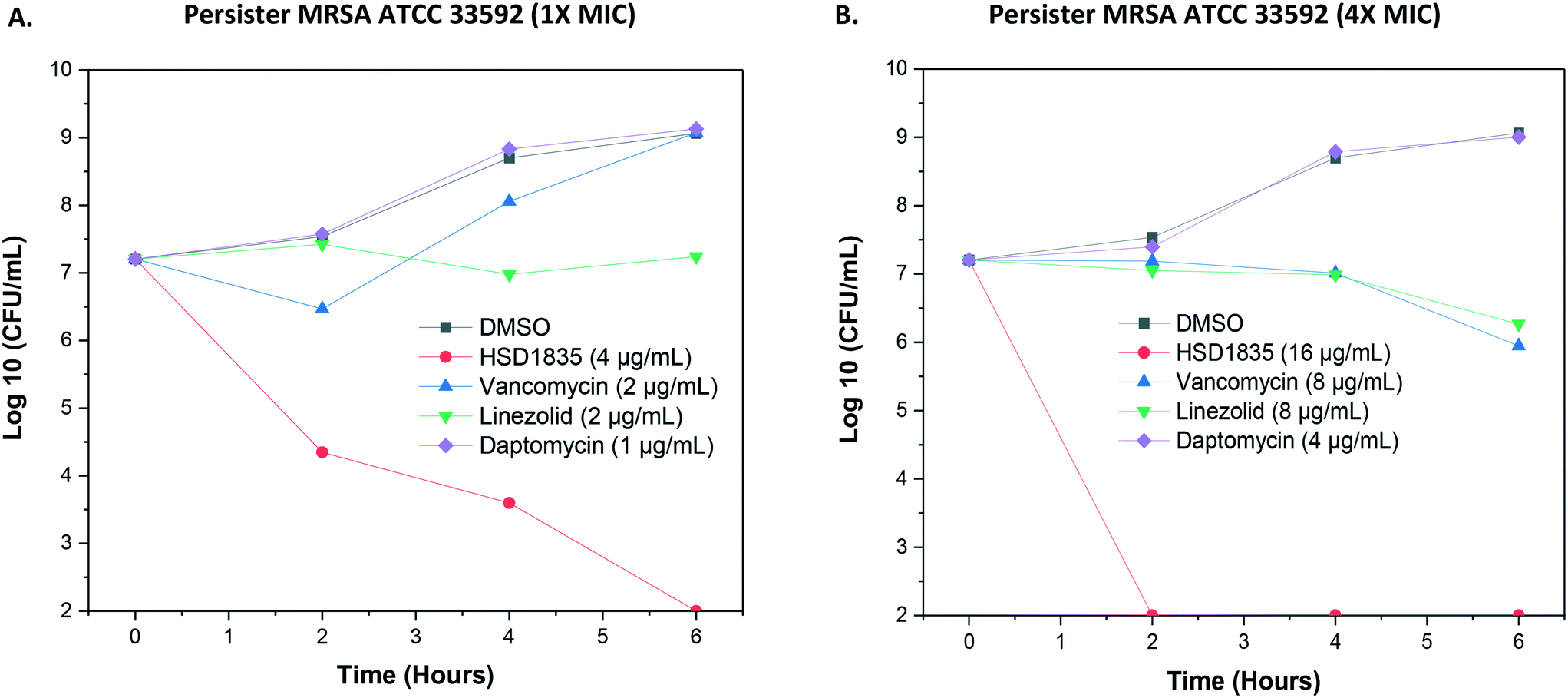
2.5 Persister MRSA ATCC 33592 are eradicated by HSD1835
As stated, S. aureus are able to easily exist in the nongrowing antibiotic-tolerant state called persisters,26 which can resist antibiotics that are dependent on metabolic energy from the bacteria for uptake, such as aminoglycosides.27 We were thus curious to know if the lead compound, HSD1835, could kill persister S. aureus. Interestingly, HSD1835 was highly active against persister MRSA ATCC 33592. HSD1835 achieved total killing of persister MRSA in 6 h. In contrast, the presently used antibiotics against MRSA (vancomycin, daptomycin and linezolid) had no activity against persister MRSA (Fig. 7A and B).
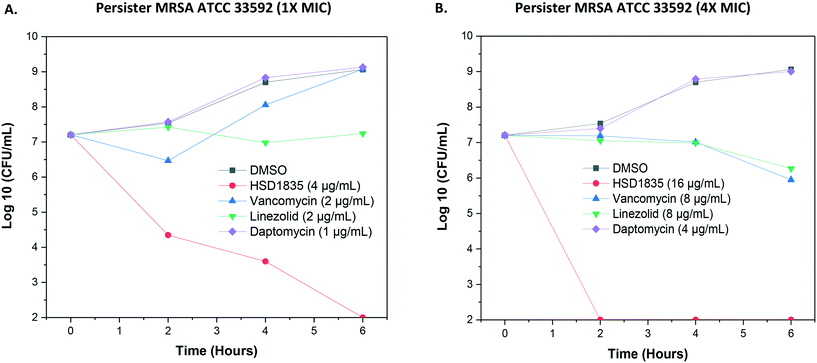 |
| | Fig. 7 Time-kill analysis of compounds against persister MRSA ATCC 33592. (A) At 1× MIC of each of the compounds, HSD1835 reduced the bacterial population by 7log. Other antibiotics had little or no effect on persister MRSA. (B) At 4× MIC, HSD1835 completely eradicated stationary phase MRSA at 6 h. Graphs plotted using Origin (Pro), Version 2020. OriginLab Corporation, Northampton, MA, USA. | |
2.6 Evaluation of the killing activity of HSD1835 against highly resistant clinical strains
Several clinical isolates of MRSA also carry other additional antibiotic resistance genes making them resistant to even newer antibiotics such as mupirocin, linezolid, new quinolones, glycopeptides, lipopeptides and pleuromutilins.28 Reports suggest that antibiotic misuse and the large-scale use of antibiotics in the livestock industry account for most of this resistance.29–32 We investigated if HSD1835 would maintain activity against nine (9) highly resistant MRSA clinical strains. These clinical strains were obtained from the Antibacterial Resistance Leadership Group (ARLG) biorepository. Besides possession of the mecA gene, which confers resistance to all beta-lactam drugs (penicillins, 1st-, 2nd- and 3rd-generation cephalosporins, carbapenems), the strains also harbored resistance genes such as MupA gene (mupirocin resistance) and Panton–Valentine leukocidin (pvl) gene and were additionally resistant to ciprofloxacin, erythromycin and clindamycin (Table 2). Outstandingly, HSD1835 had very potent activity against all the tested strains with MIC ranging from 1 to 2 μg mL−1.
Table 2 Minimum inhibitory concentration (MIC) in μg mL−1 of HSD1835 against highly resistant MRSA clinical strains
| Strain |
MRSA description |
| PFGE pattern/MLST typea |
spa type eGenomics (Ridom) |
SCCmec |
Other genetic characterizationa |
Additional non MRSA resistance (dru type)a |
HSD1835 MIC (μg mL−1) |
Oxacillin MIC (μg mL−1) |
|
ACME = arginine catabolic mobile element, PFGE = pulsed field gel electrophoresis, MLST = multilocus sequence typing, pvl = Panton–Valentin leukocidin, Ery = erythromycin, Mup = mupirocin, Cipro = ciprofloxacin, Clinda = constitutive clindamycin resistance, dru = direct repeat unit, ARLG = Antibacterial Resistance Leadership Group, NT = Not Tested.
|
| ARLG 1561 |
USA300 |
1 |
IVa |
ACME+, pvl genes+ |
Ery |
2 |
NT |
| ARLG 1567 |
USA300 |
1 |
IVa |
ACME+, pvl genes+ |
Ery, Cipro |
2 |
NT |
| ARLG 1568 |
USA300 |
1 |
IVa |
ACME+, pvl genes+, MupA gene+ |
Mup, Ery, Cipro |
2 |
NT |
| ARLG 1569 |
USA300 |
1 |
IVa |
ACME+, pvl genes+, MupA gene+ |
Mup, Ery, Cipro, Clinda |
2 |
NT |
| ARLG 1570 |
USA300 |
1 |
IVa |
ACME+, pvl genes+, MupA gene+ |
Mup, Ery, Cipro, Clinda |
2 |
NT |
| ARLG-1644 |
ST239 |
351 (t030) |
3A.1.4 |
ccrC+, dcs+, Hg-J+, mecl+ |
(dt10a) |
2 |
512 |
| ARLG-1649 |
ST1312 |
351 (t030) |
3A.1.4 |
ccrC+, dcs+, Hg-J+, mecl+ |
(dt10g) |
2 |
256 |
| ARLG-1663 |
ST239 |
351 (t030) |
16691 |
ccrC+, Hg-J+, mecl+ |
(dt9x) |
1 |
256 |
| ARLG-1665 |
ST239 |
351 (t030) |
16691 |
ccrC+, Hg-J+, mecl+ |
(dt9x) |
2 |
512 |
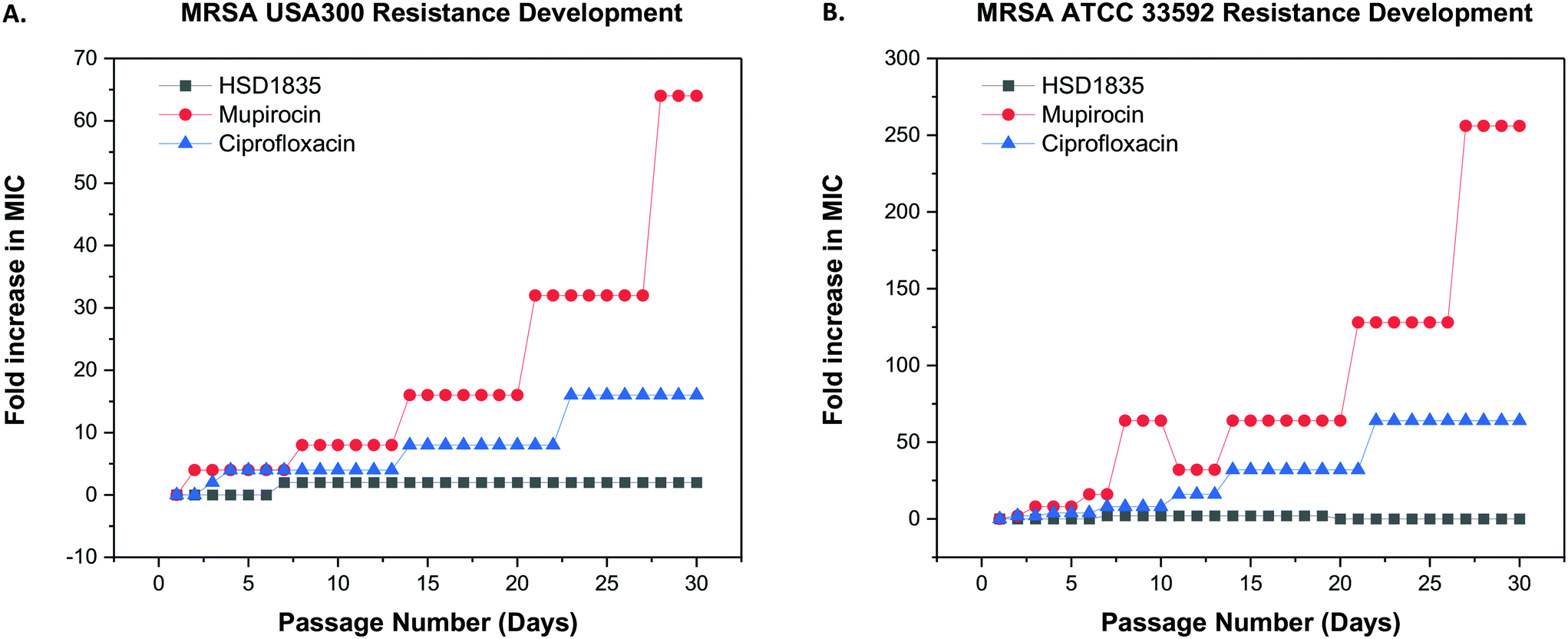
2.7 Resistance to HSD1835 did not develop in MRSA after repeated exposures
Since these compounds demonstrated bactericidal activity to both log phase and persister MRSA strains and were active against MRSA strains that had acquired other resistance genes, we proceeded to evaluate the ability of MRSA to develop resistance to HSD1835. The foremost challenge of antibiotic therapy is the development of resistance to the antibiotic after repeated exposure to the bacteria. We used the multi-step resistance selection method to stimulate MRSA to develop resistance to the test compounds. Although we observed a one-fold increase in the MIC of HSD1835 initially, no additional increase in MIC was observed up to the 30th passage in both MRSA USA300 and ATCC 33592 strains. In contrast, mupirocin and ciprofloxacin, which are currently used in the clinic to treat MRSA, rapidly developed resistance to these MRSA strains. Specifically, the MIC of ciprofloxacin, an antibiotic that inhibits the function of DNA topoisomerase, rapidly increased by 16-fold against MRSA USA300 (Fig. 8A) and by 64-fold against MRSA ATCC 33592 (Fig. 8B). Likewise, the MIC of mupirocin, an antibiotic routinely used for presurgical MRSA therapy and decontamination, increased by over 250-fold against MRSA ATCC 33592 during the course of the assay (Fig. 8B).
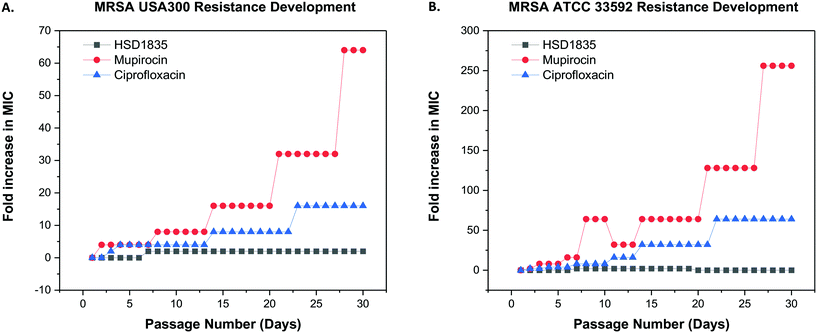 |
| | Fig. 8 Multi-step resistance selection of HSD1835, mupirocin and ciprofloxacin against MRSA strains. (A) MRSA USA300 and (B) MRSA ATCC 33592 were serially passaged daily over a 30-day period and the broth microdilution assay was used to determine the minimum inhibitory concentration (MIC) of each compound against MRSA after each successive passage. A four-fold shift in MIC would be indicative of bacterial resistance forming to the test agent. HSD1835 did not develop resistance to both MRSA ATCC 33592 and MRSA USA300 strains by the 30th passage. Graphs plotted using Origin (Pro), Version 2020. OriginLab Corporation, Northampton, MA, USA. | |
2.8
HSD1835 permeabilizes bacteria membranes
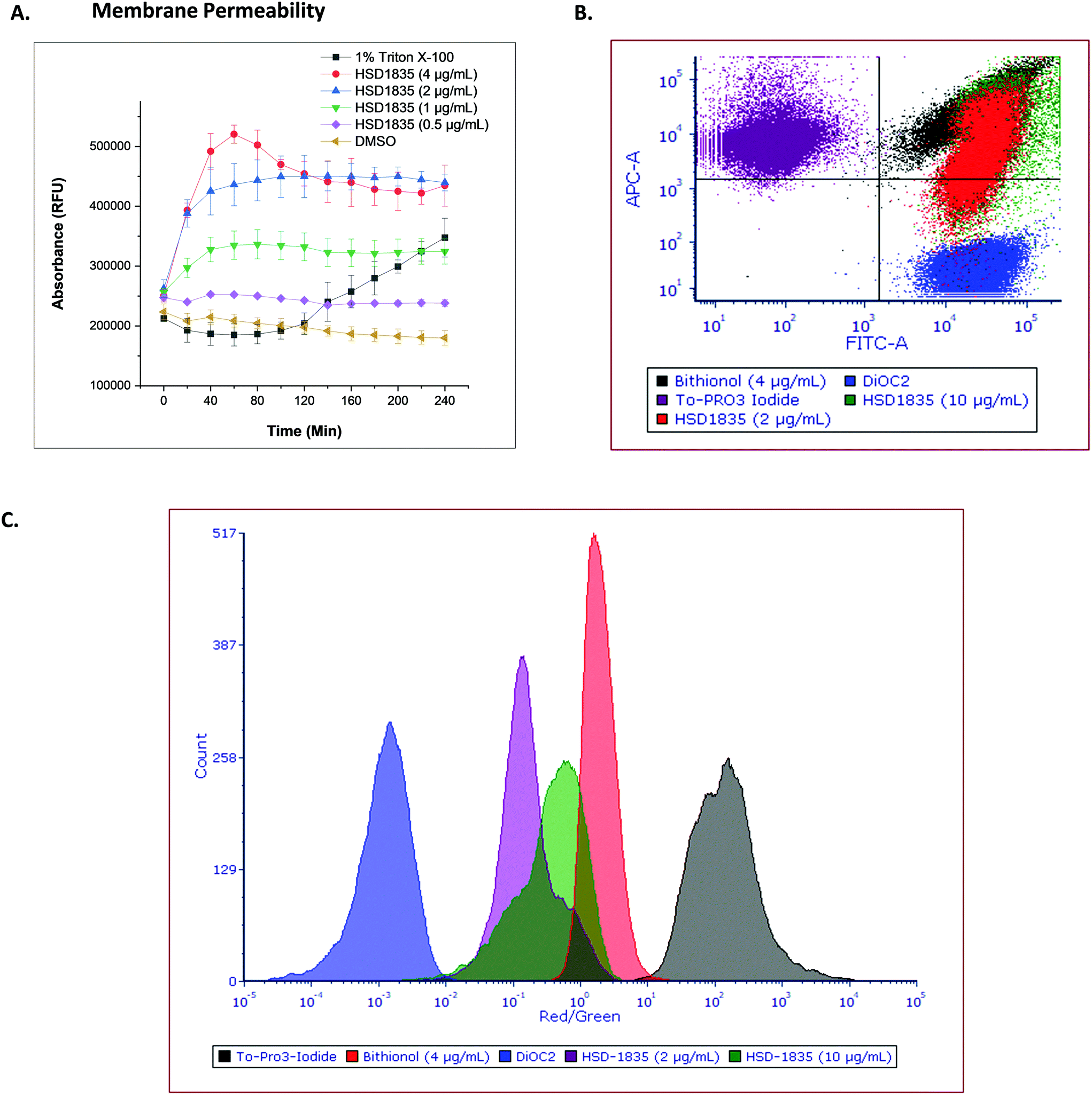
The elimination of persister bacteria remains an enormous challenge since conventional antibiotics are unable to kill them. However, reports of compounds that were able to kill persister bacteria showed that they were mostly membrane-acting compounds.19,33 For instance, Kim et al. showed that bithionol, a membrane-permeable antiparasitic agent, could eradicate persister S. aureus at 32 μg mL−1.33 Thus, the membranes of nongrowing bacteria have become attractive targets since their membranes are independent of growth. Colistin, daptomycin and several antimicrobial peptides have also recorded successes in the clinic as bacterial membrane disrupting agents which are bactericidal. Since HSD1835 is both bactericidal and is active against persister bacteria, we hypothesized that the compound might act (at least in part) on bacterial membranes. Hence, we assayed for bacterial membrane depolarization and permeabilization using the DiSC3 and Sytox green™ florescence assays, respectively. 1 μM gramicidin, a known membrane depolarization agent, was used as the positive control in the depolarization assays, whilst 1% Triton X-100 was used as the positive control in the permeabilization assay. HSD1835 did not depolarize staphylococcal bacterial membranes but permeabilized bacterial membranes at concentrations as low as 1 μg mL−1 (Fig. 9A).
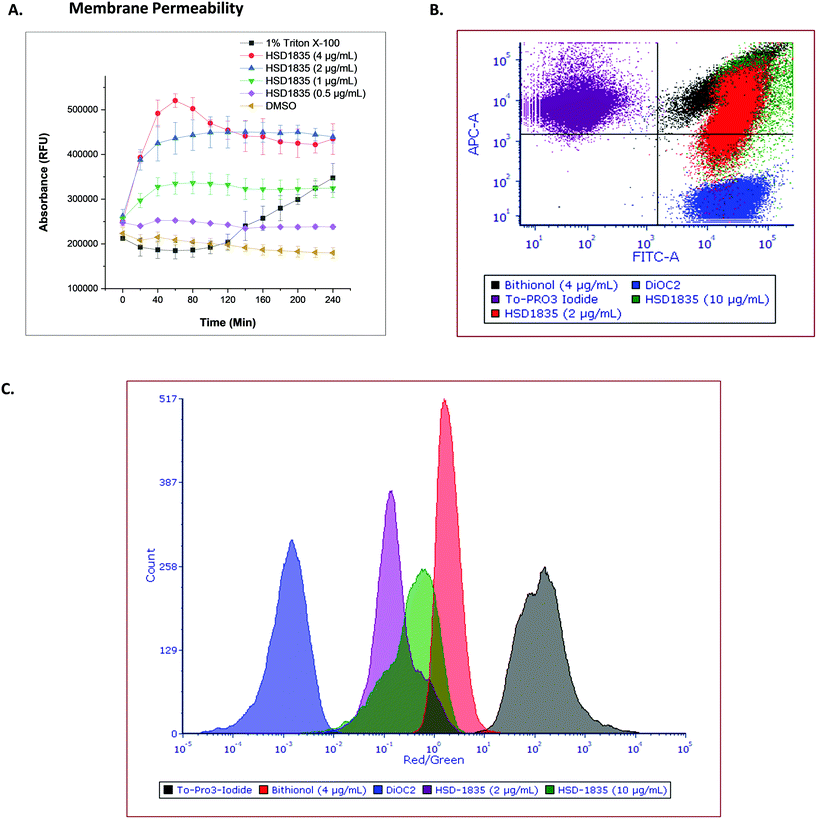 |
| | Fig. 9 Effect of HSD1835 on staphylococcal membranes. S. aureus ATCC 25923 were used in all assays. (A) Sytox green™ membrane permeability assay showing dose-dependent permeabilization of staphylococcal membranes. 1% Triton X-100 was used as the positive control. Assays were performed in triplicate and points represent their various means with corresponding standard deviation. (RFU = relative fluorescence unit). The graph was plotted using Origin (Pro), Version 2020. OriginLab Corporation, Northampton, MA, USA. (B) Flow cytometry assay showing the relative overlap of HSD1835 with bithionol (4 μg mL−1) in the same quadrant. (C) Flow cytometry assay showing the red/green ratio of HSD1835 partially overlapping with bithionol (4 μg mL−1). 10 μg mL−1HSD1835 overlapped more than a low HSD1835 concentration of 2 μg mL−1. Analysis and charts were made on FCS Express software, version 7.0. | |
To further assess the membrane permeability effect of HSD1835, we used a flow cytometry assay. To-PRO 3 iodide (red channel) was used as the membrane-permeable dye whilst DiOC2 (green channel) was the membrane-impermeable dye in the flow cytometry assay. Bithionol, a known membrane permeabilizer, was the positive control in this assay. The ratio of the red dye to the green dye is an indicator of the membrane permeabilizing effect of the compound. When compared to bithionol, it was observed that HSD1835 was in the same quadrant (Fig. 9B) and also exhibited an overlapping red/green ratio with bithionol (Fig. 9C), suggesting that HSD1835 permeabilized S. aureus membranes like bithionol. Kim et al. demonstrated that bacterial membrane-active compounds, such as bithionol, synergized with aminoglycosides such as gentamicin.33 We therefore investigated if HSD1835 also synergized with amikacin, gentamicin, tobramycin, and kanamycin but saw no synergy between HSD1835 and these drugs.
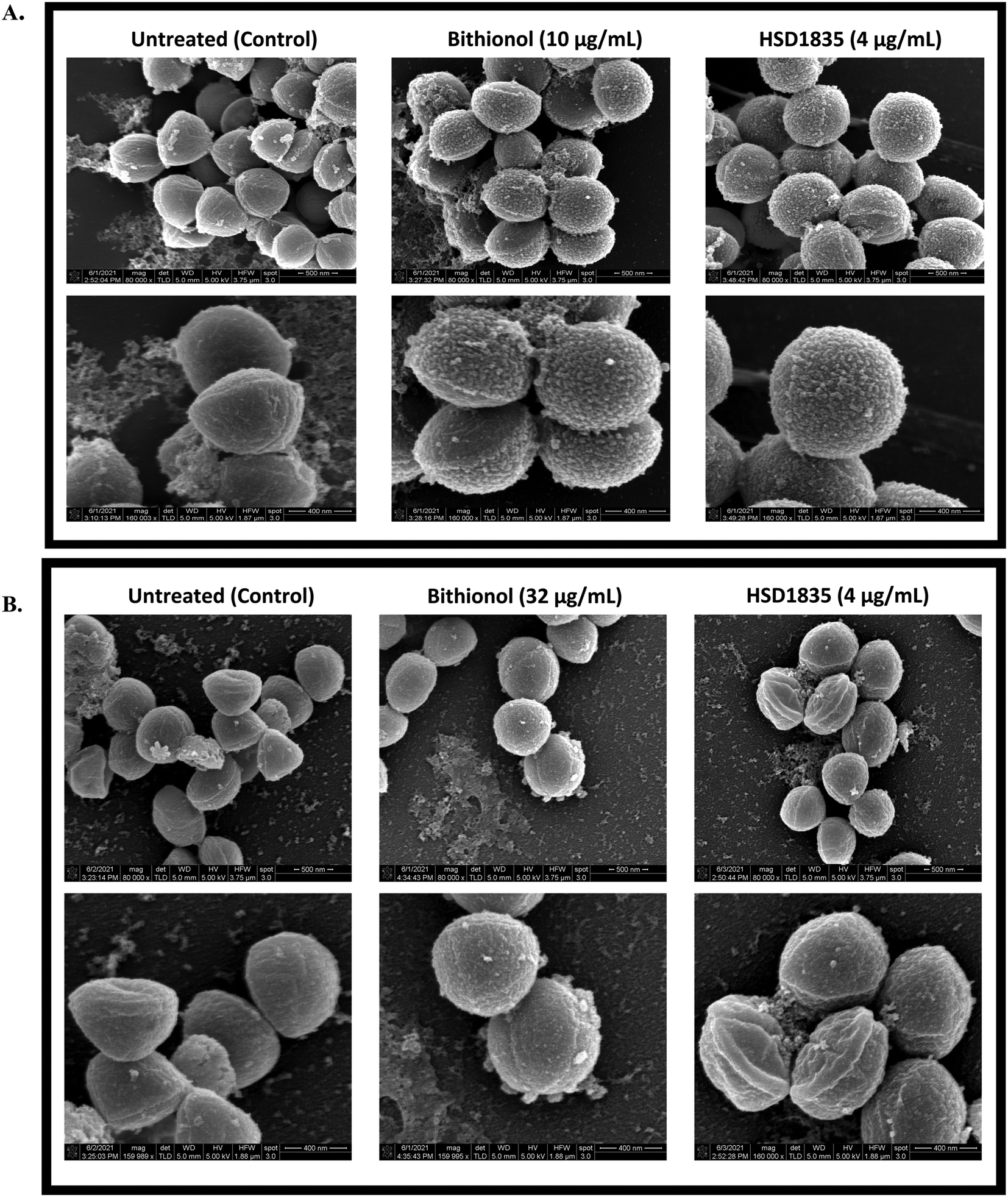
2.9 Membrane disruption effect of HSD1835 visualized by scanning electron microscopy (SEM)
To confirm and visualize the membrane effects of HSD1835 on Gram-positive bacterial membranes, scanning electron microscopy (SEM) was carried out at the Purdue Life Sciences Imaging Facility. In previous reports, SEM has been used to confirm bacterial membrane disruption.34,35 Thus, we treated exponential and stationary phase S. aureus with HSD1835 (4 μg mL−1) or bithionol. Interestingly, in the exponentially growing S. aureus samples, we found significant membrane disruption in the presence of HSD1835 (Fig. 10A). Specifically, HSD1835 or bithionol (positive control) caused formation of multiple hairy “bread crumb”-like protrusions on the surfaces of treated cells within 4 h of treatment (Fig. 10A). The untreated cells (Fig. 10A) were devoid of these protrusions. In contrast, in the stationary phase S. aureus (Fig. 10B), HSD1835-treated cells did not show the same kind of protrusions seen in the exponential phase cells. Rather, some of the treated cells had bleb-like protrusions, corrugations, rough or wrinkled surfaces and distortions. Also, in some of the bithionol-treated cells, we observed blebs at the edge of the cell with a wheel-like appearance around the circumference of the bacterial cell (Fig. 10B). Thus, with the use of SEM, the membrane disruption effects of HSD1835 were clearly visualized.
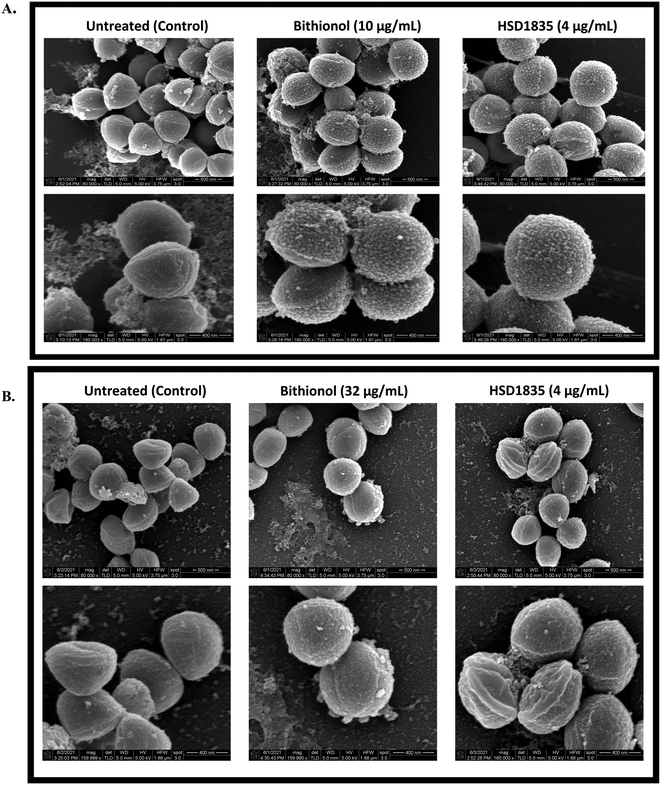 |
| | Fig. 10 SEM showing membrane disruption effects of HSD1835 on staphylococcal membranes. S. aureus ATCC 25923 were used in all assays. (A) Exponential phase S. aureus treated with either DMSO (negative control), bithionol (positive control) or HSD1835. Top panel is ×80000 magnification and bottom panel is ×160000 magnification of the same view. Multiple hairy “bread crumb”-like protrusions were prominently seen on the surfaces of treated cells within 4 h of treatment in both bithionol- and HSD1835-treated cells. (B) Stationary phase (persister) S. aureus treated with either DMSO (negative control), bithionol (positive control) or HSD1835. Top panel is ×80000 magnification and bottom panel is ×160000 magnification of the same view. Corrugations, depressions and roughening were seen on the surface of HSD1835-treated cells, while bleb-like protrusions were also seen in some of the bithionol-treated cells. SEM images were captured using an FEI Nova Nano-SEM instrument. | |
2.10
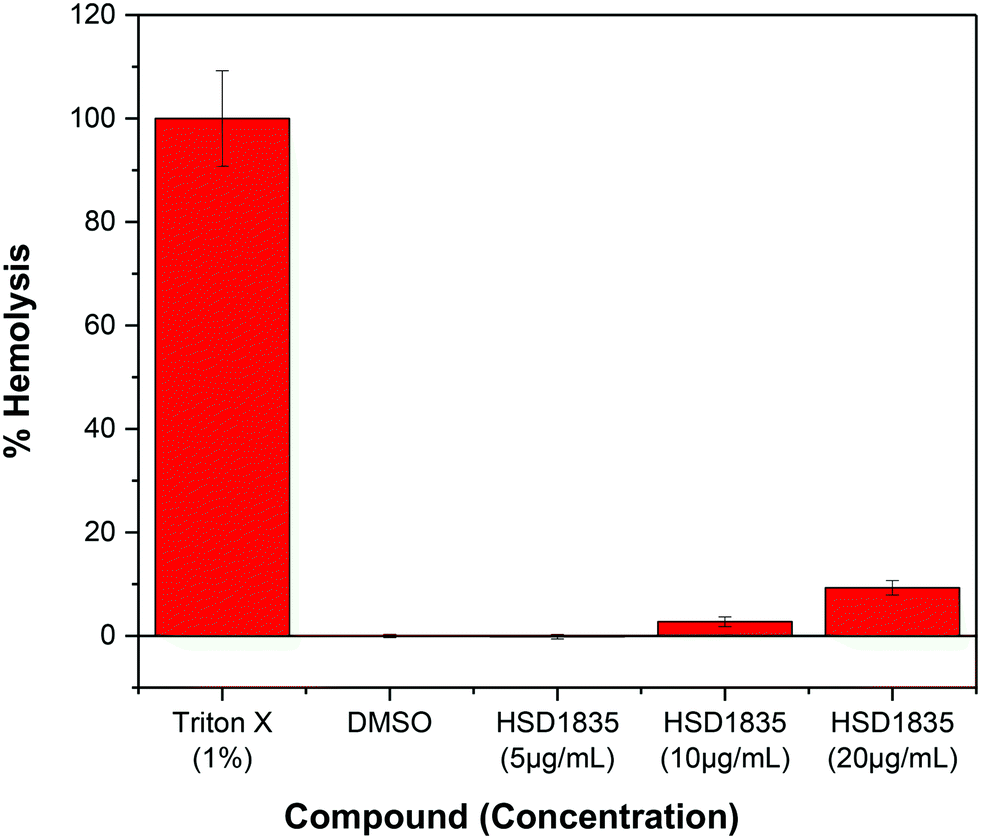
HSD1835 is non-toxic to mammalian red blood cells (RBCs)
It is challenging to achieve selectivity between mammalian membranes and bacterial membranes. To determine if HSD1835 is toxic to mammalian membranes, we exposed human red blood cells (RBCs) to higher concentrations of the compound. We observed that at concentrations as high as 20 μg mL−1 (10× MIC for MRSA), HSD1835 was relatively nontoxic to human RBCs (Fig. 11).
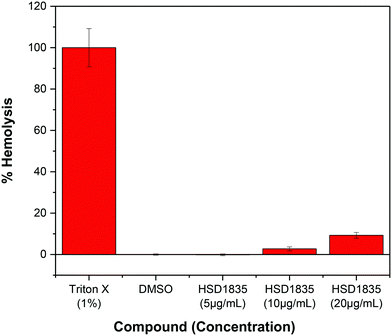 |
| | Fig. 11 Effect of HSD1835 on hemolysis of 5% human red blood cells (RBCs). All assays were performed in triplicate and bars represent their various means. | |
3.0 Conclusion
Antibiotics against bacterial persisters, which are in the stationary phase of growth, are a major therapeutic challenge in the clinic. Here, we have described the synthesis and antibacterial activities of potent SF5- and SCF3-substituted tetrahydroquinoline compounds against persister multidrug-resistant Gram-positive bacteria. Our lead compound, HSD1835, which acts via membrane disruption, was bactericidal to MRSA and VRE strains and eradicated stationary phase MRSA within 6 hours. Additionally, HSD1835 is nontoxic to mammalian RBCs and showed no potential for developing resistance after repeated exposure to MRSA strains.
4.0 Materials and methods
4.1 Bacterial strains and culture methods
Bacterial strains were obtained from various sources as listed in Table 3. Except where stated, all bacterial strains were cultured aerobically, with shaking at 250 rpm and at a temperature of 37 °C in BD™ tryptic soy broth (TSB) or Luria-Bertani (LB) broth.
Table 3 Bacterial strains, sources and characteristics
| Bacterial strain |
Source |
Characteristics |
|
ATCC = American type culture collection, GPC = Gram-positive cocci, GNB = Gram-negative bacilli, ARLG = Antibacterial Resistance Leadership Group.
|
|
Staphylococcus aureus ATCC 25923 |
ATCC |
Methicillin susceptible. Reference strain for antibiotic susceptibility testing |
| MRSA USA300 |
ATCC/BEI resources |
Methicillin resistant. Most prevalent MRSA clinical infection strain in the US36 |
| MRSA ATCC 33592 |
William Wuest, Emory University |
Methicillin resistant, gentamicin resistant |
| SCCmec: type III |
|
Enterococcus faecalis ATCC 29212 |
ATCC |
Reference strain for antibiotic susceptibility testing. Susceptible to most GPC antibiotics |
|
Enterococcus faecalis ATCC 51575 (VRE) |
ATCC |
Presence of vanB |
| Resistant to gentamicin, streptomycin, and vancomycin. Sensitive to teicoplanin |
|
Enterococcus faecium ATCC 700221 (VRE) |
ATCC |
Presence of vanA |
| Resistant to vancomycin and teicoplanin |
|
Listeria monocytogenes ATCC 19115 |
ATCC |
Reference strain for antibiotic susceptibility testing. Susceptible to most GPC antibiotics |
|
Escherichia coli ATCC 25922 |
ATCC |
Reference strain for antibiotic susceptibility testing. Susceptible to most GNB antibiotics |
|
Clinical MRSA strains
|
ARLG |
See Table 2 |
4.2 Screening of pentafluorosulfanyl- and trifluoromethylthio-containing compounds for antibacterial activity
To evaluate the antibacterial activity of the synthesized compounds, S. aureus strains were grown in 10 mL TSB while E. coli were grown in LB broth. Cultures were cultivated overnight and diluted 1:1000 in cation-adjusted Mueller–Hinton broth (MHB) for growth to early exponential phase after the overnight incubation. Stock solutions of compounds in DMSO were dispensed into sterile glass tubes to yield a final concentration of 16 μg mL−1 when bacterial suspensions are added. 1 mL aliquots of bacterial culture at the exponential phase of growth were then added into the sterile glass tubes and incubated overnight at 37 °C. After overnight incubation, 100 μL of each sample was dispensed in 96-well microtiter plates and the optical density at 600 nm (OD600) was measured on a Biotek Cytation 5 multimode reader. In order to calculate the viability of bacteria when treated with compounds, the OD600 of compound-treated cultures was normalized with that of the negative control (DMSO-treated) samples.
To obtain the percent normalized OD600 we used the equation:
where for a given compound,
X is the OD
600 of culture with the compound,
Xo is that of medium only and
XT is the OD
600 of the DMSO control.
4.3 Minimum inhibitory concentration (MIC)
The MICs of the test compounds and other antibiotics against the various bacterial strains were determined following the guidelines of the Clinical Laboratory Standards Institute (CLSI) and as previously described.21 Briefly, control antibiotics and various test compounds at 10 mg mL−1 were prepared in DMSO. Calculated concentrations of compounds or control antibiotics were then diluted serially (1:2) along the ordinate of 96-well microtiter plates. Bacteria were standardized using the 0.5 McFarland standard and diluted (1:100) prior to addition into respective wells of the microtiter plates already containing serially diluted compounds or antibiotics. The MIC was determined after 18–20 h of incubation in a static incubator at 37 °C.
4.4 Minimum bactericidal concentration (MBC)
The MBCs of the lead compound HSD1835 and control antibiotics were determined as described previously.37 Briefly, aliquots of 4 μL from wells with no growth following the MIC assay were spotted in duplicate on tryptic soy agar (TSA) plates and incubated at 37 °C in a static incubator for 20 h before recording the MBC (99.9% reduction in number of colony-forming units (CFU)).
4.5 Time-kill analysis
For the exponential phase time-kill analysis, MRSA ATCC 33592 was grown overnight in TSB at 37 °C. The overnight growth was diluted 1:1000 and incubated to grow to the exponential phase (OD600 of 0.6). Bacteria in this logarithmic growth phase were then diluted to a 1 in 10 concentration (approximately 2.92 × 106 colony-forming units per mL (CFU per mL)) in TSB and exposed to antibiotic or compound concentrations equivalent to 4× MIC or 1× MIC (in triplicate) as previously described.38 A 100 μL aliquot from each treatment vial was collected after 0, 2, 4, 8, 12, and 24 h of incubation at 37 °C, serially diluted in phosphate-buffered saline (PBS), transferred to TSA plates in duplicate and incubated at 37 °C for 18–20 h.
For stationary phase time-kill analysis, MRSA ATCC 33592 was also used. In this case, after overnight growth, bacteria were diluted to a 1 in 10 concentration (approximately 2.92 × 106 colony-forming units per mL (CFU per mL) in TSB directly from the overnight culture and exposed to concentrations equivalent to 4× MIC or 1× MIC (in triplicate) of compounds or antibiotic controls. Aliquots from each treatment vial were collected after 0, 2, 4, and 6 h of incubation at 37 °C, serially diluted in phosphate-buffered saline (PBS), transferred to TSA plates in duplicate and incubated at 37 °C for 18–24 h.
4.6 Resistance generation
The potential for MRSA to develop resistance to HSD1835 was evaluated using the previously described multi-step resistance selection method.20,38 Briefly, the MICs of compound and control antibiotics were first determined against MRSA ATCC 33592 and MRSA USA300. This was subsequently followed by daily passaging of a 5 μL aliquot of bacterial culture from wells immediately after the MIC wells (sub-inhibitory MIC of compound) with turbidity like the growth control wells. The 5 μL aliquots were diluted 1:1000 in CA-MHB and used as the inoculum for determining the MIC for the next serial passage. Bacteria were passaged for 30 days. Resistance occurs when the MIC increases more than four-fold from the starting MIC.
4.7 Bacterial membrane depolarization and permeability assays
S. aureus ATCC 25923 was grown in TSB overnight at 37 °C and sub-cultured from the overnight broth to achieve exponential phase (OD600 0.2–0.3). For membrane depolarization, cells were harvested by centrifugation at 3600g at 4 °C for 10 min and washed 3× in membrane assay buffer at pH 7.0 (5 mM glucose + 10 mM HEPES) and pellets were resuspended in the same buffer to OD600 0.2–0.3. Then, DiSC3 was added to a final concentration of 1 μM and incubated in the dark at ambient temperature for 15–30 min to allow the loading of the fluorescent dye into the cell membranes. After the loading period, potassium chloride (KCl) was added to the S. aureus cell suspension to a final concentration of 100 mM. The assay was initiated by adding the antibiotic/compound according to calculated concentrations in triplicate in a black 96 well plate. This was immediately read in a Gen5 fluorescent imager at an excitation wavelength of 615 nm and emission wavelength of 665 nm for a period of 30 min at 30 s intervals. Note that the DiSC3 was dissolved in 100% DMSO (100 μM DiSC3) and was added to the bacteria + HEPES buffer to a final concentration of 1 μM DiSC3 and 1% DMSO. Keeping the DMSO at 1% and maintaining appropriate aeration was important for good solubility and fluorescence of the dye. The microtiter plates were subjected to shaking between measurement points. 1 μM gramicidin was used as the positive control while the negative control was suspended cells + DiSC3. The baseline sample was a vial with no drug and no DiSC3.
For membrane permeability, the Sytox green™ fluorescence assay was used. S. aureus ATCC 23592 was grown as above, centrifuged, washed in 1× PBS three times and pellets resuspended in 1× PBS + 10% TSB. Sytox green™ (Invitrogen) was added to achieve a final concentration of 5 μM. This was incubated for 15–30 min in the dark for fluorescence to stabilize. 100 μL of the suspension was then aliquoted in triplicate into a 96 well microtiter plate and initially read at an excitation wavelength of 504 nm and emission wavelength of 522 nm for 10 min before controls and compounds at calculated concentrations were added and absorbance read for another 3 h at 3 min intervals using a Biotek Cytation5 image reader.
4.8 Flow cytometry
To further assess the membrane permeability effect of HSD1835, we used a flow cytometry assay using To-PRO 3 iodide (red channel) as the membrane-permeable dye and DiOC2 (green channel) as the membrane-impermeable dye as previously described. Bithionol, a known membrane permeabilizer, was used as the positive control in this assay.33 Specifically, S. aureus ATCC 25923 were grown in TSB overnight at 37 °C and sub-cultured from the overnight broth to achieve exponential phase (OD600 0.2–0.3). Exponentially growing S. aureus were then treated with two concentrations of HSD1835 (2 μg mL−1 or 10 μg mL−1) or bithionol at 4 μg mL−1. Samples were centrifuged to remove the medium, washed twice in PBS and resuspended in buffer. To-PRO 3 iodide and DiOC2 were added to the resuspended bacteria and were immediately imaged in a BD Fortessa cell analyzer in the Purdue University Flow Cytometry and Cell Separation Facility, Bindley Bioscience Centre.
4.9 Scanning electron microscopy
Scanning electron microscopy was used to visualize the membrane effects of HSD1835. Samples were prepared according to previously reported methods with slight modifications.34,35 Briefly, exponential and stationary phase S. aureus were treated with either HSD1835 (4 μg mL−1) or bithionol (10 μg mL−1 for the exponential phase, 32 μg mL−1 for the stationary phase). Bithionol at these concentrations was used as the positive control based on the reports of Kim et al.33 For the exponential phase, S. aureus were grown in TSB at 37 °C overnight, diluted 1:100 and incubated to grow to exponential phase (OD600 of 0.4). Exponentially growing S. aureus were then treated with HSD1835 or bithionol for 4 h. For the stationary phase S. aureus, after overnight culture in TSB, samples were immediately treated for 6 h. Samples were centrifuged at 3600g to collect cells, washed three times in PBS and fixed in chilled 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer. Samples were post-fixed in 1% osmium tetroxide and dehydrated in increasing concentrations of ethanol (30–95%), transferred through three changes of hexamethyldisilazane (HMDS), and allowed to dry overnight. The dried samples were coated with platinum in a Cressington 208HR sputter coater and imaged using an FEI Nova Nano-SEM 200 instrument at 5 kV and at 40000–160000× magnifications.
4.10 Hemolysis assay
Hemolysis assay to determine the toxicity of the lead compound (HSD1835) to RBCs was carried out as previously described with slight modifications.39 Briefly, 5% human RBCs (purchased from Innovative Research, Inc., Novi, MI. Catalog number: IWB3CPDA1UNIT) were incubated with concentrations of the compound ranging from 5 μg mL−1 to 20 μg mL−1 and incubated for 30 minutes at 37 °C. Subsequently, the samples were centrifuged at 10000 rpm for 2 minutes to pellet intact RBCs. The supernatants were transferred to clear 96 well microtiter plates and absorbance was spectrophotometrically measured at 540 nm. The percent of RBC disruption/hemolysis was quantified relative to positive control samples lysed with 1% Triton X-100. DMSO was used as the negative control. All samples were run in triplicate.
To obtain percent hemolysis, we used the equation:
Biological experiments were approved by the Purdue University Institutional Biosafety Committee (IBC) Protocol Ref. # 16-001-19.
4.11 Chemical analysis
General experimental methods.
All the solvents and reagents were purchased from widely available commercial sources and utilized as received. The 1H and 13C NMR spectra were obtained in deuterated NMR solvents methanol-d4, chloroform-d, or DMSO-d6 using a 500 MHz spectrometer with tetramethylsilane as the internal standard. 1H NMR data were reported as follows: chemical shift (δ ppm) (multiplicity, coupling constant (Hz), integration). Spectral chemical shifts were reported in downfield order in parts per million (δ ppm). Multiplicities are reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, or combinations thereof. Electron spray ionization (ESI) technique and TOF mass analysis were used to record high-resolution mass spectra (HRMS). All the synthesized compounds were characterized using 1H NMR, 13C NMR, and HRMS.
General procedure for the multicomponent reaction.
Aromatic amine (1 mmol) and the corresponding aldehyde (1 mmol) in 4 mL of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) were stirred for 2 h at 80 °C. After that, the reaction was cooled to room temperature followed by addition of norbornene (2 mmol) and 10 mol% scandium(III) trifluoromethanesulfonate. The reaction mixture was stirred for another 8 to 12 h at room temperature. Upon reaction completion, the reaction mixture was concentrated and purified by silica gel chromatography (hexanes/ethyl acetate 95:5 to 50:50) to give the desired cyclized compound.
6-Cyclopropyl-2-(pentafluoro-λ6-sulfanyl)-5,6,6a,7,8,9,10,10a-octahydro-7,10-methanophenanthridine (1).
Off-white solid (120 mg, 33%); 1H NMR (500 MHz, methanol-d4) δ 7.49 (t, J = 1.6 Hz, 1H), 7.30 (dd, J = 8.9, 2.6 Hz, 1H), 6.68 (d, J = 8.8 Hz, 1H), 2.68 (d, J = 8.9 Hz, 1H), 2.41 (d, J = 4.2 Hz, 1H), 2.31 (d, J = 4.0 Hz, 1H), 2.10 (t, J = 7.9 Hz, 1H), 1.99–1.92 (m, 1H), 1.74–1.64 (m, 1H), 1.64–1.56 (m, 1H), 1.50–1.42 (m, 2H), 1.42–1.34 (m, 1H), 1.05 (d, J = 9.9 Hz, 1H), 0.94–0.84 (m, 1H), 0.65–0.56 (m, 1H), 0.51–0.42 (m, 1H), 0.40–0.32 (m, 1H), 0.32–0.24 (m, 1H); 13C NMR (126 MHz, methanol-d4) δ 149.5, 143.9 (quint, J = 15.1 Hz), 126.4, 125.8, 123.6, 113.7, 60.0, 50.3, 44.6, 43.5, 41.4, 33.4, 29.3, 28.8, 16.8, 2.8, 1.4; HRMS (ESI) m/z calcd for C17H21F5NS [M + H]+ 366.1315, found 366.1309.
6-(1H-Imidazol-4-yl)-2-(pentafluoro-λ6-sulfanyl)-5,6,6a,7,8,9,10,10a-octahydro-7,10-methanophenanthridine (2).
Off-white solid (160 mg, 41%); 1H NMR (500 MHz, methanol-d4) δ 8.74 (s, 1H), 7.58 (s, 1H), 7.40–7.36 (m, 2H), 6.72 (d, J = 8.8 Hz, 1H), 4.10 (d, J = 7.9 Hz, 1H), 2.80 (d, J = 8.9 Hz, 1H), 2.51 (d, J = 4.3 Hz, 1H), 2.30 (t, J = 8.3 Hz, 1H), 2.17 (d, J = 4.2 Hz, 1H), 1.79–1.69 (m, 1H), 1.68–1.57 (m, 2H), 1.55–1.46 (m, 1H), 1.39–1.30 (m, 1H), 1.17 (d, J = 10.2 Hz, 1H). 13C NMR (126 MHz, methanol-d4) δ 148.4, 145.0 (quint, J = 16.3 Hz), 136.4, 134.1, 126.2, 125.9, 124.0, 116.3, 114.2, 50.5, 49.5, 44.7, 43.3, 41.1, 33.3, 28.9, 28.7; HRMS (ESI) m/z calcd for C17H19F5N3S [M + H]+ 392.1220, found 392.1212.
2-(pentafluoro-λ6-sulfanyl)-6-(3-(trifluoromethyl)-1H-pyrazol-4-yl)-5,6,6a,7,8,9,10,10a-octahydro-7,10-methanophenanthridine (3, HSD1835).
Off-white solid (160 mg, 35%); 1H NMR (500 MHz, chloroform-d) δ 11.74 (s, 1H), 7.60 (d, J = 3.1 Hz, 2H), 7.37 (dd, J = 8.7, 2.5 Hz, 1H), 6.52 (d, J = 8.7 Hz, 1H), 4.08 (d, J = 7.9 Hz, 1H), 4.00 (s, 1H), 2.77 (d, J = 8.8 Hz, 1H), 2.58 (d, J = 4.3 Hz, 1H), 2.23 (t, J = 8.4 Hz, 1H), 2.13 (d, J = 4.2 Hz, 1H), 1.73–1.60 (m, 3H), 1.50–1.43 (m, 1H), 1.35–1.26 (m, 1H), 1.14 (dt, J = 10.3, 1.5 Hz, 1H);13C NMR (126 MHz, chloroform-d) δ 148.0, 145.7 (quint, J = 16.3 Hz), 140.2, 129.0, 126.7, 126.6, 124.4, 123.1, 122.7 (q, J = 269.6 Hz), 114.5, 52.0, 49.6, 44.4, 44.0, 41.0, 34.0, 29.5, 29.3; 19F NMR (471 MHz, chloroform-d)) δ −61.02. HRMS (ESI) m/z calcd for C18H18F8N3S [M + H]+ 460.1094, found 460.1088.
2-(pentafluoro-λ6-sulfanyl)-6-(1H-pyrazol-4-yl)-5,6,6a,7,8,9,10,10a-octahydro-7,10-methanophenanthridine (4).
Off-white solid (133 mg, 34%); 1H NMR (500 MHz, chloroform-d) δ 7.61 (s, 3H), 7.36 (dd, J = 8.8, 2.5 Hz, 1H), 6.53 (d, J = 8.8 Hz, 1H), 3.77 (d, J = 9.4 Hz, 1H), 2.70 (d, J = 8.9 Hz, 1H), 2.62 (d, J = 4.2 Hz, 1H), 2.23–2.15 (m, 1H), 2.12 (d, J = 4.3 Hz, 1H), 1.75–1.69 (m, 1H), 1.61–1.56 (m, 2H), 1.49–1.36 (m, 1H), 1.32–1.23 (m, 1H), 1.14 (d, J = 10.2 Hz, 1H); 13C NMR (126 MHz, chloroform-d)) δ 148.8, 145.4 (quint, J = 16.3 Hz), 132.4, 126.6, 126.4, 124.2, 114.2, 52.9, 51.5, 43.8, 43.3, 40.3, 33.9, 29.5, 29.1; HRMS (ESI) m/z calcd for C17H15F5N3S [M + H]+ 388.0906, found 388.0906.
2-(pentafluoro-λ6-sulfanyl)-6-(thiophen-2-yl)-5,6,6a,7,8,9,10,10a-octahydro-7,10-methanophenanthridine (5).
Off-white solid (151 mg, 37%); 1H NMR (500 MHz, chloroform-d) 1H NMR δ 7.62 (s, 1H), 7.37 (dd, J = 8.8, 2.6 Hz, 1H), 7.30–7.24 (m, 1H), 7.05–7.02 (m, 1H), 7.00–6.96 (m, 1H), 6.54 (d, J = 8.7 Hz, 1H), 4.15 (s, 1H), 4.02 (d, J = 9.3 Hz, 1H), 2.74 (d, J = 9.0 Hz, 1H), 2.63 (d, J = 4.3 Hz, 1H), 2.26 (t, J = 9.0 Hz, 1H), 2.20 (d, J = 4.3 Hz, 1H), 1.76–1.67 (m, 1H), 1.63–1.57 (m, 2H), 1.46 (tdd, J = 11.5, 4.2, 2.0 Hz, 1H), 1.33–1.25 (m, 1H), 1.19–1.15 (m, 1H); 13C NMR (126 MHz, chloroform-d) δ 148.3, 147.0, 145.6 (quint, J = 16.3 Hz), 126.5, 126.4, 125.2, 125.0, 124.2, 114.4, 56.0, 53.6, 43.9, 43.4, 40.5, 34.0, 29.5, 29.0; HRMS (ESI) m/z calcd for C18H19F5NS2 [M + H]+ 408.0879, found 408.0871.
4-(2-(pentafluoro-λ6-sulfanyl)-5,6,6a,7,8,9,10,10a-octahydro-7,10-methanophenanthridin-6-yl)benzoic acid (6).
Off-white solid (214 mg, 48%); 1H NMR (500 MHz, DMSO-d6) δ 7.93 (d, J = 7.9 Hz, 2H), 7.57 (s, 1H), 7.52 (d, J = 8.0 Hz, 2H), 7.39 (dd, J = 8.9, 2.7 Hz, 1H), 6.78 (d, J = 8.9 Hz, 1H), 6.48 (s, 1H), 3.71 (d, J = 9.1 Hz, 1H), 2.66 (d, J = 8.9 Hz, 1H), 2.53 (d, J = 4.1 Hz, 1H), 2.09–2.04 (m, 1H), 2.03–1.99 (m, 1H), 1.65–1.54 (m, 2H), 1.49–1.36 (m, 2H), 1.15–1.02 (m, 2H); 13C NMR (126 MHz, DMSO-d6) δ 167.6, 150.6, 149.4, 143.4 (quint, J = 15.1 Hz), 130.3, 129.9, 128.4, 126.0, 125.6, 124.3, 114.7, 59.0, 52.6, 43.6, 43.5, 34.0, 29.6, 28.9; HRMS (ESI) m/z calcd for C21H21F5NO2S [M + H]+ 446.1213, found 446.1210.
6-(3-(Trifluoromethyl)-1H-pyrazol-4-yl)-5,6,6a,7,8,9,10,10a-octahydro-7,10-methanophenanthridine (7).
Off-white solid (50 mg, 15%); 1H NMR (500 MHz, chloroform-d) δ 7.68 (d, J = 1.2 Hz, 1H), 7.31–7.22 (m, 1H), 7.02 (tt, J = 7.7, 1.2 Hz, 1H), 6.84 (td, J = 7.5, 1.2 Hz, 1H), 6.58 (dd, J = 7.9, 1.3 Hz, 1H), 4.02 (d, J = 8.8 Hz, 1H), 2.76 (d, J = 8.8 Hz, 1H), 2.65 (d, J = 4.2 Hz, 1H), 2.28–2.18 (m, 1H), 2.10 (t, J = 2.6 Hz, 1H), 1.71–1.63 (m, 2H), 1.61–1.54 (m, 1H), 1.49–1.39 (m, 1H), 1.35–1.21 (m, 1H), 1.12 (dt, J = 10.2, 1.5 Hz, 1H); 13C NMR (126 MHz, chloroform-d) δ 145.9, 140.1 (q, J = 37.8 Hz), 129.4, 128.6, 127.4, 126.3, 123.6, 122.9 (q, J = 269.6 Hz), 119.9, 115.6, 52.9, 50.3, 44.0, 43.5, 40.4, 34.0, 29.6, 29.4. HRMS (ESI) m/z calcd for C18H19F3N3 [M + H]+ 334.1531, found 334.1524.
2-Fluoro-6-(3-(trifluoromethyl)-1H-pyrazol-4-yl)-5,6,6a,7,8,9,10,10a-octahydro-7,10-methanophenanthridine (8).
Off-white solid (92 mg, 26%); 1H NMR (500 MHz, chloroform-d) δ 7.68 (s, 1H), 6.98 (dd, J = 9.8, 2.9 Hz, 1H), 6.72 (td, J = 8.4, 2.9 Hz, 1H), 6.50 (dd, J = 8.6, 4.9 Hz, 1H), 2.72 (d, J = 8.8 Hz, 1H), 2.59 (d, J = 4.3 Hz, 1H), 2.22–2.15 (m, 1H), 2.09 (d, J = 4.2 Hz, 1H), 1.71–1.66 (m, 1H), 1.64–1.56 (m, 2H), 1.46–1.36 (m, 1H), 1.30–1.23 (m, 2H), 1.14–1.07 (m, 1H); 13C NMR (126 MHz, chloroform-d) δ 158.0 (d, J = 238.14 Hz), 142.1, 140.2 (q, J = 134.02 Hz), 129.3, 129.0, 123.4, 122.9 (q, J = 269.6 Hz), 116.2, 114.9 (d, J = 22.6 Hz), 113.0, (d, J = 22.6 Hz), 52.7, 50.7, 44.3, 43.4, 40.3, 34.0, 29.4, 29.4. HRMS (ESI) m/z calcd for C18H18F4N3 [M + H]+ 352.1436, found 352.1431.
6-(3-(Trifluoromethyl)-1H-pyrazol-4-yl)-5,6,6a,7,8,9,10,10a-octahydro-7,10-methanophenanthridine-2-carbonitrile (9).
Off-white solid (179 mg, 50%); 1H NMR (500 MHz, DMSO-d6) δ 7.91 (d, J = 2.1 Hz, 1H), 7.56 (s, 1H), 7.29 (dd, J = 8.3, 1.9 Hz, 1H), 6.75 (dd, J = 8.3, 2.3 Hz, 1H), 6.40 (s, 1H), 3.78 (dd, J = 8.9, 4.4 Hz, 1H), 3.15 (d, J = 5.1 Hz, 1H), 2.62 (d, J = 8.8 Hz, 1H), 2.56 (d, J = 4.1 Hz, 1H), 2.13 (t, J = 8.8 Hz, 1H), 1.95 (d, J = 4.1 Hz, 1H), 1.58 (ddt, J = 11.7, 8.6, 4.3 Hz, 1H), 1.52–1.42 (m, 2H), 1.38 (tt, J = 9.5, 5.4 Hz, 1H), 1.20–1.13 (m, 1H), 1.03 (d, J = 10.0 Hz, 1H); 13C NMR (126 MHz, DMSO-d6) δ 151.5, 138.9 (q, J = 35.2 Hz), 132.9, 130.6, 130.2, 126.8, 123.8 (q, J = 268.3 Hz), 122.5, 120.8, 115.7, 99.0, 52.5, 49.2, 49.1, 43.5, 43.1, 33.8, 29.5, 29.1; HRMS (ESI) m/z calcd for C19H18F3N4 [M + H]+ 359.1483, found 352.1478.
6-(3-(Trifluoromethyl)-1H-pyrazol-4-yl)-5,6,6a,7,8,9,10,10a-octahydro-7,10-methanophenanthridine-2-sulfonamide (10).
Off-white solid (144 mg, 35%); 1H NMR (500 MHz, methanol-d4) δ 7.73 (d, J = 16.6 Hz, 2H), 7.47 (dd, J = 8.4, 2.2 Hz, 1H), 6.72 (d, J = 8.3 Hz, 1H), 3.98 (d, J = 8.2 Hz, 1H), 2.75 (d, J = 8.8 Hz, 1H), 2.61 (d, J = 4.2 Hz, 1H), 2.24 (t, J = 8.5 Hz, 1H), 2.13–1.95 (m, 1H), 1.77–1.68 (m, 1H), 1.65–1.55 (m, 2H), 1.47 (ddd, J = 14.5, 10.2, 4.4 Hz, 1H), 1.30 (dt, J = 10.6, 5.8 Hz, 1H), 1.12 (d, J = 10.0 Hz, 1H); 13C NMR (126 MHz, methanol-d4) δ 150.3, 139.5 (q, J = 35.2 Hz), 132.3, 129.5, 128.9, 126.7, 124.3, 123.2 (q, J = 268.3 Hz), 122.8, 114.5, 52.5, 49.6, 44.0, 43.6, 40.8, 33.2, 28.9, 28.9; HRMS (ESI) m/z calcd for C18H20F3N4O2S [M + H]+ 413.1259, found 413.1253.
6-(3-(Trifluoromethyl)-1H-pyrazol-4-yl)-5,6,6a,7,8,9,10,10a-octahydro-7,10-methanophenanthridine-2-carboxamide (11).
Off-white solid (90 mg, 24%); 1H NMR (500 MHz, methanol-d4) δ 7.80 (s, 1H), 7.69 (s, 1H), 7.52 (dd, J = 8.4, 2.2 Hz, 1H), 6.68 (d, J = 8.4 Hz, 1H), 3.98 (d, J = 8.2 Hz, 1H), 2.73 (d, J = 8.8 Hz, 1H), 2.64 (d, J = 4.2 Hz, 1H), 2.21 (t, J = 8.5 Hz, 1H), 2.07 (d, J = 4.1 Hz, 1H), 1.72–1.64 (m, 1H), 1.62–1.54 (m, 2H), 1.48–1.41 (m, 1H), 1.31–1.26 (m, 1H), 1.12–1.06 (m, 1H); 13C NMR (126 MHz, methanol-d4) δ 171.4, 150.4, 139.2 (q, J = 35.2 Hz), 128.9, 128.4, 126.3, 126.0, 123.2 (q, J = 268.3 Hz), 123.0, 123.0, 114.5, 52.6, 49.7, 43.9, 43.6, 40.8, 33.3, 29.0, 29.0; HRMS (ESI) m/z calcd for C19H20F3N4O [M + H]+ 377.1589, found 377.1584.
2-(Difluoromethoxy)-6-(3-(trifluoromethyl)-1H-pyrazol-4-yl)-5,6,6a,7,8,9,10,10a-octahydro-7,10-methanophenanthridine (12).
Off-white solid (120 mg, 30%); 1H NMR (500 MHz, chloroform-d) δ 7.69–7.62 (m, 1H), 7.26 (s, 1H), 7.05 (d, J = 2.6 Hz, 1H), 6.80 (dd, J = 8.5, 2.7 Hz, 1H), 6.53 (d, J = 8.5 Hz, 1H), 6.34 (d, J = 74.8 Hz, 1H), 3.98 (d, J = 8.8 Hz, 1H), 2.73 (d, J = 8.8 Hz, 1H), 2.59 (d, J = 4.3 Hz, 1H), 2.19 (t, J = 8.9 Hz, 1H), 2.10 (d, J = 4.2 Hz, 1H), 1.74–1.53 (m, 3H), 1.48–1.38 (m, 1H), 1.33–1.22 (m, 1H), 1.16–1.09 (m, 1H); 13C NMR (126 MHz, chloroform-d) δ 144.2, 143.7, 140.2, 129.2, 128.7, 123.3, 122.8 (q, J = 269.6 Hz), 120.5, 118.4, 116.5 (t, J = 259.5 Hz), 116.1, 52.6, 50.4, 44.2, 43.6, 40.4, 34.0, 29.4, 29.4. HRMS (ESI) m/z calcd for C19H19F5N3O [M + H]+ 400.1448, found 400.1443.
2-(Trifluoromethoxy)-6-(3-(trifluoromethyl)-1H-pyrazol-4-yl)-5,6,6a,7,8,9,10,10a-octahydro-7,10-methanophenanthridine (13).
Off-white solid (129 mg, 31%);1H NMR (500 MHz, methanol-d4) δ 7.73 (d, J = 5.6 Hz, 1H), 7.10 (d, J = 5.4 Hz, 1H), 6.85 (t, J = 6.9 Hz, 1H), 6.72–6.65 (m, 1H), 3.87 (t, J = 7.7 Hz, 1H), 2.70 (t, J = 7.5 Hz, 1H), 2.58–2.52 (m, 1H), 2.24–2.18 (m, 1H), 2.07–2.01 (m, 1H), 1.73–1.66 (m, 1H), 1.62–1.55 (m, 2H), 1.48–1.39 (m, 1H), 1.31–1.22 (m, 1H), 1.10 (t, J = 8.1 Hz, 1H); 13C NMR (126 MHz, methanol-d4) δ 145.9, 141.7, 139.7 (J = 36.5 Hz), 128.9, 128.5, 123.2 (J = 269.6 Hz), 122.7, 121.7 (J = 253.2 Hz), 120.8, 118.8, 115.8, 52.8, 50.3, 43.9, 43.4, 40.3, 33.2, 29.0, 28.8; HRMS (ESI) m/z calcd for C19H18F6N3O [M + H]+ 418.1354, found 418.1348.
2-Isopropoxy-6-(3-(trifluoromethyl)-1H-pyrazol-4-yl)-5,6,6a,7,8,9,10,10a-octahydro-7,10-methanophenanthridine (14).
Off-white solid (142 mg, 36%); 1H NMR (500 MHz, methanol-d4) δ 7.76 (s, 1H), 6.82 (d, J = 2.6 Hz, 1H), 6.65–6.55 (m, 2H), 4.43 (p, J = 6.0 Hz, 1H), 3.79 (d, J = 9.6 Hz, 1H), 2.69 (d, J = 8.8 Hz, 1H), 2.61 (d, J = 4.3 Hz, 1H), 2.00 (d, J = 4.2 Hz, 1H), 1.73–1.63 (m, 1H), 1.60–1.53 (m, 2H), 1.48–1.38 (m, 1H), 1.27 (d, J = 6.0 Hz, 6H), 1.09 (d, J = 10.0 Hz, 1H); 13C NMR (126 MHz, methanol-d4) δ 151.6, 140.7, 139.8 (q, J = 36.5), 139.5, 128.8, 128.6, 123.2 (q, J = 268.3), 122.8, 116.5, 116.2, 114.7, 70.7, 53.0, 51.0, 44.2, 42.9, 40.0, 33.2, 29.0, 28.9, 21.1; HRMS (ESI) m/z calcd for C21H25F3N3O [M + H]+ 392.1950, found 392.1944.
7-(3-(Trifluoromethyl)-1H-pyrazol-4-yl)-6,7,7a,8,9,10,11,11a-octahydro-8,11-methano[1,3]dioxolo[4,5-a]phenanthridine (15).
Off-white solid (158 mg, 42%); 1H NMR (500 MHz, methanol-d4) δ 7.75 (s, 1H), 6.72 (d, J = 1.0 Hz, 1H), 6.29 (s, 1H), 5.79 (s, 2H), 3.79 (d, J = 9.5 Hz, 1H), 2.61 (d, J = 8.8 Hz, 1H), 2.53 (d, J = 4.3 Hz, 1H), 2.13–2.11 (m, 1H), 1.98 (d, J = 4.2 Hz, 1H), 1.72–1.62 (m, 1H), 1.62–1.56 (m, 1H), 1.59–1.49 (m, 1H), 1.44–1.35 (m, 1H), 1.29–1.20 (m, 1H), 1.10–1.04 (m, 1H); 13C NMR (126 MHz, methanol-d4) δ 145.7, 141.4, 141.1, 139.8 (q, J = 34.0 Hz), 128.8, 123.2 (q, J = 268.3 Hz), 122.7, 119.5, 107.5, 100.2, 97.1, 53.0, 50.9, 44.1, 43.0, 40.0, 33.0, 29.0, 28.9; HRMS (ESI) m/z calcd for C19H19F3N3O2 [M + H]+ 378.1429, found 378.1424.
6-(3-(Trifluoromethyl)-1H-pyrazol-4-yl)-2-((trifluoromethyl)thio)-5,6,6a,7,8,9,10,10a-octahydro-7,10-methanophenanthridine (16).
Off-white solid (139 mg, 32%); 1H NMR (500 MHz, chloroform-d) δ 11.45 (s, 1H), 7.62 (s, 1H), 7.51 (s, 1H), 7.28–7.25 (m, 1H), 6.58 (d, J = 8.3 Hz, 1H), 4.07 (d, J = 8.2 Hz, 1H), 2.76 (d, J = 8.8 Hz, 1H), 2.59 (d, J = 4.3 Hz, 1H), 2.26–2.19 (m, 1H), 2.12 (d, J = 4.2 Hz, 1H), 1.74–1.67 (m, 1H), 1.66–1.58 (m, 4H), 1.48–1.40 (m, 1H), 1.33–1.25 (m, 1H), 1.13 (d, J = 10.3, 1H); 13C NMR (126 MHz, chloroform-d) δ 147.8 (q, J = 21.4), 140.3 (q, J = 36.5 Hz), 137.5, 135.0, 133.4 (q, J = 308.7 Hz), 129.0, 128.5, 123.0, 122.7 (q, J = 269.6 Hz), 116.5, 113.1 52.1, 49.9, 44.2, 43.7, 40.8, 34.0, 29.4, 29.4; HRMS (ESI) m/z calcd for C19H18F6N3 [M + H]+ 434.1125, found 434.1118.
3-(Trifluoromethyl)-6-(3-(trifluoromethyl)-1H-pyrazol-4-yl)-5,6,6a,7,8,9,10,10a-octahydro-7,10-methanophenanthridine (17).
Off-white solid (104 mg, 24%); 1H NMR (500 MHz, methanol-d4) δ 7.74–7.71 (m, 1H), 7.36 (d, J = 8.0 Hz, 1H), 6.98 (dd, J = 8.0, 1.9 Hz, 1H), 6.94 (d, J = 1.9 Hz, 1H), 3.92 (d, J = 8.7 Hz, 1H), 2.75 (d, J = 8.9 Hz, 1H), 2.63 (d, J = 4.3 Hz, 1H), 2.24 (t, J = 8.8 Hz, 1H), 2.06 (d, J = 4.2 Hz, 1H), 1.77–1.67 (m, 1H), 1.63–1.55 (m, 2H), 1.49–1.41 (m, 1H), 1.33–1.25 (m, 1H), 1.12 (d, J = 10.1 Hz, 1H). 13C NMR (126 MHz, methanol-d4) δ 147.2, 139.4 (q, J = 36.5 Hz), 131.1, 130.2, 128.9, 128.6 (q, J = 31.5 Hz), 128.2, 125.5 (q, J = 270.9 Hz), 123.2 (q, J = 269.6 Hz), 122.8, 114.8, 111.5, 52.9, 50.0, 43.8, 43.6, 40.4, 33.2, 29.0, 28.9; HRMS (ESI) m/z calcd for C19H17F6N3 [M + H]+ 402.1404, found 402.1395.
2-(pentafluoro-λ6-sulfanyl)-6-(3-(trifluoromethyl)-1H-pyrazol-4-yl)-7,8,9,10-tetrahydro-7,10-methanophenanthridine (18).
In a solution of HSD1835 (0.32 mmol, 150 mg) in acetonitrile (5 mL), DDQ (1.3 equiv.) was added and the mixture was stirred at room temperature. After completion of the reaction the mixture was quenched with 2 N NaOH aqueous solution and extracted with ethyl acetate. The organic layer was dried over Na2SO4, concentrated and purified via silica gel chromatography to yield the product as an off-white solid (90 mg, 62%); 1H NMR (500 MHz, methanol-d4) δ 8.54 (d, J = 2.5 Hz, 1H), 8.16 (s, 1H), 8.12 (d, J = 9.4 Hz, 1H), 8.05 (dt, J = 9.4, 2.1 Hz, 1H), 4.23–4.18 (m, 1H), 3.65–3.60 (m, 1H), 2.24–2.16 (m, 1H), 2.16–2.06 (m, 1H), 1.99–1.93 (m, 1H), 1.78 (dt, J = 9.1, 1.6 Hz, 1H), 1.30–1.14 (m, 2H); 13C NMR (126 MHz, methanol-d4) δ 156.9, 151.1 (quint, J = 17.6 Hz), 147.8, 146.6, 141.4, 130.8, 129.8, 124.8, 122.6, 121.2, 120.6 (q, J = 269.6 Hz), 118.5, 49.6, 42.7, 41.7, 25.9, 25.0; HRMS (ESI) m/z calcd for C18H14F8N3S [M + H]+ 456.0780, found 408.0774.
Author contributions
HOS oversaw the project. ND synthesized the compounds. KIO performed antimicrobial assays. All authors contributed to the writing of the manuscript.
Conflicts of interest
There is no conflict of interest.
Acknowledgements
We thank Purdue University for funding. The research described in this publication is supported, at least in part, by a grant from the National Institutes of Health through Duke University and the findings, opinions and recommendations expressed herein are those of the author and not necessarily those of Duke University or National Institutes of Health. Bacterial isolates provided herein and identified as ARLG were provided by the Antibacterial Resistance Leadership Group.
References
- G. Hobby, K. Meyer and E. Chaffee, Exp. Biol. Med., 1942, 50, 281–285 CrossRef CAS.
- J. Bigger, Lancet, 1944, 244, 497–500 CrossRef.
- R. A. Fisher, B. Gollan and S. Helaine, Nat. Rev. Microbiol., 2017, 15, 453–464 CrossRef CAS PubMed.
- B. P. Conlon, BioEssays, 2014, 36, 991–996 CrossRef PubMed.
- B. P. Conlon, E. S. Nakayasu, L. E. Fleck, M. D. LaFleur, V. M. Isabella, K. Coleman, S. N. Leonard, R. D. Smith, J. N. Adkins and K. Lewis, Nature, 2013, 503, 365–370 CrossRef CAS PubMed.
- Y. P. Chong, S. J. Park, H. S. Kim, E. S. Kim, M. N. Kim, K. H. Park, S. H. Kim, S. O. Lee, S. H. Choi, J. Y. Jeong, J. H. Woo and Y. S. Kim, Medicine, 2013, 92, 98–108 CrossRef CAS PubMed.
- R. Khatib, L. B. Johnson, M. Sharma, M. G. Fakih, R. Ganga and K. Riederer, Scand. J. Infect. Dis., 2009, 41, 4–9 CrossRef CAS PubMed.
- D. H. Wyllie, D. W. Crook and T. E. Peto, BMJ, 2006, 333, 281 CrossRef PubMed.
- R. Khatib, L. B. Johnson, M. G. Fakih, K. Riederer, A. Khosrovaneh, M. Shamse Tabriz, M. Sharma and S. Saeed, Scand. J. Infect. Dis., 2006, 38, 7–14 CrossRef PubMed.
- V. G. Fowler, G. Sakoulas, L. M. McIntyre, V. G. Meka, R. D. Arbeit, C. H. Cabell, M. E. Stryjewski, G. M. Eliopoulos, L. B. Reller, G. R. Corey, T. Jones, N. Lucindo, M. R. Yeaman and A. S. Bayer, J. Infect. Dis., 2004, 190, 1140–1149 CrossRef CAS PubMed.
-
Centers for Disease Control & Prevention, (CDC), Threat Rep., 2013, pp. 50–52 Search PubMed.
-
World Health Organization, (WHO), Report on the Burden of Endemic Health Care-associated Infection Worldwide, 2011, pp. 1–40 Search PubMed.
- J. Enström, I. Fröding, C. G. Giske, K. Ininbergs, X. Bai, G. Sandh, U. B. Tollström, M. Ullberg and H. Fang, PLoS One, 2018, 13, e0205761 CrossRef PubMed.
- B. Allegranzi, S. Bagheri Nejad, C. Combescure, W. Graafmans, H. Attar, L. Donaldson and D. Pittet, Lancet, 2011, 377, 228–241 CrossRef.
- H. Hanberger, S. Walther, M. Leone, P. S. Barie, J. Rello, J. Lipman, J. C. Marshall, A. Anzueto, Y. Sakr, P. Pickkers, P. Felleiter, M. Engoren, J. L. Vincent and EPIC II Group of Investigators, Int. J. Antimicrob. Agents, 2011, 38, 331–335 CrossRef CAS PubMed.
- C. Westfall, A. L. Flores-Mireles, J. I. Robinson, A. J. L. Lynch, S. Hultgren, J. P. Henderson and P. A. Levin, Antimicrob. Agents Chemother., 2019, 63, 2312–2318 CrossRef PubMed.
- J. Yang, H. X. Wang, J. Xie, L. Li, J. Wang, E. C. K. Wan and X. P. Zhong, Front. Immunol., 2019, 10, 3048 CrossRef CAS PubMed.
- J. L. Copitch, R. N. Whitehead and M. A. Webber, Int. J. Antimicrob. Agents, 2010, 36, 247–251 CrossRef CAS PubMed.
- W. Kim, W. Zhu, G. L. Hendricks, D. Van Tyne, A. D. Steele, C. E. Keohane, N. Fricke, A. L. Conery, S. Shen, W. Pan, K. Lee, R. Rajamuthiah, B. B. Fuchs, P. M. Vlahovska, W. M. Wuest, M. S. Gilmore, H. Gao, F. M. Ausubel and E. Mylonakis, Nature, 2018, 556, 103–107 CrossRef CAS PubMed.
- C. Opoku-Temeng, G. A. Naclerio, H. Mohammad, N. Dayal, N. S. Abutaleb, M. N. Seleem and H. O. Sintim, Eur. J. Med. Chem., 2018, 155, 797–805 CrossRef CAS PubMed.
- N. Dayal, C. Opoku-Temeng, H. Mohammad, N. S. Abutaleb, D. Hernandez, K. I. Onyedibe, M. Wang, M. Zeller, M. N. Seleem and H. O. Sintim, ACS Infect. Dis., 2019, 5, 1820–1830 CrossRef CAS PubMed.
-
(a) G. A. Naclerio, N. S. Abutaleb, K. I. Onyedibe, M. N. Seleem and H. O. Sintim, RSC Med. Chem., 2020, 11, 102–110 RSC;
(b) G. A. Naclerio, N. S. Abutaleb, D. Li, M. N. Seleem and H. O. Sintim, J. Med. Chem., 2020, 63(20), 11934–11944 CrossRef CAS PubMed.
- N. Dayal, M. Wang and H. O. Sintim, ACS Omega, 2020, 5, 23799–23807 CrossRef CAS PubMed.
- N. A. Turner, B. K. Sharma-Kuinkel, S. A. Maskarinec, E. M. Eichenberger, P. P. Shah, M. Carugati, T. L. Holland and V. G. Fowler, Nat. Rev. Microbiol., 2019, 17, 203–218 CrossRef CAS PubMed.
- A. Shittu, O. Oyedara, F. Abegunrin, K. Okon, A. Raji, S. Taiwo, F. Ogunsola, K. Onyedibe and G. Elisha, BMC Infect. Dis., 2012, 12, 286 CrossRef PubMed.
- K. Lewis, Annu. Rev. Microbiol., 2010, 64, 357–372 CrossRef CAS PubMed.
- N. V. Ogryzko, A. Lewis, H. L. Wilson, A. H. Meijer, S. A. Renshaw and P. M. Elks, J. Immunol., 2019, 202, 494–502 CrossRef CAS PubMed.
- R. R. Watkins, M. Holubar and M. Z. David, Antimicrob. Agents Chemother., 2019, 63, e01216–e01219 CrossRef CAS PubMed.
- F. Alhomoud, Z. Aljamea, R. Almahasnah, K. Alkhalifah, L. Basalelah and F. K. Alhomoud, Int. J. Infect. Dis., 2017, 57, 3–12 CrossRef PubMed.
- M. Atif, S. Asghar, I. Mushtaq, I. Malik, A. Amin, Z. U. Babar and S. Scahill, Infect. Drug Resist., 2019, 12, 687–699 CrossRef PubMed.
- E. Gebeyehu, L. Bantie and M. Azage, PLoS One, 2015, 10, e0138179 CrossRef PubMed.
- V. Tangcharoensathien, S. Chanvatik and A. Sommanustweechai, Bull. W. H. O., 2018, 96, 141–144 CrossRef PubMed.
- W. Kim, G. Zou, T. P. A. Hari, I. K. Wilt, W. Zhu, N. Galle, H. A. Faizi, G. L. Hendricks, K. Tori, W. Pan, X. Huang, A. D. Steele, E. E. Csatary, M. M. Dekarske, J. L. Rosen, N. Q. Ribeiro, K. Lee, J. Port, B. B. Fuchs, P. M. Vlahovska, W. M. Wuest, H. Gao, F. M. Ausubel and E. Mylonakis, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 16529–16534 CrossRef CAS PubMed.
- H. Côté, A. Pichette, F. Simard, M. E. Ouellette, L. Ripoll, M. Mihoub, D. Grimard and J. Legault, Front. Microbiol., 2019, 10, 2341 CrossRef PubMed.
- J. P. de Sousa, R. E. A. Torres, G. A. de Azerêdo, R. C. Figueiredo, M. A. Vasconcelos and E. L. de Souza, Int. J. Food Microbiol., 2012, 158, 9–13 CrossRef PubMed.
- V. S. Albrecht, B. M. Limbago, G. J. Moran, A. Krishnadasan, R. J. Gorwitz, L. K. McDougal, D. A. Talan and E. I. N. S. Group, J. Clin. Microbiol., 2015, 53, 3478–3484 CrossRef CAS PubMed.
- M. ElAwamy, H. Mohammad, A. Hussien, N. S. Abutaleb, M. Hagras, R. A. T. Serya, A. T. Taher, K. A. Abouzid, M. N. Seleem and A. S. Mayhoub, Eur. J. Med. Chem., 2018, 152, 318–328 CrossRef CAS PubMed.
- H. Mohammad, W. Younis, H. G. Ezzat, C. E. Peters, A. AbdelKhalek, B. Cooper, K. Pogliano, J. Pogliano, A. S. Mayhoub and M. N. Seleem, PLoS One, 2017, 12, e0182821 CrossRef PubMed.
- L. A. Gould, A. B. Lansley, M. B. Brown, B. Forbes and G. P. Martin, J. Pharm. Pharmacol., 2000, 52, 1203–1209 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1md00211b |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *abc
*abc