
Dual-targeting SERS-encoded graphene oxide nanocarrier for intracellular co-delivery of doxorubicin and 9-aminoacridine with enhanced combination therapy†
Hui
Chen
 *,
Longqiang
Xing
,
Huiru
Guo
,
Caixia
Luo
and
Xuedian
Zhang
*,
Longqiang
Xing
,
Huiru
Guo
,
Caixia
Luo
and
Xuedian
Zhang
Key Laboratory of Optical Technology and Instrument for Medicine, Ministry of Education, University of Shanghai for Science and Technology, 200093 Shanghai, China. E-mail: chenhui@usst.edu.cn
First published on 27th September 2021
Abstract
A graphene oxide (GO)-based nanocarrier that imparts tumor-selective delivery of dual-drug with enhanced therapeutic index, is introduced. GO is conjugated with Au@Ag and Fe3O4 nanoparticles, which facilitates it with SERS tracking and magnetic targeting abilities, followed by the covalent binding of the anti-HER2 antibody, thus allowing it to both actively and passively target SKBR3 cells, human breast cancer cells expressed with HER2. Intracellular drug delivery behaviors are probed using SERS spectroscopy in a spatiotemporal manner, which demonstrates that nanocarriers are internalized into the lysosomes and release the drug in response to the acidic microenvironment. The nanocarriers loaded with dual-drug possess increased cancer cytotoxicity in comparison to those loaded with a single drug. Attractively, the enhanced cytotoxicity against cancer cells is achieved with relatively low concentrations of the drug, which is demonstrated to be involved in the drug adsorption status. These results may give us the new prospects to design GO-based delivery systems with rational drug dosages, thus achieving optimal therapeutic response of the multi-drug with increased tumor selectivity and reduced side effects.
Introduction
Conventional chemotherapy of anticancer drugs often goes along with nonspecific intracellular distribution that limit the chemotherapeutic efficacy.1,2 In addition, cancer-induced drug resistance is another serious issue involved in chemotherapy, which also reduces the therapeutic efficacy.3–5 To resolve these issues, novel therapeutic systems with specific targeting abilities have been designed to selectively deliver anticancer drugs to cancer cells efficiently.1,6–9 Moreover, two or more therapeutic agents can be exploited in the therapy to conquer the tumor-induced drug resistance.10–14In recent years, versatile nanoparticle-based targeted drug delivery systems are attractive in improving the anti-tumor therapy.15–18 Nanocarriers are rationally developed to be integrated with ligands that can specifically recognize signatures on the membranes of tumor cells. For instance, folic acid,19–21 transferrin,22,23 peptide24 and antibodies25–28 are alternatives to serve as the active targeting ligands. Advanced drug delivery systems with recognition abilities can improve the specific cellular uptake of anti-tumor drugs, and simultaneously decrease nonspecific accumulation in normal tissue sites, which are beneficial for limiting the serious side effects. In addition to this active targeting, a passive magnetic targeting strategy is also employed to regulate the drug delivery by guiding the drug nanocarriers directly into the desired tumor site under an external magnetic field.29–31 Through the combination of both active and passive targeting strategies, drug delivery systems can be directly transferred to tumor sites and, subsequently, internalized into cancer cells by recognition with cell surface receptors. This is superior in the demands of enhanced therapeutic efficacy as well as decreased side effects.
As a new type of carbon material, GO is growing to be a hot topic in biomedical research recently. Accompanied by the application potential is the great concern about its biosafety. Varied physicochemical properties, including types, size, layer number and surface functionalization, influence the biosafety of GO. Manipulation of these properties offers the opportunity to regulate the toxicity of GO. Among them, the proper surface modification can be effective, since multi-functionalized GO can be obtained through its abundant oxygen groups on the surface, such as hydroxyl and carboxylic groups.23 As such, despite there being still a long journey to study the health risks of GO materials, we can foresee the attractive opportunities of GO as drug nanocarriers.32–34 Moreover, GO-based nanocarrier is advantageous for high drug loading capacity and pH-sensitive drug release.35 Multi-drug loading on GO may be operable. Graphene-based drug nanocarriers have been proven to provide a promising platform for efficient drug delivery with good therapeutic efficacy.36–38 For instance, GO has been utilized as an innovative nanocarrier to deliver anti-cancer drugs, antibiotics, DNA, RNA, peptides as well as genes, which offer advanced transport frameworks of a more broadened range of therapeutic agents.38–41 However, further innovations to substantially hoist their current abilities are still needed, which are urgent for the practical transformation of nanomedicine. For example, the precise imaging ability and monitoring of therapeutic progress is very challenging, especially in the complex cellular microenvironment. Lately, surface-enhanced Raman scattering spectroscopy (SERS) is emerging as an attractive technique for the detection and imaging of various biological molecules based on the vibrational spectroscopy.42,43 SERS spectroscopy has ultra-high sensitivity and excellent photostability, which can be utilized for real-time sensing and imaging.44–46 These characteristics make SERS grow in popularity for cancer diagnosis and therapy. Meanwhile, some important factors, such as the reproducibility and biocompatibility of SERS constructs are still needed to be carefully evaluated. With respect to biocompatibility, the bio-degradability as wells as toxicity triggered by nanoparticles need to be addressed properly before the translation of SERS technology. Despite these hurdles, SERS holds great opportunities for the development of a more effective and precise theranostic platform.47 Drug nanocarriers with the SERS-optical-tracking ability are expected to be powerful for the noninvasive and rapid investigation of their intracellular behaviors.48–50 Although the thriving nanotechnology development paves the way for the design of drug delivery systems with multifunctional biomedical applications, it is a great challenge to develop one nanoscale system integrated with the above-mentioned demands in a rational manner.
Herein, the synthesis of a functional GO-based nanocarrier featured SERS tracking and dual-targeting is reported. As illustrated in Scheme 1, Au@Ag and Fe3O4 nanoparticles were attached to GO, rendering it excellent SERS activity and magnetic targeting ability. Afterward, the anti-HER2 antibody was conjugated to target the cancer cells expressed with HER2. Furthermore, dynamic monitoring of the targeted delivery of anticancer drugs was investigated using SERS and fluorescence joint spectroscopy. 9AA and DOX, as model drugs, were co-delivered by the nanocarrier to generate enhanced combinational cytotoxicity against the cancer cells. The enhanced cytotoxicity is found to occur only when the loaded amount of drug is under a certain threshold value. To our knowledge, this is the first report on multi-drug-loaded graphene-based nanocarriers.
 | ||
| Scheme 1 Schematic illustration of the GO-based dual-targeting SERS-tracking nanosystem. | ||
Experimental section
Materials
Graphite powder, ascorbic acid (AA), H2SO4 (98%), H2O2 (30 wt%), KMnO4 (AR) were purchased from Sinopharm Chemical Reagent Co., Ltd. Ammonia solution (NH3·H2O), trisodium citrate dihydrate (TSCD) were purchased from Shanghai Zhongshi Chemical Co., Ltd. Hydrogen tetrachloroaurate(III) trihydrate (HAuCl4·3H2O), silver nitrate (AgNO3), sodium borohydride (NaBH4), and cetyltrimethylammonium bromide (CTAB) were purchased from Alfa Aesar. Poly(acrylic acid) (PAA, MW1800), poly(allylamine hydrochloride) (PAH, MW15000), 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), and N-hydroxysuccinimide (NHS) were purchased from Sigma-Aldrich. Ferric chloride hexahydrate (FeCl3·6H2O), ferrous chloride tetrahydrate (FeCl2·4H2O), 9-aminoacridine (9AA), doxorubicin (DOX), and chitosan (CTS) were purchased Aladdin Reagent Co., Ltd. In all experiments, deionized water (Millipore Milli-Qgrade) with a resistivity of 18.2 MΩ cm was used.Synthesis of GO
Graphite oxide was synthesized from graphite powder using a modified Hummer's method.51 Typically, 1.0 g of graphite powder was added to 23 mL of H2SO4 (98%) and the mixture was under stirring for 12 h. Afterward, while keeping the mixture in an ice bath, 4.0 g of KMnO4 was added. After 1 h, the mixture was stirred at 40 °C for 30 minutes. Next, the heat was increased to 100 °C for 30 min. Then, the mixture was diluted to 100 mL and kept stirring for another 30 min. 10 mL of 30% H2O2 solution was subsequently added and the color of the mixture rapidly changed to bright yellow. The resulting mixture was washed several times, first with 5% HCl solution and then with deionized water, to make the solution become neutral. The obtained oxidized graphite solution was treated with an ultrasonic probe at 400 W for 30 min, which was then centrifuged at 7378g for 30 min. GO was obtained in the supernatant, which was stable and did not precipitate for several months.Chitosan functional GO
CTS solution of 1.0 wt% was prepared by dissolving CTS in 0.5% (v/v) aqueous acetic acid solution. Then, 4 mL of the prepared chitosan solution was added to 8 mL GO solution, followed by stirring for 24 h. CTS-GO was obtained by centrifugation at 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 252g for 20 min.
252g for 20 min.
Fe3O4 and Au@Ag NPs attached CTS functional GO (GO-Fe3O4/Au@Ag)
Au@Ag NPs were synthesized according to the previously published literature.52 10 mL of Au@Ag NPs was first mixed with 50 μL of 10 mM DTNB and stirred for 4 h, which was followed by adding 2 mL of 10 mg mL−1 PAH (in 10 mM NaCl) solution and stirred for 2 h. The mixture was centrifuged to remove excess PAH at 4722g for 30 min. The precipitate was then dispersed in 10 mL of 15 mg mL−1 PAA solution and stirred for another 2 h. The mixture was centrifuged to remove excess PAA. The obtained precipitate was re-dispersed in 4 mL of water.Fe3O4 nanoparticles were prepared by the co-precipitation method.53 Briefly, 1.0 g of FeCl2·4H2O and 2.7 g of FeCl3·6H2O were mixed in 40 mL of water under argon in a three-necked flask and the mixture was heated to 80 °C. While keeping the mixture stirring, 6 mL of NH3·H2O was added, and the heating was continued for 30 min. Afterwards, 4.0 g of TSCD in 10 mL water was introduced, and the temperature was increased to 90 °C. The mixture was stirred for 90 min. The Fe3O4 precipitate was then collected by centrifugation at 10625g for 10 min. After washing it several times, the precipitate was re-dispersed in 10 mL of water.
4 mL of the pre-prepared Au@Ag NPs and 800 μL of Fe3O4 nanoparticles were added to 2 mL solution of GO-CTS, and the mixture was gently stirred overnight. Finally, the product was obtained by centrifugation at 4722g for 20 min.
GO-Fe3O4/Au@Ag-anti-HER2
GO-Fe3O4/Au@Ag (5 mg) was dispersed in 4 mL 0.1 M PBS (pH = 7.4) solution and followed by adding 200 μL of 10 mM EDC and 40 μL of 100 mM NHS. After the mixture was stirred for 15 min, 500 μL of 20 mg mL−1 anti-HER2 antibody was added and the mixture was kept stirring for 2 h. The product was obtained by magnetic separation.DOX loading of GO-Fe3O4/Au@Ag-anti-HER2
100 μL of 10 mg mL−1 DOX aqueous solution was added to 5 mL of GO-Fe3O4/Au@Ag-anti-HER2 and kept stirring overnight in dark. The product was obtained by magnetic separation. Unloaded DOX was removed by magnetic separation. The amount of unloaded DOX was calculated according to a calibration curve recorded with known concentrations. The drug loading efficiency (LE) was calculated using the equation:DOXi is the initial amount (mg) of DOX, DOXf is the unloaded DOX.

For dual drug loading, 9AA was first loaded onto the nanocarrier by mixing the 9AA solution with the nanocarriers and stirring for 12 h. DOX was then added and stirred for another 12 h. After the unbound drugs were removed by centrifugation, dual drug-loaded nanocarriers were obtained. In this process, the concentration of the 9AA solution was fixed. By adjusting the concentration of DOX, the equal loading capacity of 9AA and DOX was obtained.
Cell culture and in vitro experiments
Human breast cancer cells (SKBR3) were purchased from China Type Culture Collection. SKBR3 cells were cultured in DMEM supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin–streptomycin (Nanjing KeyGen Biotech. Co., Ltd). The culture was always kept under the standard culture condition (5% CO2, 37 °C).To test the targeting capability of the cancer cell targeting nanocarrier, SKBR3 cells were first seeded into culture dishes and incubated for 24 h. SKBR3 cells were incubated by targeting the nanocarrier solution and with anti-HER2 free nanocarrier solution for 2 h at 37 °C, respectively (volume ratio of nanocarrier solution/culture media = 1:5). By fixing a magnet under the culture dish, SKBR3 cells in another culture dish were incubated with the targeting nanocarrier for 2 h at 37 °C. Then, the culture media were removed and the culture dishes were gently washed with PBS three times and subjected to SERS and fluorescence measurements.
To investigate the intracellular drug delivery of nanocarriers, SKBR3 cells were seeded in a culture dish and incubated for 24 h. Then, by fixing a magnet under the culture dish, the drug-loaded targeting nanocarriers were added to the cell culture dish and incubated for different time periods (volume ratio of nanocarriers solution/culture media = 1:5). 0.8 μL of LysoTracker Green DND-26 (1 mM) was added 10 min before the preassigned time. After incubation, the culture media were discarded and the culture dishes were gently washed with PBS solution 3 times before SERS and fluorescence measurements.
The viability of SKBR3 cells was tested by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide) assay. SKBR3 cells were first seeded into 96-well plates (100 μL per well) and incubated for 24 h under standard conditions. Then, the solutions of the nanocarriers loaded with drugs or without drugs were added, respectively. After 48 h, 50 μL of MTT solution was added to each well and cultured for another 4 h (volume ratio of MTT buffer to dilution buffer 1:4). After removing the supernatant medium, 150 μL of DMSO was then added to each well. The absorbance of each well at 490 nm was measured using a microplate reader (Bio-Rad model 680). SKBR3 cells without being treated with nanocarriers were used as a control.
Instruments
TEM images were collected on an FEI Tecnai G2T20 electron microscope operating at 200 kV. Extinction spectra were recorded using a Shimadzu UV-3600 PC spectrophotometer. Photoluminescence emission spectra were recorded on an Edinburgh FLS920 spectrofluorometer. Zeta potential measurements were performed using a Zetasizer Nano instrument (ZS90, Malvern). Each sample was analyzed in triplicate and each measurement was an average of three 30 s runs. Intracellular SERS and fluorescence images were recorded using confocal microscopy (FV1000, Olympus). Fluorescence images of the cells were recorded at the excitation of 488 nm. SERS spectra were measured at 633 nm excitation. Rayleigh scattering light was removed by a holographic notch filter. The Raman scattering light was then directed to an Andor shamrock spectrograph equipped with a charge-coupled device (CCD).Results and discussion
Characterization of the nanocarriers
CTS is reported to be beneficial in drug delivery because of its superior biocompatibility, biodegradability, and positively charged characteristics. Generally, the surface of GO is negatively charged with a zeta potential of −38.2 mV. As such, the positively charged CTS can be introduced to the surface of GO through electrostatic interactions. After the treatment of CTS, the zeta potential of GO is 23.8 mV, which can be attributed to the adsorption of CTS onto GO. Fe3O4 and Au@Ag NPs were then attached to CTS-functionalised GO through the reaction between the amino groups of CTS and a carboxyl group on the surface of Fe3O4 and Au@Ag. After the attachment of Fe3O4 and Au@Ag nanoparticles, some amino groups in chitosan were neutralized and the zeta potential decreased to 14.5 mV (Fig. S1, ESI†). The morphology of GO and GO-based composites was analyzed using TEM as shown in Fig. 1. It can be observed that the GO sheet is irregular and curly with a lateral size of 1–2 μm (Fig. S2, ESI†). As shown in Fig. 1B and C, Fe3O4 and Au@Ag NPs are found to be decorated onto the surface of GO. A STEM analysis of GO-Fe3O4/Au@Ag was also utilized to reveal the distribution of elements, which is shown in Fig. 1D–I. Element mapping of GO-Fe3O4/Au@Ag shows the spatial distribution of C, Fe, O, Au and Ag, respectively. As shown in Fig. 1E, the sheet is filled with a uniform and intense C signals. Meanwhile, Fe, Au, Ag elements are located on a part of the sheet, and the location of Au overlaps with Ag, which indicates the successful attachment of Fe3O4 and Au@Ag NPs. The absorbance spectrum of GO with a peak at 230 nm is shown in Fig. 2A, which originates from the π-plasmon of carbon. After GO was treated with CTS, it shows a similar curve in the absorbance spectrum because no obvious absorption occurs from CTS. Then, the extinction spectrum of GO-Fe3O4/Au@Ag was detected. A band from 400 to 600 nm appears in the spectrum, which is attributed to Au@Ag NPs (no obvious absorption peak from Fe3O4). EDX spectrum of GO-Fe3O4/Au@Ag is presented in Fig. S3 (see ESI†) along with the percentages of C, O, Fe, Ag and Au. The result indicates the successful loading of Au@Ag NPs onto the GO sheets. In the absence of a magnetic field, GO-Fe3O4/Au@Ag composites were well-dispersed in an aqueous solution and the suspension was kept stable for a long duration (at least 48 h). When an external magnetic field was applied, as shown in Fig. 2A inset, these GO-Fe3O4/Au@Ag composites were aggregated within 20 min, which demonstrates the superparamagnetic characteristic of GO-Fe3O4/Au@Ag. | ||
| Fig. 1 (A–C) TEM images of GO, GO-Fe3O4, GO-Fe3O4/Au@Ag; (D) dark-field STEM image of GO-Fe3O4/Au@Ag composites. The mapping images of elements in the composites, including C (E), Fe (F), O (G), Au (H) and Ag(I). | ||
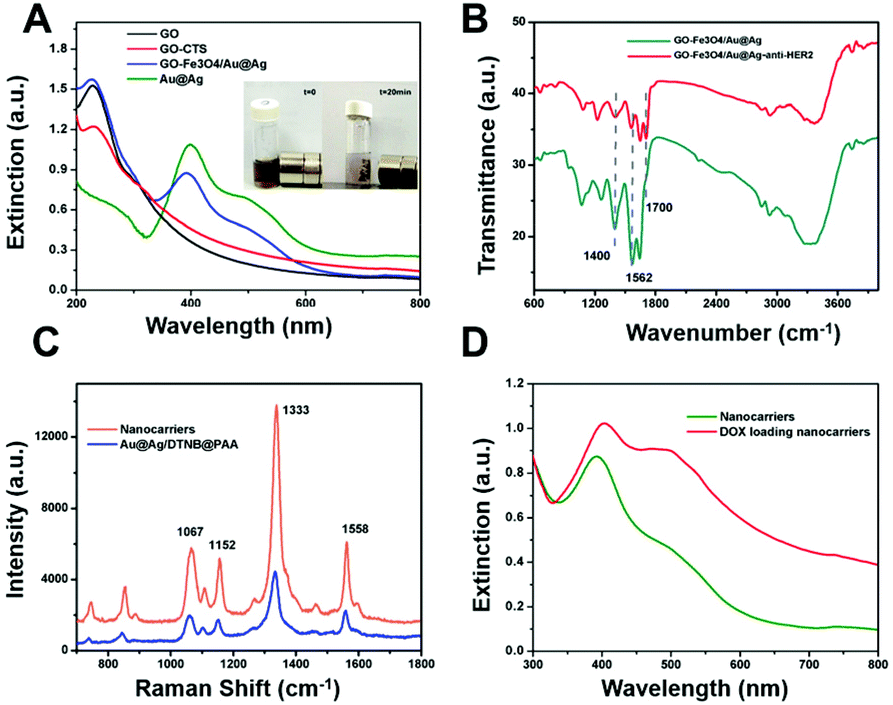 | ||
| Fig. 2 (A) Absorbance spectra of GO and GO-based nanocomposites. Inset: Photograph showing magnetic behavior under an extra magnet. (B) FTIR spectra of GO-Fe3O4/Au@Ag and nanocarriers (GO-Fe3O4/Au@Ag@Ag-anti-HER2). (C) SERS spectra of DTNB tagged PAA coated Au@Ag NPs and nanocarriers. (D) Absorbance spectra of the nanocarriers and DOX-loaded nanocarriers. | ||
Then, the anti-HER2 antibody was linked to the GO-Fe3O4/Au@Ag composite through the formation of amide bonds between the amino groups of the antibody and the carboxyl groups on Fe3O4 and PAA coated DTNB tagged Au@Ag in the presence of EDC and NHS, rendering a more positive zeta potential of 37.6 mV (Fig. S1, ESI†). The increase in zeta potential was attributed to the presence of positive anti-HER2.54 The EDX spectrum of GO-Fe3O4/Au@Ag-anti-HER2 (Fig. S3, ESI†) exhibited higher percentages of C, O, N and S in comparison to that of GO-Fe3O4/Au@Ag, which can be attributed to anti-HER2. This is in agreement with the previous reports.55 In the FTIR spectrum of GO-Fe3O4/Au@Ag (Fig. 2B), two representative peaks at 1702 cm−1, 1400 cm−1 are assigned to the carbonyl of –COOH and C–N bond, while a strong band at 3356 cm−1 originates from the O–H group.56,57 After the antibody is linked, it was observed that the peak at 1700 cm−1 assigned to the carbonyl of –COOH is almost disappeared and much stronger peaks at 1562 cm−1 of N–H in the amide group and 1400 cm−1 of C–N bond are present, indicating the successful attachment of anti-HER2 antibody. The quantity ratio of HER2 antibody to GO-Fe3O4/Au@Ag in the final nanocomposites was calculated to be 1.57 (Fig. S4, ESI†). Thus, the designed multifunctional nanocarriers GO-Fe3O4/Au@Ag-anti-HER2 are completely constructed.
Finally, DOX molecules were loaded on the nanocarrier mainly through π–π stacking as reported previously. The unloaded drug was removed by centrifugation. The UV/Vis spectra of nanocarriers and drug-loaded nanocarriers were recorded (Fig. 2D). In contrast to the spectrum of nanocarriers, the absorption band at 480 nm in the spectrum of DOX-loaded nanocarriers was much more intense. This can be attributed to the loading of DOX because DOX exhibited strong absorption around 480 nm. The zeta potential of DOX-loaded nanocarriers was observed to be 39.3 mV (Fig. S1, ESI†). The slightly increased zeta potential can be attributed to the addition of amino groups in DOX. The drug loading capacity was calculated to be 0.65 mg mg−1 with the weight ratio of DOX to GO-Fe3O4/Au@Ag-anti-HER2 at 2:1. The corresponding DOX release is pH-responsive as shown in Fig. 3, which can be attributed to the weakened π–π stacking interaction between DOX and GO in acidic condition.
 | ||
| Fig. 3 DOX release profiles at given pH values. | ||
SERS characteristics of nanocarriers
Au@Ag NPs were first tagged with the Raman reporter DTNB and then coated with PAA. The PAA-coated layer isolated the Raman reporter thus keeping the SERS signal stable and reliable, and simultaneously served as the conjugate agent to CTS functional GO. The nanocarrier was designed to generate SERS signals for tracing the intracellular distribution. Fig. 2D shows the SERS spectrum of the nanocarriers. The assignments of Raman vibrational modes for DTNB have been reported previously. The representative Raman modes of DTNB are at 1333, 1152, 1067, and 1558 cm−1, assigned to the symmetric stretch of the nitro groups, the C–H deformation modes, the succinimidyl N–C–O stretch overlapping with aromatic ring modes and the aromatic ring C–C stretching modes, respectively.53 The prepared nanocarrier was further applied to in vitro studies. The dynamic process of nanocarriers being internalized into cells can be monitored through SERS signals. In contrast with GO-based nanocarriers labeled with fluorescence, SERS, inherited from Raman spectroscopy characteristics, yields higher sensitivity, spatial and spectral resolution as well as photostability, while also presenting high multiplexing potential. This is superior for online continuous monitoring of nanocarriers in the complex biological microenvironment.Magnetic and receptor-mediated targeting abilities
To achieve increased tumor selectivity, a prerequisite is the high cellular uptake efficacy of drug nanocarriers for tumor cells specifically. Therefore, anti-HER-2 antibody and Fe3O4 NPs were chemically conjugated to render the nanocarriers dual targeting recognition. Both, magnetic targeting and receptor-mediated targeting behaviors can bring much improved specific cellular uptake efficiency of nanocarriers, which is integrated with SERS optical tracking property. A conclusion can be drawn that improved internalized nanocarriers result in increased detectable SERS signals. Thus, dual targeting behaviors internalized in tumor cells can be studied utilizing SERS. Herein, we used SKBR3 as an in vitro model cell because of the overexpression of the HER2 receptor on the membrane. SERS signals of the carriers inside the cells were then collected to reflect the uptake of drug carriers in SKBR3 cells (Fig. 4).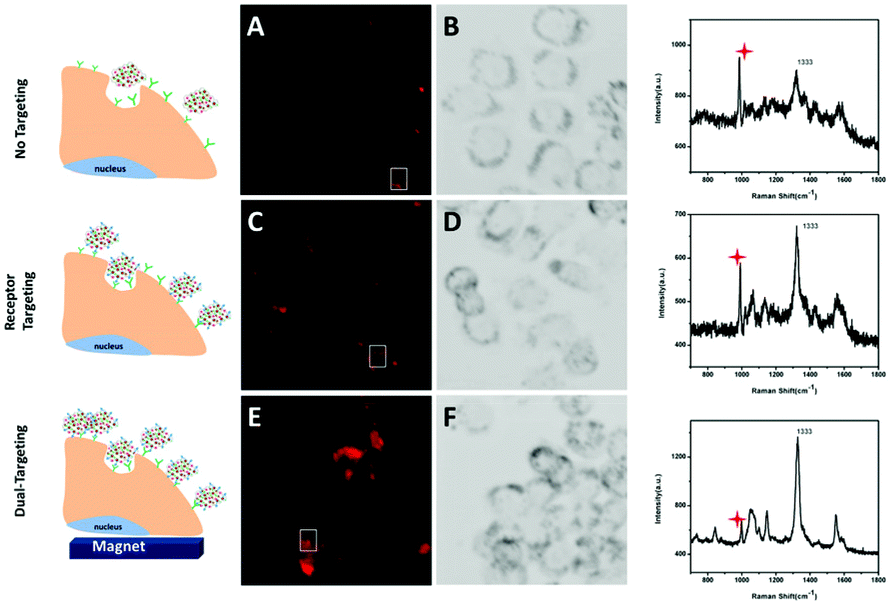 | ||
| Fig. 4 Images and spectra of SKBR3 cells incubated with GO-Fe3O4/Au@Ag (A and B), GO-Fe3O4/Au@Ag-anti-HER2 (C and D) and GO-Fe3O4/Au@Ag-anti-HER2 (E and F) under an extra magnetic field. The Raman shift at 996 cm−1 indicated by the red mark is assigned to the culture dish bottom. | ||
Receptor-targeting NPs are usually internalized in cancer cells by a receptor-mediated endocytosis process. To investigate if the antibody-conjugated nanocarriers can be more efficiently internalized into HER2 overexpressed SKBR3 cells, a group of control experiments was carried out. SKBR3 cells were cultured with nanocarriers conjugated with or without anti-HER2 for 2 h under the same conditions. SERS-mapping images of SKBR3 cells are depicted in Fig. 4A–D, more nanocarriers conjugated with anti-HER2 were internalized to SKBR3 cells exhibiting a strong targeting effect. These results showed that the binding and uptake of anti-HER2 conjugated nanocarriers into SKBR3 cells could be improved due to the special interaction between anti-HER2 and overexpressed HER2 receptors on SKBR3 cells. Thus, the antibody-conjugated nanocarriers can target SKBR3 cells sensitively. Next, for the study of magnetic targeting behavior, a magnet was placed under the culture dish where SKBR3 cells were incubated with anti-HER2 conjugated nanocarriers. The nanocarriers were guided to the culture bottom under the magnetic field, which makes SKBR3 cells exposed to more nanocarriers. Thus, nanocarriers were internalized into SKBR3 cells through both surface-receptor-targeting and magnetic targeting. As depicted in the SERS mapping image (Fig. 4E), the SERS signal of SKBR3 cells is much enhanced in the magnetic field active region compared to that in Fig. 4C, exhibiting excellent magnetic targeting ability. Therefore, compared to the SERS mapping image in Fig. 4A, the significantly increased traces of SERS signals in SKBR3 cells in Fig. 4E prove that these presented dual-targeting nanocarriers can sensitively target SKBR3 cells, which holds great promise for improved tumor-selective therapy.
Intracellular drug release
After the cellular internalization of nanocarriers, the drug-release behavior of GO-Fe3O4/Au@Ag-anti-HER2 was investigated using confocal laser scanning microscopy (Fig. 5). The intracellular distribution of nanocarriers can be tracked through SERS signals while the released DOX can be monitored by fluorescence measurement. The green Lysotracker dye was utilized to stain the acidic organelles in SKBR3 cells. Intracellular DOX fluorescence and SERS signals of GO-Fe3O4/Au@Ag-anti-HER2 in SKBR3 cells were observed after treatment with the drug-loaded nanocarriers for different time durations (as shown in Fig. 5). It was observed that after a short incubation time of 0.5 h, no obvious SERS and fluorescence signals were detected inside the cells (Fig. 5A–E), suggesting that DOX-loaded nanocarriers had not entered into the cells sufficiently. When the incubation time was increased to 1 h, weak SERS signals inside the cells were observed, which demonstrated that some DOX-loaded nanocarriers were internalized by the cells. From the merged image (Fig. 5J), the co-localization of SERS signals (red) and fluorescence of the lysosomes (green) demonstrated that nanocarriers were mainly distributed in the acidic lysosomes. It can be speculated that DOX-loaded nanocarriers are internalized to SKBR3 cells through HER-2 receptor-mediated endocytosis and accumulated at the intracellular lysosomes.58 The acidic lysosomal pH consequently triggered DOX release, which was evidenced by the detected fluorescence of DOX inside the cells (Fig. 5G). As shown in Fig. 5K–O, more GO-Fe3O4/Au@Ag-anti-HER2 was efficiently internalized by SKBR3 cells after 3 h of incubation. These nanocarriers distributed inside the lysosomes were observed to be yellow in colour, which was caused by the overlapping of green (Lysotracker) and red (nanocarriers) signals. Significantly enhanced fluorescence of DOX was found to be distributed in both the cytoplasm and nucleus (Fig. 5L) due to the DOX release that was triggered by the acidic lysosomal pH. Nevertheless, as the incubation time was further increased to 4 h, some of the GO-Fe3O4/Au@Ag-anti-HER2 composites were observed to escape from the lysosome. Fig. 5T showed that some GO-Fe3O4/Au@Ag-anti-HER2 composites were distributed in the cytoplasm. The results demonstrate that the presented dual-targeting drug nanocarriers can enhance the uptake efficacy by specific cancer cells, and efficiently release the drug into cancer cells.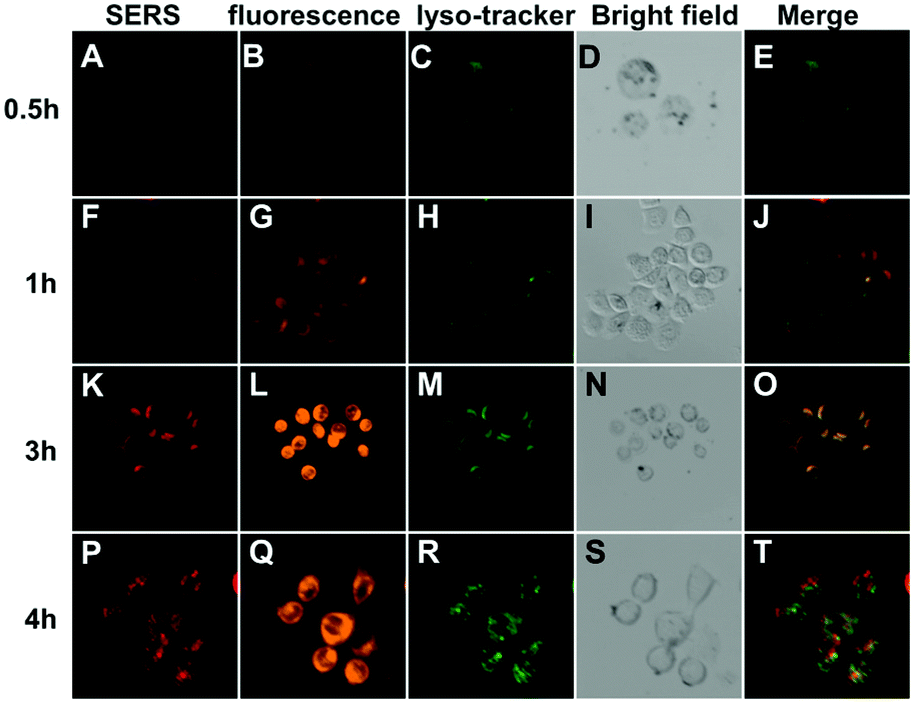 | ||
| Fig. 5 CLSM images of SKBR3 cells incubated with GO-Fe3O4/Au@Ag-anti-HER2 for different time durations under an extra magnetic field. SERS mapping images of intracellular nanocarriers (A, F, K and P); fluorescence images of intracellular DOX (B, G, L and Q); fluorescence images of the lysosomes (C, H, M and R); bright-field images of SKBRS3 cells (D, I, N and S); merge images of the nanocarriers and lysosomes (E, J, O and T). | ||
Anticancer efficacy of dual drug-loaded nanocarriers
The anti-proliferation of the drug-loaded nanocarriers was investigated by the MTT assay was conducted in SKBR3 cells (Fig. 6A). The results revealed that nanocarriers exhibited no obvious cytotoxicity to SKBR3 cells, even at a high concentration of 500 μg mL−1. However, when the cells were cultured with DOX-loaded nanocarriers, significantly enhanced inhibition of the cell growth was observed. SKBR3 cells were treated with free DOX, DOX-loaded nanocarriers and DOX-GO-Fe3O4/Au@Ag for 48 h, with an equivalent DOX concentration of 10 μg mL−1 (Fig. 6A). Cytotoxic efficacy of DOX-loaded nanocarriers in SKBR3 cells was higher than that of DOX-GO-Fe3O4/Au@Ag, which proved that the drug-loaded nanocarriers with targeting ability increased the cellular internalization through receptor-mediated endocytosis, thus leading to a high concentration of intracellular DOX. Compared to free DOX, the relatively low cytotoxicity of DOX-GO-Fe3O4/Au@Ag-anti-HER2 in SKBR3 cells was due to the controlled release of DOX by nanocarriers.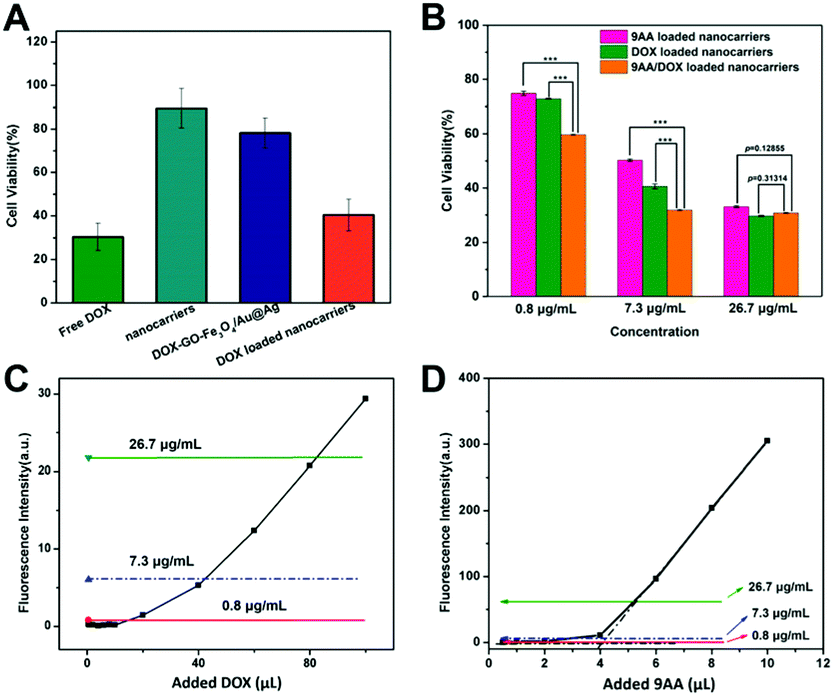 | ||
| Fig. 6 (A) Cell viability of SKBR3 treated with nanocarriers, DOX-loaded nanocarriers, DOX-GO-Fe3O4-Au@Ag and free DOX for 48 h. (B) Cell viability of SKBR3 after treatment with DOX or 9AA loaded nanocarriers and dual-drug loaded nanocarriers. ***p < 0.001. Calibration curves of fluorescence intensity of DOX (C) and 9AA (D) as a function of the amount of DOX and 9AA added to 5 mL of nanocarriers. Lines marked in (C) and (D) indicated the measured fluorescence of 9AA/DOX-GO-Fe3O4/Au@Ag-anti-HER2 with a concentration of 0.8 μg mL−1 (red), 7.3 μg mL−1 (blue) and 26.7 μg mL−1 (green). The intersection of black dotted lines was defined as the turning point. | ||
To evaluate the combined cytotoxicity of DOX and 9AA loading on DOX-GO-Fe3O4/Au@Ag-anti-HER2 in vitro, the cytotoxicity of 9AA/DOX-GO-Fe3O4/Au@Ag-anti-HER2 (loading ratio of 9AA:DOX was about 1:1) was compared with that of 9AA-GO-Fe3O4/Au@Ag-anti-HER2 and DOX-GO-Fe3O4/Au@Ag-anti-HER2. The composites with three different loading weight ratios of the drug to nanocarriers were subjected to MTT assay as shown in Fig. 6B. It was found that the DOX-loaded nanocarriers with a concentration of 0.8 μg mL−1 showed about 75% cell viability. A similar result was also observed for 9AA loaded nanocarriers with a 9AA concentration of 0.8 μg mL−1. When 9AA and DOX were loaded together in the nanocarriers, notably, 9AA/DOX-GO-Fe3O4/Au@Ag-anti-HER2 (concentrations of both 9AA and DOX approximately 0.8 μg mL−1) displayed obvious lower cell viability (about 60%). When loaded 9AA and DOX were increased to a higher concentration of 7.3 μg mL−1, dual drug-loaded nanocarriers also exhibited lower cell viability, indicating enhanced combined chemotherapy. However, when loaded 9AA and DOX were increased to a concentration of 26.7 μg mL−1, dual drug-loaded nanocarriers showed similar cytotoxicity compared with that of DOX or 9AA loaded nanocarriers and no enhanced cytotoxicity was observed. Hence, we postulate that the cytotoxicity of dual drug-loaded nanocarriers is dependent on the drug loading concentration. This hypothesis was then studied in detail through fluorescence spectroscopy. As shown in Fig. 6C, in the beginning, the fluorescence intensity of DOX was quenched due to energy transfer to GO and was maintained at a low level. Then, with the further increase in the added amount of DOX, the fluorescence intensity increased suddenly and then linearly with the added DOX. This phenomenon can be explained as follows. DOX molecules first continuously adsorbed onto GO through π–π stacking and the fluorescence was quenched significantly, which has been reported previously.59–61 Then, with further addition of DOX, they began to form a monolayer on GO until the turning point was reached. At the turning point, the surface of GO was occupied. The further added DOX molecules started to stack as multi-layers and the fluorescence was unable to be quenched efficiently anymore, which showed linear growth after the turning point. That is to say, the multilayer adsorbed molecules made a great contribution to the recovered fluorescence after the turning point. Similar results were obtained for 9AA (as shown in Fig. 6D). On the basis of this presented drug-adsorbed model, the above-mentioned drug-loaded nanocarriers with different loading concentrations for MTT assay were subjected to fluorescence measurements, which are indicated by lines marked in Fig. 6C and D. It was observed that 9AA/DOX-GO-Fe3O4/Au@Ag-anti-HER2 with a drug concentration of 0.8 μg mL−1 or 7.3 μg mL−1 was the monolayer-adsorbed model. Noteworthily, for 9AA/DOX-GO-Fe3O4/Au@Ag-anti-HER2 with a drug concentration of 7.3 μg mL−1, the measured fluorescence of DOX indicated that the adsorbed DOX exceeded the turning point very slightly, which can still be recognized as an adsorbed monolayer. While 9AA/DOX-GO-Fe3O4/Au@Ag-anti-HER2 with a drug concentration of 26.7 μg mL−1 was the multilayer adsorbed model. Thus, the multilayer adsorbed model of the drug may affect the release behavior of the drug, thus suppressing the cytotoxicity. This hypothesis is consistent with the observation in the MTT result. When the loaded drug onto GO is in the multilayer adsorbed status, the cytotoxicity of 9AA and DOX-loaded nanocarriers was no longer increased compared with that of 9AA or DOX-loaded nanocarriers. Therefore, the results may give us the new method to obtain an optimal therapeutic response of the multi-drug. Finally, the IC50 values (as shown in Fig. S5, ESI†) were determined as 7.34 μg mL−1 for 9AA-loaded nanocarriers, 4.90 μg mL−1 for DOX-loaded nanocarriers and 1.96 μg mL−1 for 9AA/DOX-loaded nanocarriers, exhibiting the enhanced anti-tumor efficiency induced by dual-drug.
Conclusions
In summary, we have presented the synthesis of a multi-functional GO as a nanocarrier, and its application to the targeted delivery of multiple drugs. Dynamic cellular uptake was tracked by SERS and fluorescence joint spectroscopy, which demonstrated the efficient and dual-targeted delivery of drugs into cancer cells by GO-based nanocarriers through receptor-mediated endocytosis and external magnetic field. Moreover, nanocarriers loaded with two anticancer drugs, 9AA and DOX, presented much higher cytotoxicity to cancer cells than those loaded with only one single drug. The enhanced combinational cytotoxicity occurred at low concentrations of anticancer drugs, which can be determined by drug adsorption status. The way DOX and 9AA are adsorbing on the GO surface plays an important role in their anti-proliferation effect in living cells, which may offer the opportunity for the optimal therapeutic response of the multi-drug. These findings break the cognition of an irrational endeavor to high drug loading capacity while ignoring its real therapeutic efficacy. Thus, this work gives a new perspective and method for the development of GO-based nanoplatforms for targeted multi-drug delivery, which may have prosperous clinical advantages with improved therapeutic efficacy.Author contributions
Conceptualization: Hui Chen; data curation: Hui Chen, Longqiang Xing, Huiru Guo; formal analysis: Hui Chen, Caixia Luo; funding acquisition: Hui Chen; investigation: Hui Chen, Longqiang Xing; methodology: Hui Chen, Longqiang Xing; writing – original draft: Hui Chen; writing – review & editing: Xuedian Zhang.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (no. 61805143).Notes and references
- S. D. Perrault, C. Walkey, T. Jennings, H. C. Fischer and W. C. W. Chan, Nano Lett., 2009, 9, 1909–1915 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. FaroKhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- M. M. Gottesman, Annu. Rev. Med., 2002, 53, 615–627 CrossRef CAS PubMed.
- A. Singh and J. Settleman, Oncogene, 2010, 29, 4741–4751 CrossRef CAS PubMed.
- I. Pastan, M. Gottesman, C. R. Kahn, J. Flier and P. Eder, N. Engl. J. Med., 1987, 316, 1388–1393 CrossRef CAS PubMed.
- Z. Cheng, A. Al Zaki, J. Z. Hui, V. R. Muzykantov and A. Tsourkas, Science, 2012, 338, 903–910 CrossRef CAS PubMed.
- B. Du, X. Cao, F. Zhao, X. Su, Y. Wang, X. Yan, S. Jia, J. Zhou and H. Yao, J. Mater. Chem. B, 2016, 4, 2038–2050 RSC.
- L. Chen, L. Zhou, C. Wang, Y. Han, Y. Lu, J. Liu, X. Hu, T. Yao, Y. Lin, S. Liang, S. Shi and C. Dong, Adv. Mater., 2019, 31, 1904997 CrossRef CAS PubMed.
- X.-Z. Chen, M. Hoop, N. Shamsudhin, T. Huang, B. Ozkale, Q. Li, E. Siringil, F. Mushtaq, L. Di Tizio, B. J. Nelson and S. Pane, Adv. Mater., 2017, 29, 1605458 CrossRef PubMed.
- Y. Chen, S. R. Bathula, J. Li and L. Huang, J. Biol. Chem., 2010, 285, 22639–22650 CrossRef CAS PubMed.
- L. Fan, F. Li, H. Zhang, Y. Wang, C. Cheng, X. Li, C.-h. Gu, Q. Yang, H. Wu and S. Zhang, Biomaterials, 2010, 31, 5634–5642 CrossRef CAS PubMed.
- Y. Patil, T. Sadhukha, L. Ma and J. Panyam, J. Controlled Release, 2009, 136, 21–29 CrossRef CAS PubMed.
- H. He, Y. Wang, H. Wen and X. Jia, RSC Adv., 2014, 4, 3643–3652 RSC.
- W. Scarano, P. de Souza and M. H. Stenzel, Biomater. Sci., 2015, 3, 163–174 RSC.
- W. Cui, J. Li and G. Decher, Adv. Mater., 2016, 28, 1302–1311 CrossRef CAS PubMed.
- C. Peters, M. Hoop, S. Pane, B. J. Nelson and C. Hierold, Adv. Mater., 2016, 28, 533–538 CrossRef CAS PubMed.
- W. Zhu, J. Guo, Y. Ju, R. E. Serda, J. G. Croissant, J. Shang, E. Coker, J. O. Agola, Q.-Z. Zhong, Y. Ping, F. Caruso and C. J. Brinker, Adv. Mater., 2019, 31, 1806774 CrossRef PubMed.
- T. Ji, Y. Ding, Y. Zhao, J. Wang, H. Qin, X. Liu, J. Lang, R. Zhao, Y. Zhang, J. Shi, N. Tao, Z. Qin and G. Nie, Adv. Mater., 2015, 27, 1865–1873 CrossRef CAS PubMed.
- L. Ao, B. Wang, P. Liu, L. Huang, C. Yue, D. Gao, C. Wu and W. Su, Nanoscale, 2014, 6, 10710–10716 RSC.
- A. Gabizon, H. Shmeeda, A. T. Horowitz and S. Zalipsky, Adv. Drug Delivery Rev., 2004, 56, 1177–1192 CrossRef CAS PubMed.
- P. Huang, C. Xu, J. Lin, C. Wang, X. Wang, C. Zhang, X. Zhou, S. Guo and D. Cui, Theranostics, 2011, 1, 240–250 CrossRef CAS PubMed.
- Y. Guo, L. Wang, P. Lv and P. Zhang, Oncol. Lett., 2015, 9, 1065–1072 CrossRef PubMed.
- G. Liu, H. Shen, J. Mao, L. Zhang, Z. Jiang, T. Sun, Q. Lan and Z. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 6909–6914 CrossRef CAS PubMed.
- F. Q. Chen and D. Gerion, Nano Lett., 2004, 4, 1827–1832 CrossRef CAS.
- H. Chen, J. Gao, Y. Lu, G. Kou, H. Zhang, L. Fan, Z. Sun, Y. Guo and Y. Zhong, J. Controlled Release, 2008, 128, 209–216 CrossRef CAS PubMed.
- S. H. Chiu, N. T. Ueno and R. J. Lee, J. Controlled Release, 2004, 97, 357–369 CrossRef CAS PubMed.
- S. Dou, Y.-D. Yao, X.-Z. Yang, T.-M. Sun, C.-Q. Mao, E.-W. Song and J. Wang, J. Controlled Release, 2012, 161, 875–883 CrossRef CAS PubMed.
- E. W. Song, P. C. Zhu, S. K. Lee, D. Chowdhury, S. Kussman, D. M. Dykxhoorn, Y. Feng, D. Palliser, D. B. Weiner, P. Shankar, W. A. Marasco and J. Lieberman, Nat. Biotechnol., 2005, 23, 709–717 CrossRef CAS PubMed.
- B. Chertok, B. A. Moffat, A. E. David, F. Yu, C. Bergemann, B. D. Ross and V. C. Yang, Biomaterials, 2008, 29, 487–496 CrossRef CAS PubMed.
- S. Mornet, S. Vasseur, F. Grasset and E. Duguet, J. Mater. Chem., 2004, 14, 2161–2175 RSC.
- L. H. Reddy, J. L. Arias, J. Nicolas and P. Couvreur, Chem. Rev., 2012, 112, 5818–5878 CrossRef CAS PubMed.
- A. H. Hung, R. J. Holbrook, M. W. Rotz, C. J. Glasscock, N. D. Mansukhani, K. W. MacRenaris, L. M. Manus, M. C. Duch, K. T. Dam, M. C. Hersam and T. J. Meade, ACS Nano, 2014, 8, 10168–10177 CrossRef CAS PubMed.
- Y.-L. Su, K.-T. Chen, Y.-C. Sheu, S.-Y. Sung, R.-S. Hsu, C.-S. Chiang and S.-H. Hu, ACS Nano, 2016, 10, 9420–9433 CrossRef CAS PubMed.
- C. L. Weaver, J. M. LaRosa, X. Luo and X. T. Cui, ACS Nano, 2014, 8, 1834–1843 CrossRef CAS PubMed.
- J. Liu, L. Cui and D. Losic, Acta Biomater., 2013, 9, 9243–9257 CrossRef CAS PubMed.
- M. Daniyal, B. Liu and W. Wang, Curr. Med. Chem., 2020, 27, 3665–3685 CrossRef CAS PubMed.
- A. A. Ghawanmeh, G. A. M. Ali, H. Algarni, S. M. Sarkar and K. F. Chong, Nano Res., 2019, 12, 973–990 CrossRef CAS.
- X.-M. Han, K.-W. Zheng, R.-L. Wang, S.-F. Yue, J. Chen, Z.-W. Zhao, F. Song, Y. Su and Q. Ma, Am. J. Transl. Res., 2020, 12, 1515–1534 CAS.
- M. Hoseini-Ghahfarokhi, S. Mirkiani, N. Mozaffari, M. A. A. Sadatlu, A. Ghasemi, S. Abbaspour, M. Akbarian, F. Farjadian and M. Karimi, Int. J. Nanomed., 2020, 15, 9469–9496 CrossRef CAS PubMed.
- F. Farjadian, S. Abbaspour, M. A. A. Sadatlu, S. Mirkiani, A. Ghasemi, M. Hoseini-Ghahfarokhi, N. Mozaffari, M. Karimi and M. R. Hamblin, ChemistrySelect, 2020, 5, 10200–10219 CrossRef CAS.
- N. Karki, H. Tiwari, C. Tewari, A. Rana, N. Pandey, S. Basak and N. G. Sahoo, J. Mater. Chem. B, 2020, 8, 8116–8148 RSC.
- J. J. Niu, M. G. Schrlau, G. Friedman and Y. Gogotsi, Small, 2011, 7, 540–545 CrossRef CAS PubMed.
- W. Zhou, X. Gao, D. Liu and X. Chen, Chem. Rev., 2015, 115, 10575–10636 CrossRef CAS PubMed.
- J. Huang, C. Zong, H. Shen, M. Liu, B. Chen, B. Ren and Z. Zhang, Small, 2012, 8, 2577–2584 CrossRef CAS PubMed.
- A. Huefner, W.-L. Kuan, R. A. Barker and S. Mahajan, Nano Lett., 2013, 13, 2463–2470 CrossRef CAS PubMed.
- J. Yang, Y. Cui, S. Zong, R. Zhang, C. Song and Z. Wang, Mol. Pharm., 2012, 9, 842–849 CrossRef CAS PubMed.
- D. Cialla-May, X. S. Zheng, K. Weber and J. Popp, Chem. Soc. Rev., 2017, 46, 3945–3961 RSC.
- B. Kann, H. L. Offerhaus, M. Windbergs and C. Otto, Adv. Drug Delivery Rev., 2015, 89, 71–90 CrossRef CAS PubMed.
- X. Qian, X.-H. Peng, D. O. Ansari, Q. Yin-Goen, G. Z. Chen, D. M. Shin, L. Yang, A. N. Young, M. D. Wang and S. Nie, Nat. Biotechnol., 2008, 26, 83–90 CrossRef CAS PubMed.
- W. Xie, L. Wang, Y. Zhang, L. Su, A. Shen, J. Tan and J. Hu, Bioconjugate Chem., 2009, 20, 768–773 CrossRef CAS PubMed.
- H. Chen, Z. Wang, S. Zong, L. Wu, P. Chen, D. Zhu, C. Wang, S. Xu and Y. Cui, ACS Appl. Mater. Interfaces, 2014, 6, 17526–17533 CrossRef CAS PubMed.
- S. Zong, Z. Wang, H. Chen, J. Yang and Y. Cui, Anal. Chem., 2013, 85, 2223–2230 CrossRef CAS PubMed.
- H. Chen, Z. Wang, X. Ma, S. Zong and Y. Cui, Talanta, 2013, 116, 978–984 CrossRef CAS PubMed.
- A. Loiseau, L. Zhang, D. Hu, M. Salmain, Y. Mazouzi, R. Flack, B. Liedberg and S. Boujday, ACS Appl. Mater. Interfaces, 2019, 11, 46462–46471 CrossRef CAS PubMed.
- A. H. Haghighi, M. T. Khorasani, Z. Faghih and F. Farjadian, Heliyon, 2020, 6, e03677 CrossRef PubMed.
- L. Zhang, Z. Lu, Q. Zhao, J. Huang, H. Shen and Z. Zhang, Small, 2011, 7, 460–464 CrossRef CAS PubMed.
- S. Y. Qin, J. Feng, L. Rong, H. Z. Jia, S. Chen, X. J. Liu, G. F. Luo, R. X. Zhuo and X.-Z. Zhang, Small, 2014, 10, 599–608 CrossRef CAS PubMed.
- D. Zhu, Z. Wang, S. Zong, H. Chen, X. Wu, Y. Pei, P. Chen, X. Ma and Y. Cui, Nanoscale, 2014, 6, 8155–8161 RSC.
- F. Pahang, P. Parvin and A. Bavali, Spectrochim. Acta, Part A, 2020, 229, 117888 CrossRef CAS PubMed.
- X. Zhao, Z. Wei, Z. Zhao, Y. Miao, Y. Qiu, W. Yang, X. Jia, Z. Liu and H. Hou, ACS Appl. Mater. Interfaces, 2018, 10, 6608–6617 CrossRef CAS PubMed.
- Y. Luo, X. Cai, H. Li, Y. Lin and D. Du, ACS Appl. Mater. Interfaces, 2016, 8, 4048–4055 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1an01237a |
| This journal is © The Royal Society of Chemistry 2021 |