
HIV detection from human serum with paper-based isotachophoretic RNA extraction and reverse transcription recombinase polymerase amplification†
Andrew T.
Bender
 a,
Benjamin P.
Sullivan
a,
Jane Y.
Zhang
a,
David C.
Juergens
b,
Lorraine
Lillis
c,
David S.
Boyle
c and
Jonathan D.
Posner
*abd
a,
Benjamin P.
Sullivan
a,
Jane Y.
Zhang
a,
David C.
Juergens
b,
Lorraine
Lillis
c,
David S.
Boyle
c and
Jonathan D.
Posner
*abd
aDepartment of Mechanical Engineering, University of Washington, Seattle, USA. E-mail: jposner@uw.edu
bDepartment of Chemical Engineering, University of Washington, Seattle, USA
cPATH, Seattle, USA
dFamily Medicine, School of Medicine, University of Washington, Seattle, USA
First published on 6th April 2021
Abstract
The number of people living with HIV continues to increase with the current total near 38 million, of which about 26 million are receiving antiretroviral therapy (ART). These treatment regimens are highly effective when properly managed, requiring routine viral load monitoring to assess successful viral suppression. Efforts to expand access by decentralizing HIV nucleic acid testing in low- and middle-income countries (LMICs) has been hampered by the cost and complexity of current tests. Sample preparation of blood samples has traditionally relied on cumbersome RNA extraction methods, and it continues to be a key bottleneck for developing low-cost POC nucleic acid tests. We present a microfluidic paper-based analytical device (μPAD) for extracting RNA and detecting HIV in serum, leveraging low-cost materials, simple buffers, and an electric field. We detect HIV virions and MS2 bacteriophage internal control in human serum using a novel lysis and RNase inactivation method, paper-based isotachophoresis (ITP) for RNA extraction, and duplexed reverse transcription recombinase polymerase amplification (RT-RPA) for nucleic acid amplification. We design a specialized ITP system to extract and concentrate RNA, while excluding harsh reagents used for lysis and RNase inactivation. We found the ITP μPAD can extract and purify 5000 HIV RNA copies per mL of serum. We then demonstrate detection of HIV virions and MS2 bacteriophage in human serum within 45-minutes.
1. Introduction
The number of people infected with HIV globally continues to steadily increase, with the current total over 36 million.1 Since the advent of highly effective antiretroviral therapy (ART), almost 20 million HIV-positive people are on treatment, which requires routine viral load monitoring to assess successful viral suppression.2 Additionally, early infant detection of HIV infections is not possible with typical lateral flow-based antibody tests, so highly sensitive nucleic acid amplification testing (NAATs) must be used to detect HIV nucleic acids.3 The majority of nucleic acid testing for HIV in low- and middle-income countries (LMICs) is carried out on dried blood spots shipped to central laboratories, where expensive automated tests quantitate viral titers.4 There have been increased efforts to scale-up decentralized HIV molecular testing in LMICs, and several point-of-care (POC) viral load tests have reached market to address this need.5 Yet effective scale-up efforts have been hampered by the platform and per-test costs, as well as the operational complexity of current viral load tests. For widespread use in LMICs, POC HIV viral load tests must be rapid, robust, low-cost, simple to use, and capable of sensitive detection of HIV virions in blood–the World Health Organization recommends a limit of detection (LOD) of 1000 copies per mL of plasma.6The majority of current commercial HIV POC tests have miniaturized and automated gold-standard approaches to molecular testing of RNA from blood samples.5 A primary roadblock for simplifying these tests for POC use is sample preparation due to the susceptibility of RNA to degradation and the complexity of blood, which contains immunoglobulins, cellular debris, and nucleases that inhibit downstream molecular assays.7,8 Endogenous blood RNases are particularly problematic because they are exceptionally stable enzymes and capable of rapidly degrading exogenous free RNA in blood in the order of seconds.9,10 Traditional sample preparation methods for bloodborne RNA targets use high concentrations of chaotropic salts, toxic disulfide reducing chemicals (e.g. β-mercaptoethanol), and harsh anionic detergents to lyse virions and inactivate blood RNases.11–13 Highly effective nucleic acid extraction and purification from the lysate is required to prevent chemicals from interfering with downstream amplification assays. Solid phase extraction is commonly used for purification, but this necessitates repeated buffer exchanges to separate, wash, and elute nucleic acids.14 For example, a gold standard product for viral RNA extraction is the QIAamp Viral RNA Mini Kit (Qiagen), and it requires 6 manual pipetting steps and 5 centrifugations, totaling 30 minutes to an hour of hands-on time – according to the product handbook. Commercial POC tests automate these steps using robotics, pumps, valves, and other methods for fluidic exchanges.14 Automating extensive fluidic manipulation has required complicated engineering designs. This approach faces a practical barrier to lowering the overall platform costs of molecular testing in blood samples, which has prompted researchers to find new sample preparation approaches for POC use.
Isotachophoresis (ITP) is an electrophoretic separation and concentration technique that can extract and purify nucleic acids from complex biological samples.15 ITP is capable of extracting nucleic acids from blood samples and removing contaminants, such as hemoglobin and immunoglobulin G, that inhibit downstream amplification assays.8,16 This separation process requires no physical manipulations, buffer exchanges, or other intermediate user steps, but rather automates nucleic acid purification using an applied electric field and simple buffers. ITP leverages a discontinuous buffer system with a leading electrolyte (LE) and trailing electrolyte (TE) to develop an electric field gradient that focuses charged species based on their electrophoretic mobilities.17 Analytes with mobilities less than the LE and greater than the TE are focused into a concentrated plug at the interface of the two electrolytes. Kondratova et al. were the first to use ITP in agarose gels for DNA extraction from human blood samples, but their work was not well-suited for POC diagnostics because it required lengthy deproteinization and dialysis pretreatment steps.15,18 Microchannel-based ITP has since emerged as a promising sample preparation approach for extracting DNA from blood specimens and amplifying with off-chip quantitative polymerase chain reaction (qPCR).19–22 Notably, Eid et al. detected DNA from Listeria monocytogenes cells in 2.5 μL of whole blood using alkaline and proteinase K lysis, microchannel ITP purification, and recombinase polymerase amplification for detection.23
In moving towards molecular diagnostics that are appropriate for POC use in LMICs, there are continuing efforts to implement ITP in microfluidic paper-based analytical devices (μPADs). μPADs are well-suited for POC diagnostics due to their wicking properties, ease of reagent deposition and storage, low material cost, and established methods for high-volume manufacturing.24 There are a number of ITP μPADs that have investigated extraction and concentration of analytes (e.g. fluorophores, DNA, indicator dyes) from pure buffer systems.25–30 Our group has developed an ITP μPAD with integrated whole blood fractionation and DNA extraction.31 We have also demonstrated simultaneous DNA extraction with ITP and on-chip DNA amplification using recombinase polymerase amplification (RPA).32
There are significant technical challenges that need to be overcome in order for ITP μPADs to be implemented for POC molecular testing of bloodborne pathogens, especially RNA viruses. Sample preparation for nucleic acid testing of HIV and other bloodborne RNA viruses requires lysis of the viral envelope, deactivation of blood RNases, and RNA purification. Traditional lysis and RNase inactivation leverage high concentrations of guanidine (4 to 6 M), which rapidly destroy viral envelopes and inactivate endogenous blood RNases.11 They are easily paired with silica-based columns or other substrates for nucleic acid purification via solid phase extraction.14 However, guanidine is difficult to pair with ITP systems for nucleic acid purification because high salt samples significantly disrupt the electric field gradient, hindering rapid ITP separation.16 The only study we are aware of that extracted RNA from blood samples was an assay targeting bacterial rRNA using alkaline-based lysis and a glass microchannel for the ITP separation. The study suffered LOD issues due to a low sample volume (∼1 nL) and incomplete inactivation of exogenous and endogenous RNases.22 There have been no reported ITP-based extractions of viral RNA from blood or serum.
In this paper, we report a method for HIV detection from human serum with an MS2 bacteriophage internal process control using a novel lysis and RNase inactivation method, paper-based ITP, and duplexed reverse transcription recombinase polymerase amplification (RT-RPA). A previous study from our group examined varied enzymatic and chemical approaches for immobilizing blood RNases.33 We build on this work to develop a novel 15-minute protocol for off-chip viral lysis, RNase inactivation, and serum protein digestion. We design a specialized ITP system to focus RNA into a characteristic ITP plug, while excluding proteinase K and anionic detergent present in the lysate. We determine the LOD of the ITP μPAD for RNA extraction by processing digested serum spiked with known RNA concentrations and amplifying with off-chip RT-RPA. We then demonstrate detection of HIV virions and MS2 bacteriophage in human serum within 45-minutes. To our knowledge, this is the first example of an ITP-based assay for detecting RNA viruses from blood samples. We propose that our assay may ultimately be implemented in a fully integrated POC HIV viral load test with no intermediate user steps using innovations that have previously been reported, such as integrated plasma separation, in-membrane protein digestion, and simultaneous ITP and RPA.31,32
2. Materials and methods
2.1 Biological samples
All experiments in this study were performed in a Biosafety Level (BSL) 2 with BSL-3 practices laboratory under a Biological Use Authorization approved by the Institutional Biosafety Committee at the University of Washington. Human serum used in this study was obtained from Sigma-Aldrich (St Louis, MO, USA). According to the manufacturer's specifications, pooled blood samples from males with blood type AB were centrifuged and resulting plasma was clotted via calcium addition. We chose to perform all experiments using this pooled serum rather than plasma for the sake of experimental consistency and ease of use, as serum has no anticoagulants or risk of clotting. Pooled serum is identical to plasma, except with clotting factors removed. Fluorescently labeled DNA was a 70 base pair (bp) double stranded DNA sequence modified with a single Alexa Fluor 488 molecule (Integrated DNA Technologies, Coralville, IA, USA).Purified HIV RNA was prepared from HIV-1 supernatant as previously detailed by Lillis et al.34 HIV-1 supernatant (Group M, Subtype A, NCBI accession number: JX140650) was obtained from the External Quality Assurance Program Oversight Laboratory at Duke University.35 Viral RNA was extracted and purified using the QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's standard protocol. RNA was then quantified with quantitative real-time PCR based on the method described by Rouet et al. using Superscript® III one-step RT-PCR system (Life Technologies, Carlsbad, CA, USA).36
Experimental work on HIV virion detection from human serum used a non-infectious HIV strain to reduce laboratory safety risks. HIV detection work employed a cultured HIV-1 subtype B (8E5) virus (SeraCare, Milford, MA, USA). The 8E5 HIV contains a single base addition in its RNA genome at the pol gene, creating a reverse transcription-defective virus with no infectivity. The 8E5 HIV was supplied in a concentrated supernatant and then diluted with serum for experimental work. Bacteriophage MS2 was the internal process control for the HIV assay. The phage was grown and isolated using an established protocol.37 MS2 stock solution was diluted with phosphate buffered saline and stored at −80 °C.
2.2 Lysis, RNase inactivation, and protein digestion
We employed a specialized chemistry for combined viral lysis, inactivation of blood RNases, and digestion of serum proteins. This chemistry was based on a previous study from our group investigating various methods for inactivating blood RNases.33 We incubated serum with a combination of 0.5% sodium dodecyl sulfate (Sigma-Aldrich), 1 mg mL−1 of proteinase K (Thermo Fisher Scientific, Waltham, MA, USA), and 10 mM dithiothreitol (Sigma-Aldrich). Working stock reagent concentrations were high, such that serum was only diluted 10% (i.e. a 40 μL sample contained 36 μL serum and 4 μL lytic reagents).We conducted a series of experiments extracting HIV RNA from pre-digested serum. For these RNA extraction experiments, we incubated serum with SDS and proteinase K in a water bath for 1 hour at 50 °C. Following this incubation, we spiked known concentrations of purified HIV RNA into the digested serum. For experiments detecting HIV virions in serum, we incubated serum spiked with 8E5 HIV for 15-minutes at 65 °C.
2.3 ITP device construction and buffer composition
ITP extractions were performed in single-use, disposable ITP μPADs consisting of a plastic Petri dish (Thermo Fisher Scientific), acrylic reservoirs, and 22-gauge titanium wire electrodes (McMaster Carr, Elmhurst, IL, USA). Reservoirs were cut with a CO2 laser cutter (Universal Laser Systems, Scottsdale, AZ, USA) and adhered to the Petri dish bases with double-sided tape (3M, Maplewood, MN, USA). The ITP strip spanned the two reservoirs and was constructed from Fusion 5 membrane (GE Healthcare, Chicago, IL, USA), which is made with a proprietary method to maximize porosity and minimize adsorption of biomolecules. Membranes were cut into a teardrop shape (40 mm long with maximum width of 8 mm and minimum width of 3 mm) with an electronic cutter machine (Cameo 3, Silhouette, UT, USA). The membranes were not washed or blocked, and they were stored at room temperature.For the ITP system, the TE buffer consisted of 70 mM Tris, 70 mM serine, and 0.1% w/v polyvinylpyrrolidone (PVP). The LE buffer in the ITP membrane was 135 mM Tris, 90 mM HCl, 50 mM KCl, 0.1% w/v PVP, and 100 ng mL−1 poly(A) carrier RNA. The LE buffer in the anode reservoir contained 240 mM Tris, 160 mM HCl, 10 mM KCl, and 0.1% PVP. Although the pH remained constant, the LE in the reservoir employed a higher concentration of Tris to maximize its buffer capacity, which is common in ITP systems to prevent pH changes from hydrolysis at the electrodes.38 All chemicals were obtained from Sigma-Aldrich. Buffers were prepared with molecular biology grade reagents, RNase-free water (Thermo Fisher Scientific), and in PCR-grade microcentrifuge tubes (Eppendorf AG, Hamburg, Germany) to limit introductions of exogenous RNases.
2.4 ITP extraction
The ITP μPAD processes a 40 μL sample of serum pretreated with proteinase K, SDS, and DTT. Experiments in this work used serum samples with either spiked RNA, spiked HIV, or no analyte. The first step in ITP extraction is pipetting 40 μL of sample onto the porous membrane in the widened sample region proximal to the TE reservoir. 1 μL of fluorescently labeled DNA is also added to the sample region for monitoring the location of the ITP during the separation. Then 40 μL of LE buffer is added to wet the remainder of the membrane. 250 μL of LE and TE buffers are added to their respective reservoirs. Initial locations of ITP buffers and sample are depicted in Fig. 1.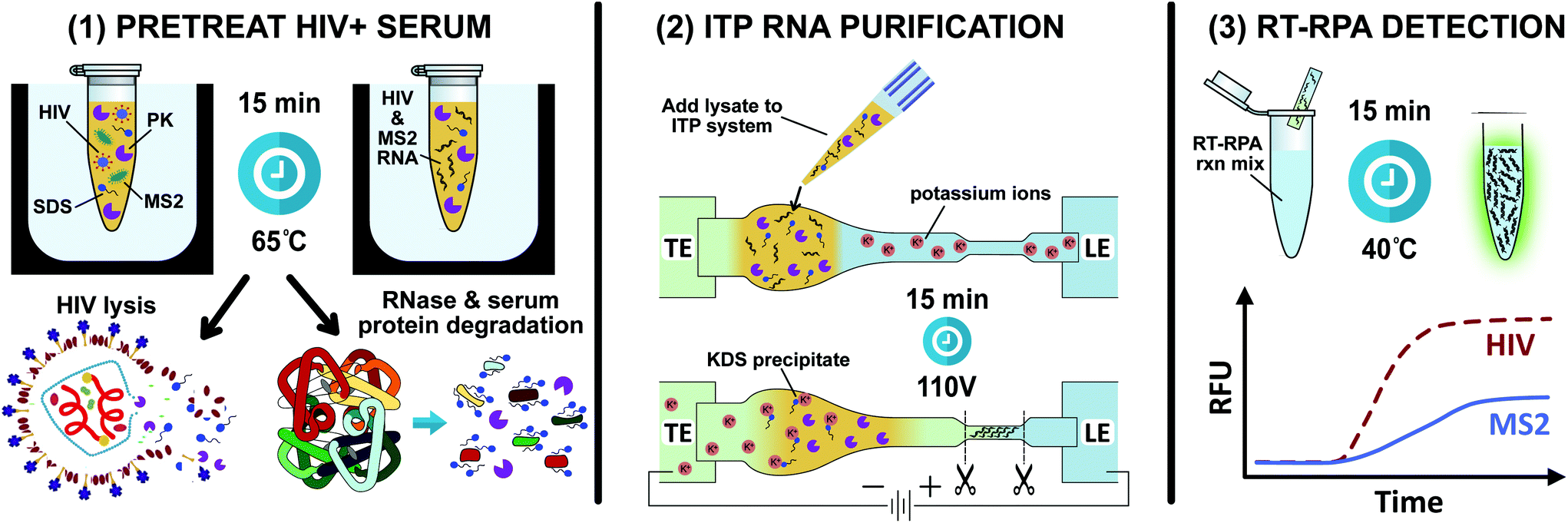 | ||
| Fig. 1 Schematic of the three-step process for detecting HIV and MS2 bacteriophage from human serum. (1) HIV+ serum spiked with MS2 phage is pretreated with proteinase K, SDS, and DTT at 65 °C for 15 minutes. SDS and proteinase K simultaneously lyse HIV and degrade endogenous blood RNases. (2) Free HIV and MS2 RNA are extracted and purified with ITP from serum components, proteinase K, and SDS. Potassium ions in the leading electrolyte precipitate potassium dodecyl sulfate, preventing the anionic detergent from focusing in the ITP plug. (3) A duplexed RT-RPA reaction directly from the cut portion of the paper strip simultaneously amplifies HIV and MS2. | ||
ITP is initiated with a constant 110-volt bias across the ITP strip applied with a source meter (model 2410, Keithley Instruments, OH, USA). The ITP plug location is indicated by the fluorescently labeled DNA. We collected fluorescence images of the separation membrane with a microscope (AZ-100, Nikon, USA) equipped with a 0.5× (NA = 0.05) objective. Light supplied by a mercury lamp light source (X-Cite Exacte, Excelitas Technologies Corp., Waltham, MA, USA) passed through an epifluorescence filter cube set (Omega Optics, Austin, TX, USA) with peak excitation and emission wavelengths of 488 nm and 518 nm, respectively. A 16-bit cooled electron multiplying charge-coupled device camera (Cascade II, Photometrics, Tucson, AZ, USA) collected grayscale images of ITP extractions.
When the ITP plug reaches the center of the narrow extraction zone of the strip, the voltage bias is removed, and this region of the strip is cut out. For RNA extraction experiments, the extraction zone of the paper strip is placed in a 0.5 mL plastic tube with a small hole at the bottom. The 0.5 mL plastic tube is placed inside a 1.5 mL plastic tube and centrifuged, removing the contents of the ITP plug from the paper (∼4 μL of eluate). This ITP eluate is pipetted directly into an RT-RPA reaction. For HIV detection experiments from serum, we do not use a centrifuge, but instead add the extraction zone of the paper strip directly to an off-chip RT-RPA reaction for duplexed detection of HIV and MS2, as illustrated in Fig. 1. We treated the benchtop, pipettes, forceps, and scissors with 10% bleach solution between each experiment in order to prevent cross contamination of nucleic acids.
2.5 RT-RPA amplification and detection
The RT-RPA primers and probe for HIV detection were developed by Lillis et al. and can be used to amplify HIV-1 RNA across multiple subtypes.34 The RT-RPA HIV detection assay consists of a lyophilized pellet of RPA reagents from the TwistAmp exo kit (TwistDx, UK), 29.5 μL rehydration buffer, 14 mM magnesium acetate, 540 nM forward primer (Integrated DNA Technologies), 540 nM reverse primer, 120 nM FAM-labeled probe (LGC Biosearch Technologies, Hoddesdon, UK), 0.2 U μL−1 RNasin Plus Ribonuclease Inhibitor (Thermo Fisher), 0.5 U μL−1 reverse transcriptase (AffinityScript, Agilent, Santa Clara, CA, USA), and 1% w/v Triton X-100 (Sigma-Aldrich). The duplexed RT-RPA assay for HIV and MS2 employed the reagents listed above as well as 216 nM MS2 forward primer, 216 nM MS2 reverse primer, and 48 nM Fluor Red 610-labeled probe.Experiments examining ITP plug purity used 4 μL of ITP extraction liquid and 2.5 μL of HIV RNA in the RT-RPA reactions. Experiments studying RNA extraction from digested serum used 4 μL of ITP extraction liquid in RT-RPA reactions. For HIV detection experiments, we added the cutout paper ITP extraction zone (containing ∼4 μL of liquid) directly to RT-RPA reaction tubes. We used RNase-free water to bring all RT-RPA reactions to a total volume of 50 μL per tube. A standalone fluorometer specifically designed for point-of-care testing applications (T16-ISO, Axxin, Australia) heated and measured fluorescence of the RT-RPA reactions. Reaction tubes were removed after 5 minutes of incubation, briefly mixed, and returned to the fluorometer for another 10 minutes. The baseline fluorescence at 3 minutes was subtracted from fluorescence values at all measurement time points for each respective reaction tube. For the HIV assay, we used a threshold of 100 arbitrary fluorescence units in the FAM detection channel to differentiate between successful and unsuccessful amplification. The MS2 assay fluorescence threshold was 50 arbitrary fluorescence units in the ROX detection channel.
2.6 RNase detection assay
We employed the RNaseAlert Substrate Detection System (Integrated DNA Technologies) for testing RNase activity in serum samples. We prepared the RNaseAlert experiments in a lidded 96-well plate with black walls and clear bottom (Corning Incorporated, Corning, NY, USA). The total assay volume for each well was 100 μL. Each RNase detection assay contained 10 μL of RNaseAlert substrate, 10 μL of 10× RNaseAlert buffer, 60 μL of RNase-free water, and 20 μL of sample. We used a 12-channel pipette to concurrently add serum samples to each well. The plate was immediately loaded into a plate reader (SpectraMax iD3, Molecular Devices, San Jose, CA, USA). The excitation and emission wavelengths were 485 nm and 535 nm, respectively. The gain was set to “low” with an exposure of 140 ms. The heating block in the plate reader was set to 37 °C. The instrument agitated the plate and measured the fluorescence in the wells every 2 minutes over a 30-minute incubation time.3. Results and discussion
3.1 Assay design and considerations
The HIV detection assay with an MS2 bacteriophage internal control requires serum pretreatment, RNA purification with our ITP μPAD, followed by duplexed RT-RPA. This diagnostic scheme is illustrated in Fig. 1. Serum pretreatment via SDS and proteinase K is necessary for viral lysis, RNase inactivation, and serum protein degradation. SDS is a powerful protein denaturant that has long been used in lysis chemistries while proteinase K is a broad spectrum protease that degrades proteins into a corresponding assortment of polypeptides.10 Proteolytic digestion is a crucial serum pretreatment step in ITP-based extractions. It has been widely reported that extraction of nucleic acids with ITP is inhibited by nonspecific binding with blood proteins.20,23,32,39 Extensive protein degradation reduces nucleoprotein complex formation and allows for electromigration of nucleic acids.We designed our ITP system to separate RNA from inhibitors of downstream RT-RPA and achieve high analyte accumulation in the ITP plug. Blood contains a variety of inhibitors of nucleic acid amplification assays, including undesirable salts and interfering proteins (e.g. hemoglobin, immunoglobulin, lactoferrin).8,40 Both SDS and proteinase K are potent inhibitors of RT-RPA because they inactivate the enzymes and proteins required in the amplification mechanism. SDS is an anionic detergent, so dodecyl sulfate carries the same negative charge as nucleic acids in most buffer conditions. Therefore, it is challenging to electrophoretically separate dodecyl sulfate from nucleic acids using ITP. To address this we instead removed dodecyl sulfate from the lysate with precipitation mediated by a potassium salt, leveraging the very low solubility of potassium dodecyl sulfate (KDS) in water.41,42 We employed potassium chloride in the leading electrolyte in the ITP strip and in the reservoir. Upon application of the electric field, potassium cations migrated from the LE buffer toward the cathode in the TE reservoir. Potassium cations therefore encountered dodecyl sulfate in this migration path forming KDS precipitate, as illustrated in Fig. 1.
Proteinase K in the serum lysate can be purified away from viral RNA with ITP based on its charge. Proteinase K has an isoelectric point of 8.9 and therefore has a net positive charge in buffers less than pH 8.9.10 In electrokinetic systems that maintain pH less than 8.9, proteinase K electromigrates in the opposite direction of nucleic acids due to their contrasting charges. ITP systems must be carefully designed because there can be sharp pH gradients, depending on the selection of TE, LE, and buffering counterion. We used numerical simulations to guide our design of the ITP system and select proper electrolytes for purifying and extracting RNA from serum. We used an open-source electrophoretic modeling tool, the Stanford Public Release Electrophoretic Separation Solver (SPRESSO), to approximate concentration and pH profiles resulting from various ITP buffers and plot the simulation outputs in Fig. 2. We do not go into depth on the equations and assumptions of the simulations here, but details can be found in the original SPRESSO report.43
 | ||
| Fig. 2 Simulations describing the ion concentration and pH profiles of ITP systems. (A) SPRESSO simulation results of concentration profiles with a TE buffer compromised of 70 mM Tris and 70 mM serine and a LE buffer of 240 mM Tris, 160 mM HCl, and 10 mM KCl. As the ITP plug migrates into the region previously containing LE, an adjusted trailing electrolyte (ATE) zone develops directly adjacent to the ITP plug. (B) Simulation results of pH profiles of three different TE selections: HEPES, serine, and β-alanine. All TE, LE, and counterion concentrations are the same as in (A). The pH of the ATE differs from 8.26 to 9.31 depending on the TE selection. When the pH of the ATE zone is less than 8.9, proteinase K is positively charged and will not electromigrate with negatively charged RNA. | ||
In Fig. 2A, we show simulated concentration profiles of distinct ionic species for an ITP system with a leading electrolyte comprised of 160 mM HCl and 240 mM tris paired with a trailing electrolyte of 70 mM tris and 70 mM serine. As ITP progresses with an applied electric field, three distinct zones are formed: the original TE zone, the adjusted TE (ATE) zone, and the LE zone. The ATE zone is a region with TE ions which was previously occupied by the LE. As seen in Fig. 2A, the ATE has increased concentrations of serine and tris compared with the original TE, elevating its pH. We found that using serine as the TE and an LE comprised of HCl with tris as the counterion maintained a pH less than 8.9 in all regions (see Fig. 2B). Therefore, our ITP system was designed for proteinase K to be positively charged and electrophoretically separated from RNA.
We found that serine (pKa = 9.33, fully ionized electrophoretic mobility of 34.3 × 10−9 m2 V−1 s−1) was a highly effective TE for its suitability for proteinase K removal and obtaining sufficient nucleic acid extraction. It has been reported in the literature that lowering the TE conductivity is a key mechanism for increasing analyte accumulation in the ITP plug.44 Low conductivity creates high electric fields in the TE region of the ITP system, resulting in faster electromigration of analytes in this zone and enhanced accumulation in the ITP plug. We found that under a pH of 9.0, serine has a very low electrophoretic mobility, resulting in low conductivities in the TE and ATE zones. A majority of the ITP studies on nucleic acid extraction from blood use HEPES (pKa = 7.66, mobility of 21.8 × 10−9 m2 V−1 s−1) as the TE.16 As shown in Fig. 2B, HEPES maintains relatively low pH in the system and is well-suited for removing proteinase K. However, HEPES has a higher electrophoretic mobility than serine at pH less than 9, so simulations showed that HEPES could not generate as high of electric field strength in the ATE zone as serine (∼0.5× lower). In experimentation, we found that we could not achieve efficient nucleic acid extraction from serum using HEPES as TE. The lower electrophoretic mobility of serine is useful in focusing HIV RNA that may have reduced mobility in our ITP system due to the tortuosity of the porous membrane and polypeptides that may bind or interact with RNA.45 There have been reports of ITP nucleic acid extractions from blood samples using β-alanine (pKa = 10.24, mobility of 30.8 × 10−9 m2 V−1 s−1) as the TE which offers very high electric field strength in the ATE zone and extraction efficiencies up to 93%.15,39 However, we found that the high pKa of β-alanine resulted in a higher alkaline ATE than serine, making it ineffective for proteinase K removal (see Fig. 2B). We experimented with different counterions, which can be used for pH control, but we found that using tris as the counterion resulted in ITP plugs near pH 8 that were highly compatible with RT-RPA reaction conditions which are also tris-buffered (∼pH 8). We also performed experimental validations of pH profiles generated in SPRESSO using pH paper (Fig. S1†).
3.2 Nucleic acid extraction visualization
We examined the nucleic acid extraction performance of our ITP μPAD using fluorescence imaging of labeled DNA. We found that labeled DNA was a convenient analyte for optimizing experimental conditions and studying ITP dynamics. Our ITP device employs a uniquely shaped porous membrane with a wide sample region that holds a 40 μL volume, as shown in Fig. 3A. Over a 15-minute period, nucleic acids electromigrate into an extraction region containing approximately 4 μL volume, which can be cut out and directly added to an RT-RPA reaction. This concentration step from a large sample volume to a 10-fold smaller extraction volume is advantageous for detecting HIV which may contain less than hundreds of RNA copies in 40 μL of serum. | ||
| Fig. 3 Experimental fluorescence images of ITP extraction of labeled DNA from proteolyzed serum. (A) The ITP system consists of a paper strip spanning two acrylic reservoirs within a plastic Petri dish. Nucleic acids are extracted from a 40 μL serum lysate into a 4 μL extraction zone. (B) DNA labeled with Alexa Fluor 488 is mixed with digested human serum and is initially located in the wide sample zone of the Fusion 5 membrane. DNA focuses into a concentrated plug in the straight portion of the strip (t = 10 min) before entering the extraction zone (∼t = 15 min). Pixel intensities of the images are y-averaged, creating normalized fluorescence distribution with respect to distance along the membrane for each time point (0, 5, and 10 minutes). | ||
In Fig. 3B, we present experimental images of an ITP extraction of DNA labeled with Alexa Fluor at a concentration of 100 nM in 40 μL of digested serum. We show images of the early electromigration and focusing into a concentrated plug over the first 10 minutes of ITP. We plot the y-averaged intensity as a function of strip length at 0, 5, and 10 minutes. Before the electric field is applied (t = 0) the DNA is diffusely distributed over the sample zone with a low average fluorescent intensity. Labeled DNA electromigrate out of the sample zone into the straight region of the strip, forming a concentrated plug between the LE and ATE. After 10 minutes, the DNA has electromigrated across the majority of the strip length, reaching a distance approximately 23 mm from the middle of the sample zone. An additional 5 minutes of migration centers the ITP plug in the extraction region of the strip. We measured the extraction efficiency of the ITP system using fluorescence quantification of labeled DNA (see Fig. S2†). We observed extraction efficiencies ranging from 70 to 81% with a 40 μL digested serum sample. We observed improved extraction efficiencies, up to greater than 90%, when processing diluted serum samples. For reference, Qiagen's laboratory-based QIAamp Viral RNA Mini Kit self-reports RNA extraction yields greater than 90% from clinical specimens. We calculated the extraction efficiency of the ITP system using 70 bp labeled DNA, and we expect this yield is similar for HIV RNA which is ∼10 kilobase pairs in length. Previous studies have shown that DNA and RNA have a similar electrophoretic mobility, and nucleic acid length does not significantly change its free solution mobility.46,47 Therefore, both short DNA and longer viral RNA should focus into the ITP plug of our system. It is possible that the mobility of the HIV RNA may be reduced by the porous media due to its greater length, but we do not expect this to significantly affect the yield.
We observe several interesting phenomena in extraction experiments with our ITP μPAD. The data suggest DNA concentration profiles are Gaussian, as predicted by peak-mode ITP literature.44 We observe electroosmotic flow of the system causes slight dispersion of the plug, widening the DNA distribution and reducing the maximum peak intensity. Electroosmotic dispersion is common in electrokinetic systems and has been extensively studied in isotachophoresis.48,49 A region of low-level fluorescence is evident trailing the ITP plug. We hypothesize this fluorescence is from DNA that has formed complexes with polypeptides in the proteolyzed serum proteins, reducing its electrophoretic mobility and preventing stacking. This phenomenon has been previously observed in ITP studies and is supported by the propensity of nucleic acids to nonspecifically interact with proteins in biological samples.20,50 It is also possible that KDS precipitate may accumulate within the pore structure and impede the electromigration of nucleic acids. We also see a small amount of residual fluorescence remain in the sample region of the ITP strip during extraction. We believe this is due to a trace amount of target DNA adsorbing to the porous membrane. We screened various membranes to identify the optimal substrate for the ITP μPAD, and we found that Fusion 5 resulted in the least DNA adsorption or entanglement (Fig. S3†). In experiments using pure buffer systems, we did not encounter any issues with analyte loss during the ITP extraction (Fig. S4 and S5†). We found that we were able to successfully electromigrate 10 copies of synthetic DNA and 50 copies of HIV RNA across a 30 mm Fusion 5 strip using RPA for detection.
3.3 HIV RNA extraction
Our initial experimental efforts to extract HIV RNA spiked into serum were challenged by rapid RNA degradation in serum samples with no RNase control measures, which is consistent with previous reports of extensive RNA degradation in serum on the order of seconds.9 To mitigate issues with RNases, we leveraged previous work that developed an RNase inactivation method for human serum via incubation with 0.5% SDS, 1 mg mL−1 proteinase K, and 10 mM DTT for 1 hour at 50 °C.33 Our ITP system was designed to remove SDS and proteinase K in the resulting lysate from focusing in the plug. We performed a set of experiments to assess the purity of the ITP plug by observing its effect on RT-RPA reactions. For these experiments, we processed a 40 μL serum lysate with no added HIV RNA with our ITP system. The extraction zone of the strip was centrifuged to dewater the membrane. The resulting ITP plug eluate (∼4 μL) was added to an RT-RPA reaction with 500 copies of HIV RNA to determine if the eluate inhibited the amplification reaction. Fig. 4A shows fluorescence amplification curves detecting HIV RNA which indicate the compatibility of ITP plugs with RT-RPA. Positive control reactions with only HIV RNA are provided for comparison. We found that the contents of ITP plugs in extractions including KCl in the LE did not significantly impact RT-RPA performance. In ITP extractions with no KCl, the contents of the ITP plug inhibited RT-RPA such that no amplification was detected. This indicates that the potassium-mediated SDS precipitation removed enough of the anionic detergent to enable RT-RPA. Our results also indicate that our ITP system was successful in preventing proteinase K from focusing in the ITP plug. This supports the simulations of pH conditions in Fig. 2B that found the ITP system pH was less than the isoelectric point of proteinase K (8.9). We tested an alternate ITP system that was not designed for proteinase K removal (β-alanine as TE) and found that the resulting ITP plugs contained proteinase K and completely inhibited RT-RPA (Fig. S6†).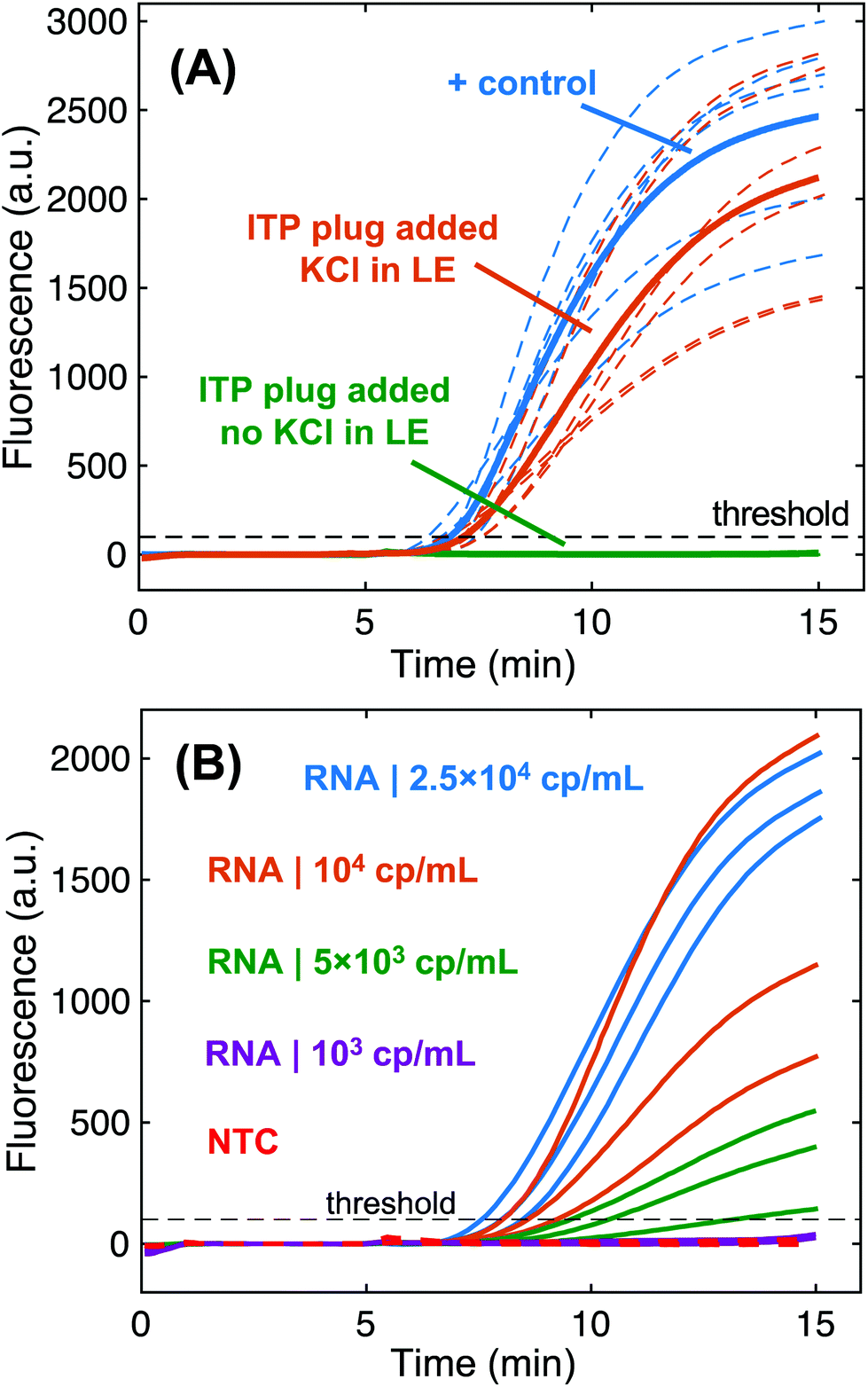 | ||
| Fig. 4 Purification and extraction of nucleic acids from serum samples via paper-based ITP. (A) Fluorescence measurements of RPA assays assessing the level of inhibitors present in ITP plugs. Experiments evaluating ITP purification have 500 copies of HIV-1 RNA with 4 μL of ITP plug eluate added into them. Positive control reactions contain only 500 copies of RNA. We plot the replicate amplification curves (N = 6 for each) with a dashed line and respective averages with a solid line. Two different ITP systems were evaluated: one containing potassium chloride in the leading electrolyte to precipitate dodecyl sulfate and the other with no potassium chloride. Positive control experiments (N = 3) simply include nuclease free water. (B) 5000 HIV-1 RNA copies per mL of digested serum (200 copies in 40 μL of serum) were consistently extracted and amplified over the threshold fluorescence value with RT-RPA (N = 3). No template control (NTC) experiments (N = 3 for each assay) did not increase in fluorescence. | ||
We analyzed the performance of the ITP μPAD for RNA extraction using pre-digested serum spiked with known concentrations of HIV-1 RNA. Fig. 4B presents amplification curves for extracted HIV-1 RNA at different input concentrations in serum. This assay successfully detects 5000 copies of HIV RNA per mL of serum, corresponding to 200 RNA copies per 40 μL of processed serum. As expected, we found that amplification is much more robust when extracting and detecting higher concentrations of HIV in serum. We found that our sample pretreatment protocol for digesting serum proteins and inactivating endogenous RNases was crucial for ITP RNA extractions. We also tested the ITP μPAD performance with a robust, synthetic DNA target (200 bp in length) and found an order of magnitude improved sensitivity of 500 cp per ml (20 copies per 40 μL sample) compared with HIV RNA (Fig. S7†). One primary consideration is that the RPA assay used for DNA detection (nearly single copy sensitivity) was approximately 10-fold more sensitive than the RT-RPA assay for HIV RNA (∼25 copy sensitivity). There may be multiple reasons why lower detection limits were observed with DNA, but we hypothesize that direct amplification from DNA targets is significantly more efficient than first synthesizing complementary DNA templates from the viral RNA via reverse transcription before RPA can begin.
3.4 Detection of HIV-positive serum
We employed our RNase inactivation chemistry for viral lysis and serum protein digestion, followed by isotachophoretic extraction on our μPAD to detect HIV virions in human serum. We built upon previous work to identify a protocol for rapid RNase inactivation that is better suited for POC testing applications. We used a commercial RNase detection assay to evaluate the serum pretreatment conditions for rapid and complete RNase inactivation and plotted the results in Fig. 5A. Incubations of serum with proteinase K alone did not remove RNase activity. Serum treatment with 0.5% SDS alone temporarily inactivated RNases, but activity was restored when the sample was diluted into the detection assay. The combination of 0.5% SDS, 1 mg mL−1 proteinase K, and 10 mM DTT was able to permanently reduce serum RNase activity to negligible levels when incubated at 50 °C for 1 hour (Fig. 5A). A significantly reduced incubation time of 15 minutes also achieved nearly complete RNase inactivation when heated to a higher temperature of 65 °C.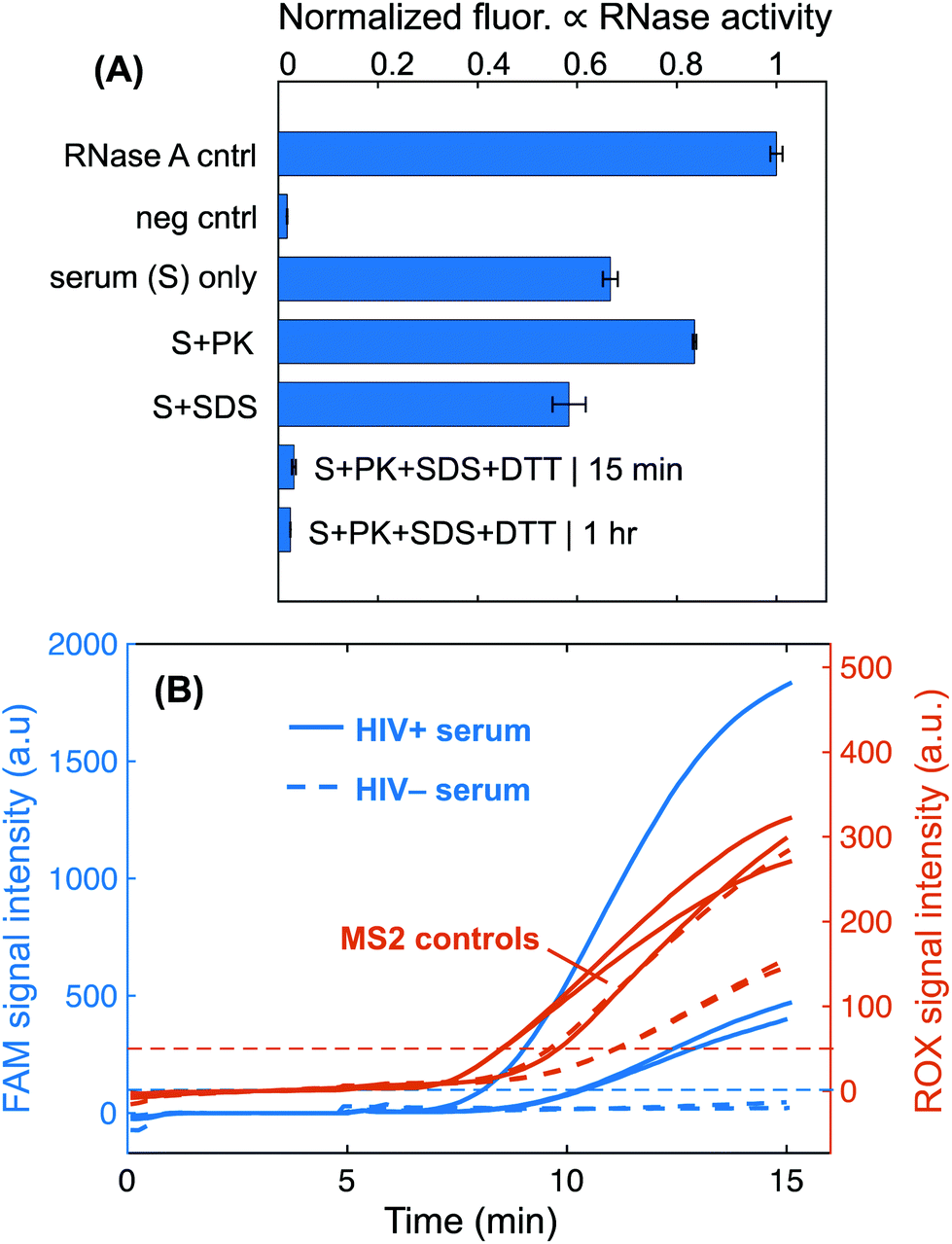 | ||
| Fig. 5 (A) RNase activity of serum samples pretreated with proteinase K and/or SDS. A commercial RNase detection assay, RNaseAlert, measures RNase activity by means of fluorescence intensity increase. High fluorescence denotes high RNase activity. We found that incubation of serum with 0.5% SDS, 1 mg mL−1 proteinase K at 65 °C for 15 minutes resulted in negligible RNase activity in the lysate. Experiments were run in triplicate and one standard deviation around the mean is plotted for each. RNase A (1.5 U L−1) is the positive control, and the negative control is RNase-free water add to the RNaseAlert assay. (B) Detection of HIV-1 virions and MS2 phage from human serum. Fluorescence intensities of the two different emission spectra used to simultaneously detect HIV and MS2 amplification. FAM signal intensity indicates successful amplification of target HIV-1, while ROX signal reports amplification of an MS2 region. Experiments with HIV-positive serum amplify while those with HIV-negative serum do not. All MS2 controls amplify for each respective experiment. | ||
The SDS and proteinase K leveraged in the 15-minute RNase inactivation protocol are both commonly used reagents in lysis protocols,51 so we hypothesized that this chemistry would by effective for HIV viral lysis. SDS is a powerful anionic detergent that is effective for protein denaturation and lipid membrane disruption.52,53 Proteinase K is a broad-spectrum protease that lyses cells and pathogens via degradation of structural proteins.10,54 We pretreated HIV+ serum with our specialized protocol and then extracted RNA from the lysate with our ITP μPAD. Off-chip duplexed RT-RPA detected HIV and MS2 internal control RNA. Similar to HIV, MS2 bacteriophage is a single-stranded RNA virus and consequently acts as an internal process control for viral lysis, RNase inactivation, RNA extraction, reverse transcription, and RPA. We were able to detect HIV in serum at 5 × 104 cp per mL using our assay. Tests with HIV-negative serum did not amplify although the MS2 internal control was still detected. Our assay protocol requires 15 minutes for serum pretreatment, 15 minutes for ITP, and 15 minutes for RT-RPA, which totals a 45-minute test runtime.
4. Concluding remarks
We report on a diagnostic assay for HIV detection from human serum within 45-minutes using a novel sample pretreatment chemistry, an ITP μPAD, and RT-RPA. We demonstrate several advancements in the use of ITP for POC nucleic acid-based tests. We identified a protocol for viral lysis, RNase inactivation, and serum protein digestion using a short incubation with proteinase K, SDS, and DTT. This chemistry is unique from previous sample pretreatments in ITP studies which did not adequately address serum RNases. Our pretreatment method is also distinct from typical solid phase extraction lysis buffers which rely on high concentrations of guanidinium salts. We designed a specialized ITP μPAD that can directly process 40 μL of serum lysate. This is the largest volume of serum that has been used in ITP nucleic acid extractions, to our knowledge. We controlled the pH of our system to remove proteinase K and leveraged potassium chloride to precipitate SDS in the lysate. We confirmed that the resulting ITP plug was free of inhibitors of RT-RPA and found the ITP μPAD could extract 5000 copies of HIV RNA per mL of proteolyzed serum. We then demonstrated that our assay detects HIV in human serum within 45-minutes. Our assay features an MS2 bacteriophage for an internal process control of lysis, RNA extraction, reverse transcription, and amplification. This work is the first example of an ITP-based detection assay for RNA viruses in human serum.Our work describes a potential sample preparation method leveraging paper-based ITP that may be used in POC molecular testing for HIV. While our primary focus was detecting bloodborne viruses, the ITP μPAD may also be an effective for sample preparation tool for viral pathogens in other complex samples, such as SARS-CoV-2 in saliva or respiratory specimens. We seek to eliminate the need for numerous buffer exchanges, highly concentrated chaotropic agents, and toxic chemicals found in typical viral RNA sample preparation methods. For example, solid-phase extraction employs high-molarity guanidine thiocyanate which forms harmful cyanide gas when combined with bleach, complicating safe disposal of test materials.55 Lysis buffers with SDS, proteinase K, and DTT are relatively safe for handling by untrained users and feature easy disposal in resource-limited health care settings. Future work is needed to ensure that our lysis protocol results in sample lysate that is safe to handle and free of culturable virus.
We demonstrate HIV detection from serum at a viral load of 5 × 104 cp per mL, which is within the clinically relevant range for HIV. Among people living with HIV, there is a significant population who are either not on ART or who have not achieved viral suppression due to adherence issues or a strain of HIV that is resistant to a particular drug regimen. People with unsuppressed HIV infections may have viral loads as high as 107 cp per mL.56 The WHO has recommended that POC tests for viral load monitoring of HIV-positive patients on drug therapies have an LOD of 1000 cp per mL in order to maximize treatment failure detection.6 While RT-RPA is not as effective as RT-PCR for viral load quantification, there is an established approach for a semi-quantitative test that indicates whether a sample has exceeded the recommended 1000 cp per mL threshold.57,58 This approach is leveraged by a commercial product, the SAMBA II, which qualitatively indicates whether an HIV viral load is above or below the test's LOD of 1000 cp per mL.58 Ongoing efforts in our lab are focused on improving our device's LOD to this recommended value by increasing the sample volume, minimizing RNA degradation, and improving the ITP extraction efficiency.
The ITP μPAD features convenient sample addition, low-cost materials, and rapid results, yet there are several aspects of an effective POC test that we do not address, such as plasma separation and on-chip integration of assay steps. Our research group and others have previously demonstrated that plasma can be generated from whole blood samples with asymmetric polymer membranes that are readily integrated with paper-based receiving membranes.31,59,60 We have also reported a method for integrating paper-based ITP and RPA for simultaneous extraction and amplification of nucleic acids.32 Future work is focused on advancing our ITP μPAD to integrate all assay operations on-chip, as we move towards a fully integrated point-of-care HIV viral load monitoring test that is well suited for LMICs.
Conflicts of interest
The authors have declared no conflict of interest.Acknowledgements
We thank the funding and support from the National Institute of Biomedical Imaging and Bioengineering (R01EB022630) of the National Institutes of Health (NIH) and a National Science Foundation (NSF) Graduate Research Fellowship (A.T.B.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or NSF.References
- WHO | Data and statistics, http://www.who.int/hiv/data/en/, (accessed March 11, 2020).
- World Health Organization (WHO), Guideline on when to start antiretroviral therapy and on pre-exposure prophylaxis for HIV, http://apps.who.int/iris/bitstream/10665/186275/1/9789241509565_eng.pdf.
- World Health Organization, WHO recommendations on the diagnosis of HIV infection in infants and children, World Health Organization, and Department of HIV/AIDS, 2010 Search PubMed.
- T. Roberts, J. Cohn, K. Bonner and S. Hargreaves, Clin. Infect. Dis, 2016, 62, 1043–1048 CrossRef PubMed.
- P. K. Drain, J. Dorward, A. Bender, L. Lillis, F. Marinucci, J. Sacks, A. Bershteyn, D. S. Boyle, J. D. Posner and N. Garrett, Clin. Microbiol. Rev., 2019, 32, 25 CrossRef PubMed.
- World Health Organization, Consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection: recommendations for a public health approach, 2016 Search PubMed.
- A. Niemz, T. M. Ferguson and D. S. Boyle, Trends Biotechnol., 2011, 29, 240–250 CrossRef CAS PubMed.
- M. Sidstedt, J. Hedman, E. L. Romsos, L. Waitara, L. Wadsö, C. R. Steffen, P. M. Vallone and P. Rådström, Anal. Bioanal. Chem., 2018, 410, 2569–2583 CrossRef CAS PubMed.
- N. B. Y. Tsui, E. K. O. Ng and Y. M. D. Lo, Clin. Chem., 2002, 48, 1647–1653 CrossRef CAS.
- Enzymes of molecular biology, ed. M. M. Burrell, Humana Press, Totowa, N.J, 1993 Search PubMed.
- R. Boom, C. J. Sol, M. M. Salimans, C. L. Jansen, P. M. Wertheim-van Dillen and J. van der Noordaa, J. Clin. Microbiol., 1990, 28, 495–503 CrossRef CAS PubMed.
- P. Chomczynski and N. Sacchi, Anal. Biochem., 1987, 162, 156–159 CrossRef CAS PubMed.
- E. J. Wood, Biochem. Educ., 1983, 11, 82–82 CrossRef.
- N. Ali, R. de C. P. Rampazzo, A. D. T. Costa and M. A. Krieger, BioMed Res. Int., 2017, 2017, 9306564 Search PubMed.
- V. Kondratova, O. Serd'uk, V. Shelepov and A. Lichtenstein, BioTechniques, 2005, 39, 695–699 CrossRef CAS PubMed.
- A. Rogacs, L. A. Marshall and J. G. Santiago, J. Chromatogr., A, 2014, 1335, 105–120 CrossRef CAS PubMed.
- L. G. Longsworth, J. Am. Chem. Soc., 1953, 75, 5705–5709 CrossRef CAS.
- V. N. Kondratova, I. V. Botezatu, V. P. Shelepov and A. V. Lichtenstein, Biochemistry (Moscow), 2009, 74, 1285–1288 CrossRef CAS PubMed.
- L. A. Marshall, A. Rogacs, C. D. Meinhart and J. G. Santiago, J. Chromatogr., A, 2014, 1331, 139–142 CrossRef CAS PubMed.
- A. Persat, L. A. Marshall and J. G. Santiago, Anal. Chem., 2009, 81, 9507–9511 CrossRef CAS PubMed.
- Y. Qu, L. A. Marshall and J. G. Santiago, Anal. Chem., 2014, 86, 7264–7268 CrossRef CAS PubMed.
- A. Rogacs, Y. Qu and J. G. Santiago, Anal. Chem., 2012, 84, 5858–5863 CrossRef CAS PubMed.
- C. Eid and J. G. Santiago, Analyst, 2017, 142, 48–54 RSC.
- M. M. Gong and D. Sinton, Chem. Rev., 2017, 117, 8447–8480 CrossRef CAS PubMed.
- B. Y. Moghadam, K. T. Connelly and J. D. Posner, Anal. Chem., 2014, 86, 5829–5837 CrossRef CAS PubMed.
- X. Li, L. Luo and R. M. Crooks, Lab Chip, 2015, 15, 4090–4098 RSC.
- F. Schaumburg, P. A. Kler, C. S. Carrell, C. L. A. Berli and C. S. Henry, Electrophoresis, 2020, 41, 562–569 CrossRef CAS PubMed.
- T. Rosenfeld and M. Bercovici, Lab Chip, 2018, 18, 861–868 RSC.
- B. Y. Moghadam, K. T. Connelly and J. D. Posner, Anal. Chem., 2015, 87, 1009–1017 CrossRef CAS PubMed.
- T. Rosenfeld and M. Bercovici, Lab Chip, 2014, 14, 4465–4474 RSC.
- B. P. Sullivan, A. T. Bender, D. N. Ngyuen, J. Y. Zhang and J. D. Posner, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2020, 122494 Search PubMed.
- A. T. Bender, M. D. Borysiak, A. M. Levenson, L. Lillis, D. S. Boyle and J. D. Posner, Anal. Chem., 2018, 90, 7221–7229 CrossRef CAS PubMed.
- A. T. Bender, B. P. Sullivan, L. Lillis and J. D. Posner, J. Mol. Diagn., 2020, 22, 1030–1040 CrossRef CAS PubMed.
- L. Lillis, D. A. Lehman, J. B. Siverson, J. Weis, J. Cantera, M. Parker, O. Piepenburg, J. Overbaugh and D. S. Boyle, J. Virol. Methods, 2016, 230, 28–35 CrossRef CAS PubMed.
- A. M. Sanchez, C. T. DeMarco, B. Hora, S. Keinonen, Y. Chen, C. Brinkley, M. Stone, L. Tobler, S. Keating, M. Schito, M. P. Busch, F. Gao and T. N. Denny, J. Immunol. Methods, 2014, 409, 117–130 CrossRef CAS PubMed.
- F. Rouet, M.-L. Chaix, E. Nerrienet, N. Ngo-Giang-Huong, J.-C. Plantier, M. Burgard, M. Peeters, F. Damond, D. K. Ekouevi, P. Msellati, L. Ferradini, S. Rukobo, V. Maréchal, N. Schvachsa, L. Wakrim, C. Rafalimanana, B. Rakotoambinina, J.-P. Viard, J.-M. Seigneurin, C. Rouzioux and for the A. N. de R. sur le S. A. W. Groups, JAIDS, 2007, 45, 380–388 CAS.
- J. Dreier, M. Störmer and K. Kleesiek, J. Clin. Microbiol., 2005, 43, 4551–4557 CrossRef CAS PubMed.
- A. Persat, M. E. Suss and J. G. Santiago, Lab Chip, 2009, 9, 2454–2469 RSC.
- V. N. Kondratova, I. V. Botezatu, V. P. Shelepov and A. V. Lichtenstein, Anal. Biochem., 2011, 408, 304–308 CrossRef CAS PubMed.
- W. A. Al-Soud and P. Rådström, J. Clin. Microbiol., 2001, 39, 485–493 CrossRef CAS PubMed.
- H. Suzuki and T. Terada, Anal. Biochem., 1988, 172, 259–263 CrossRef CAS PubMed.
- L. Boiso and J. Hedman, Forensic Sci. Int.: Genet., 2017, 29, e16–e18 CrossRef CAS PubMed.
- M. Bercovici, S. K. Lele and J. G. Santiago, J. Chromatogr., A, 2009, 1216, 1008–1018 CrossRef CAS PubMed.
- T. K. Khurana and J. G. Santiago, Anal. Chem., 2008, 80, 6300–6307 CrossRef CAS PubMed.
- N. C. Stellwagen and E. Stellwagen, J. Chromatogr. A, 2009, 1216, 1917–1929 CrossRef CAS PubMed.
- N. C. Stellwagen, C. Gelfi and P. G. Righetti, Biopolymers, 1997, 42, 687–703 CrossRef CAS PubMed.
- I. C. Yeh and G. Hummer, Biophys. J., 2004, 86, 681–689 CrossRef CAS PubMed.
- S. Bhattacharyya, P. P. Gopmandal, T. Baier and S. Hardt, Phys. Fluids, 2013, 25, 022001 CrossRef.
- G. Garcia-Schwarz, M. Bercovici, L. A. Marshall and J. G. Santiago, J. Fluid Mech., 2011, 679, 455–475 CrossRef.
- E. Jankowsky and M. E. Harris, Nat. Rev. Mol. Cell Biol., 2015, 16, 533–544 CrossRef CAS PubMed.
- D. Goldenberger, I. Perschil, M. Ritzler and M. Altwegg, PCR Methods Appl., 1995, 4, 368–370 CrossRef CAS PubMed.
- M. Johnson, Mater. Methods, 2013, 3, 163 Search PubMed.
- R. B. Brown and A. Julie, J. R. Soc., Interface, 2008, 5, S131–S138 CrossRef CAS PubMed.
- R. McGookin, Methods Mol. Biol., 1985, 2, 109–112 CAS.
- S. Paik and X. Wu, Chem. Health Saf., 2005, 12, 33–38 CrossRef CAS.
- C. D. Pilcher, G. Joaki, I. F. Hoffman, F. E. Martinson, C. Mapanje, P. W. Stewart, K. A. Powers, S. Galvin, D. Chilongozi, S. Gama, M. A. Price, S. A. Fiscus, M. S. Cohen and M. for the UNC Project, AIDS, 2007, 21, 1723–1730 CrossRef PubMed.
- L. M. Luft, M. J. Gill and D. L. Church, Int. J. Infect. Dis., 2011, 15, e661–e670 CrossRef PubMed.
- A. V. Ritchie, I. Ushiro-Lumb, D. Edemaga, H. A. Joshi, A. De Ruiter, E. Szumilin, I. Jendrulek, M. McGuire, N. Goel, P. I. Sharma, J.-P. Allain and H. H. Lee, J. Clin. Microbiol., 2014, 52, 3377–3383 CrossRef PubMed.
- A. Nabatiyan, Z. A. Parpia, R. Elghanian and D. M. Kelso, J. Virol. Methods, 2011, 173, 37–42 CrossRef CAS PubMed.
- S. Hin, M. Loskyll, V. Klein, M. Keller, O. Strohmeier, F. von Stetten, R. Zengerle and K. Mitsakakis, Microelectron. Eng., 2018, 187–188, 78–83 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0an02483j |
| This journal is © The Royal Society of Chemistry 2021 |