
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystallization of colourless hexanitratoneptunate(IV) with anhydrous H+ countercations trapped in a hydrogen bonded polymer with diamide linkers†
Koichiro Takao *a,
Juliane Märzb,
Moe Matsuokaa,
Takanori Mashitaa,
Hiroyuki Kazamaa and
Satoru Tsushimaabc
*a,
Juliane Märzb,
Moe Matsuokaa,
Takanori Mashitaa,
Hiroyuki Kazamaa and
Satoru Tsushimaabc
aLaboratory for Advanced Nuclear Energy, Institute of Innovative Research, Tokyo Institute of Technology, 2-12-1 N1-32, O-okayama, Meguro-ku, 152-8550 Tokyo, Japan. E-mail: ktakao@lane.iir.titech.ac.jp
bInstitute of Resource Ecology, Helmholtz-Zentrum Dresden-Rossendorf (HZDR), Bautzner Landstraße 400, 01328 Dresden, Germany. E-mail: j.maerz@hzdr.de; s.tsushima@hzdr.de
cTokyo Tech World Research Hub Initiative (WRHI), Institute of Innovative Research, Tokyo Institute of Technology, 2-12-1, O-okayama, Meguro-ku, 152-8550 Tokyo, Japan. E-mail: tsushima.s.ab@m.titech.ac.jp
First published on 7th February 2020
Abstract
Colourless crystalline compounds of centrosymmetric [Np(NO3)6]2− were yielded from 3 M HNO3 aq in the presence of double-headed 2-pyrrolidone derivatives (L). In the obtained crystal structures, H+ was also involved as a countercation to compensate for the negative charge of [Np(NO3)6]2−, where the initial hydration around H+ was fully removed during crystallization despite it having the strongest hydration enthalpy. Instead, this anhydrous H+ was captured by L to form a [H+⋯L]n hydrogen bonded polymer. In [Np(NO3)6]2−, the Np4+ centre is twelve-coordinated with 6 bidentate NO3−, and therefore, present in an icosahedral geometry bearing inversion centre. In such a centrosymmetric system, any f–f transitions stemming from the 5f3 electronic configuration of Np4+ are electric-dipole forbidden. This is the reason why the compounds currently obtained were colourless unlike ordinary Np(IV) species, which are olive-green.
Introduction
The coordination chemistry of actinides is highly relevant to nuclear chemical engineering, especially for reprocessing of spent nuclear fuels and geological disposal of radioactive wastes.1 Aqueous HNO3 is most popularly employed to dissolve the spent nuclear fuels and to separate U and Pu from fission products and minor actinides with solvent extraction. Therefore, actinide coordination chemistry of nitrato complexes is the most essential to systematically understand the separation behaviour of actinides.2 Among the various oxidation numbers available for actinide elements relevant to nuclear fuel recycling, the tetravalent state, An(IV), is highly important especially in terms of Pu4+ separation by solvent extraction with tri-n-butyl phosphate (TBP). The limiting An4+ complexes with NO3− are [An(NO3)6]2− (An = Th,3–7 U,8,9 Np,10,11 Pu12–14), which usually crystallize with various countercations having relatively low hydration enthalpy like heavy alkali metals and quaternary ammonium ions (NR4+, R = H, alkyl).15 To our best knowledge, all the reported compounds of An(IV) usually exhibit original colours defined by their respective 5f electronic configurations.In contrast, we recently demonstrated that H+ showing the highest hydration enthalpy3 also has a potential to make crystalline compounds with [U(NO3)6]2− and NO3− by coupling with diamide linker molecules appropriately selected to allow strong hydrogen bond showing little potential barrier along atomic coordinate of H+ between hydrogen bond donor and acceptor, where we employed double-headed 2-pyrrolidone derivatives like L1 and L2 shown in Fig. 1.16,17 The resulting U(IV) compounds have a general formula of (HL)2[U(NO3)6] (L = L1 (1), L2 (2)). In these crystal structures, H+ countercations are fully dehydrated despite deposition from the aqueous HNO3 solutions. Instead, these anhydrous H+ are trapped into a cavity between two carbonyl O atoms of the neighbouring L molecules to form a unique hydrogen bond polymer, [H+⋯L]n. Interestingly, 1 and 2 do not exhibit characteristic green colour of U(IV), but are nearly colourless. Thanks to separation of U4+ centre from H+ (>5.8 Å), U4+ in these compounds are located in icosahedral geometries with nearly perfect Th-symmetry. In such a centrosymmetric system, the f–f transitions stemming from its 5f2 electronic configuration becomes electric-dipole (i.e., Laporte) forbidden, making 1 and 2 nearly colourless. This assumption was further corroborated by complete active space self-consistent field (CASSCF) calculation including spin–orbit coupling.
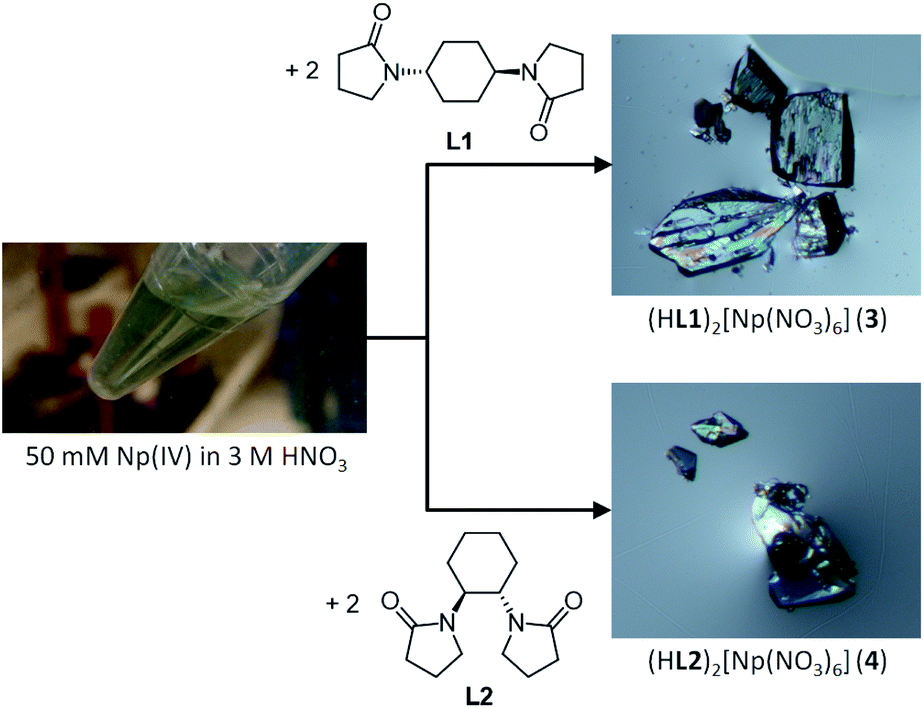 | ||
| Fig. 1 Reaction scheme to prepare colourless crystalline compounds, (HL1)2[Np(NO3)6] (3) and (HL2)[Np(NO3)6] (4) from Np4+ with L1 and L2 in 3 M HNO3 aq. An (S,S)-isomer of L2 is only shown here, whereas its racemate has been used in this work. | ||
To our understanding, crystallization of [U(NO3)6]2− with anhydrous H+ is quite exceptional from usual trend of [An(NO3)6]2−. Furthermore, formation of nearly colourless U(IV) is also unprecedented because An(IV) usually exhibit characteristic colour due to visible-light absorption arising from its own f–f transitions. There remains open questions whether crystallization of [An(NO3)6]2− as anhydrous H+ salts like 1 and 2 is peculiar to U4+ or common in An(IV), and whether 5f orbitals remain separated enough from those of NO3− to still make [An(NO3)6]2− colourless. To answer these points, we decided to employ Np4+ as another An4+. In this paper, we report details about synthesis and structural characterization of [Np(NO3)6]2− crystallized together with H+ in aid of L1 (3) and L2 (4).
Experimental
Materials
Caution! 237Np is a radioactive isotope (specific activity: 2.63 × 107 Bq g−1 with T1/2 = 2.14 × 106 years) and an alpha emitter. It has to be handled in dedicated facilities with appropriate equipment for radioactive materials to avoid health risks caused by radiation exposure.All the operations to handle Np have been done in a dedicated glove box in the control area of HZDR. Reaction scheme is shown in Fig. 1. A stock solution of Np4+ (50 mM) was prepared by dissolving NpCl4(DME)2 (13.9 mg)18 in 3 M HNO3 aq. The concentration of Np4+ in this stock solution was determined by γ-ray spectrometry. The diamide linker molecules (L1 and L2) were prepared through a method reported elsewhere.19–21 The Np4+ stock solution (50 mM, 50 μL) was layered with 3 M HNO3 aq (30 μL) and 3 M HNO3 solution of 0.50 M diamide (L1 or L2, 10 μL) in a ϕ5 mm glass test tube. Slow diffusion of Np4+ and L resulted in deposition of colourless crystals of (HL)2[Np(NO3)6] (L = L1 (3), L2 (4)) in 88% and 91% yield, respectively.
Methods
The deposited crystals were covered with mineral oil, followed by being mounted on a MicroMount. Single crystal X-ray diffraction patterns were recorded by D8 VENTURE diffractometer (Bruker) with micro-focused Mo Kα radiation (λ = 0.71073 Å). The frames were integrated with the Bruker SAINT software package using a narrow-frame algorithm. Absorption correction by SADABS22 was applied which resulted in transmission factors described in the crystallographic information file of each compound. The obtained data were processed by Olex2.1.2 software package23 suited with SHELX program.24 The structures of 3 and 4 were solved by direct method, SHELXS,25 and expanded using Fourier techniques. All non-hydrogen atoms were anisotropically refined by SHELXL-2017/1.24 Anhydrous H+ in each compound were isotropically refined, whereas all other hydrogen atoms were refined using the riding model. The final cycle of full-matrix least-squares refinement on F2 was based on observed reflections and parameters, and converged with unweighted and weighted agreement factors, R and wR, respectively. Crystallographic data of 3 and 4 were summarized in Table 1, and compared with those of U(IV) analogues, (HL)2[U(NO3)6] (L = L1 (1), L2 (2)) we reported previously16 in Tables S1 and S2 (ESI†), respectively.| (HL1)2[Np(NO3)6], 3 | (HL2)2[Np(NO3)6], 4 | |
|---|---|---|
| Formula | C28H46N10NpO22 | C28H46N10NpO22 |
| fw | 1113.76 | 1113.76 |
| Crystal size (mm) | 0.075 × 0.098 × 0.157 | 0.080 × 0.098 × 0.110 |
| Cryst. system | Monoclinic | Monoclinic |
| Space group | C2/c (#15) | P21/n (#14) |
| a (Å) | 18.1413 (10) | 9.8264 (10) |
| b (Å) | 10.9944 (6) | 10.7164 (11) |
| c (Å) | 21.6888 (12) | 19.517 (2) |
| β (°) | 109.931 (2) | 103.175 (4) |
| V (Å3) | 4066.8 (4) | 2001.1 (4) |
| Z | 4 | 2 |
| T (K) | 100 | 100 |
| Dcalcd (g cm−3) | 1.814 | 1.848 |
| μ (mm−1) | 2.652 | 2.695 |
| Obsd data (all) | 4312 | 4249 |
| R (I > 2σ) | 0.0192 | 0.0188 |
| wR (all) | 0.0420 | 0.0691 |
| GOF | 1.071 | 1.340 |
| Δρmax (e− Å−3) | 0.474 | 0.975 |
| Δρmin (e− Å−3) | −0.523 | −1.196 |
The γ-ray analysis of 237Np in the stock solution and supernatants after the deposition of 3 and 4 have been performed at VKTA Dresden. Each solution sample (0.5 or 1 μL) was loaded into a ϕ5 mm glass test tube, followed by loading to the γ-ray detector. The γ-ray emission from the sample was counted for 20 min real time plus 0.05% dead time.
For colour component analysis, the photomicrographs of Np(IV) crystalline compounds shown in Fig. 1 were processed by ImageJ (ver. 1.52a)26 to extract colour appearance parameters, hue, saturation, and brightness in 8 bit grey scale.
Theoretical calculations
Electronic absorption spectra of Np(IV) complexes were calculated at the CASSCF (3,7) level using ORCA program version 4.0.27 The coordinates of [Np(NO3)6]2− were taken from the crystal structures of 3, while those of aqua species [Np(H2O)n]4+ (n = 8, 9)28 were preliminarily optimized by DFT calculations (Gaussian 16 B.01)29 at the B3LYP level in water. Energies and wavefunctions were calculated by the CASSCF/sc-NEVPT2 (strongly contracted n-electron valence state perturbation theory) approach. CASSCF calculations using ORCA were performed following the protocol provided by Prof. Frank Neese and his coworkers.30,31 An active space considering the seven 5f orbitals was employed in the calculations whereas the number of roots was set to include all possible states stemming from 5f3 configuration of both doublet and quartet states. For Np, segmented all-electron relativistically-contracted basis sets of valence triple-zeta quality with polarization functions adapted to the Douglas–Kroll–Hess Hamiltonian (SARC-DKH-TZVP) were used. For all other atoms, scalar relativistically recontracted Karlsruhe valence triple-zeta basis sets were employed. Spin–orbit coupling (SOC) effects are included by quasi-degenerate perturbation theory (QDPT), where the multiplets stemming from the S = Ms CASSCF states are mixed by the spin–orbit mean field (SOMF) operator. Calculated spectra do not include vibronic progression to the electronic transitions.Results and discussion
In accordance with reaction scheme shown in Fig. 1, Np4+ reacted with L1 and L2 in 3 M HNO3 aq. In a glass test tube (∼5 mm O.D.), this Np(IV) stock solution (50 μL, Np4+: 2.5 μmol) was carefully layered with 3 M HNO3 aq (30 μL), and 3 M HNO3 solution of L (0.50 M, 10 μL, L: 5.0 μmol). These reaction mixtures were stored at silent place in an Np-dedicated glove box overnight to allow slow diffusion of Np4+ and L. As a result, crystalline compounds 3 and 4 grew up from the reaction mixtures of L1 and L2, respectively. Interestingly, the characteristic olive colour of Np(IV) (see Fig. 1) remarkably faded upon crystal growth, while the deposited compounds were also colourless. According to γ-ray spectrometry, the Np(IV) stock solution contained 315 kBq mL−1 of 237Np, whereas only 20.6 kBq mL−1 and 15.2 kBq mL−1 were found in the supernatants of the L1 and L2 systems after crystal deposition, respectively. Thus, Np precipitated as colourless crystals 3 and 4 from the reaction mixtures in 88% and 91% yield, respectively. Due to strong radioactivity of 237Np (2.63 × 107 Bq g−1), further characterization methods like elemental analysis, IR, and powder XRD are unavailable at present. However, taking into account our recent results of well-characterized U(IV)-analogues, (HL)2[U(NO3)6] (L = L1 (1), L2 (2)),16 the most probable identities of 3 and 4 are crystalline salts of a centrosymmetric hexanitratoneptunate(IV), [Np(NO3)6]2−.Indeed, the single crystal X-ray analysis resulted in the precise molecular and crystal structures of (HL)2[Np(NO3)6] (L = L1 (3), L2 (4)) as shown in Fig. 2. As predicted from the U(IV)-analogues (1, 2), these compounds consist of [Np(NO3)6]2− together with two L molecules. As shown in Tables S1 and S2 (ESI†), lattice parameters of 3 and 4 are almost identical with those of the corresponding U(IV)-analogues 1 and 2,16 respectively, indicating that the obtained Np compounds are isomorphic and isostructural to these U(IV)-analogues. These facts are strong evidence that the Np centre in both 3 and 4 still remains tetravalent. The selected structural parameters of 3 and 4 are summarized in Table 2 together with those of the U(IV)-analogues, 1 and 2.16 Bidentate manner of NO3− resulted in dodeca-coordination around the Np4+ centre to make an icosahedral geometry. Interatomic distances between Np and O of NO3− are 2.49–2.51 Å in 3 and 2.48–2.52 Å in 4. These bond lengths seem to be nearly the same with those in bipyridinium dication salts of [Np(NO3)6]2− (2.48–2.53 Å, 2.48–2.54 Å),10,11 while tend to be slightly shorter than those found in [U(NO3)6]2− of 1 (2.51–2.53 Å) and 2 (2.50–2.53 Å), respectively.16 The latter contrast can be ascribed to a typical trend of the actinide contraction.2 Considering similarity in chemical behaviour frequently observed in the actinide series, formation of isostructural compounds of Np with U is quite reasonable.
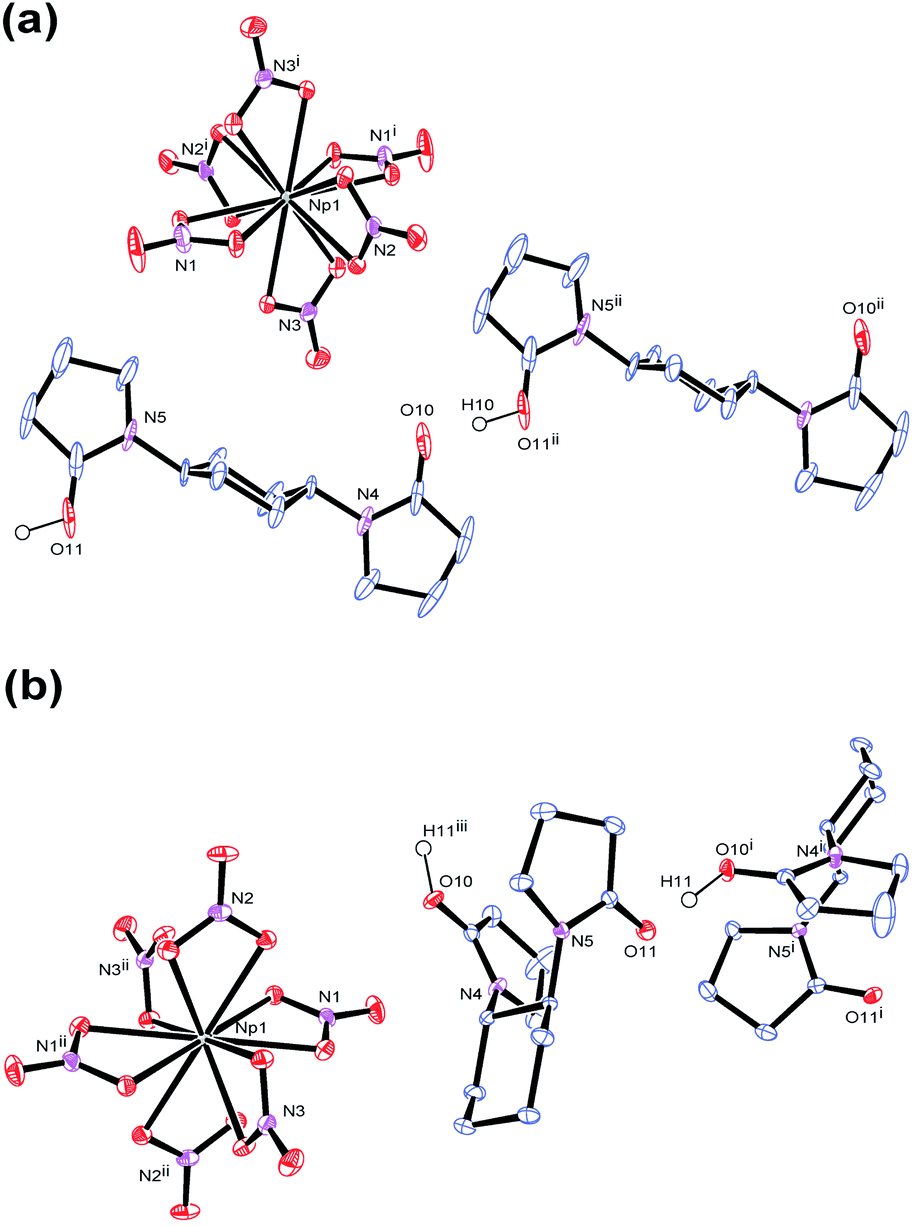 | ||
| Fig. 2 ORTEP views of (a) (HL1)2[Np(NO3)6] (3) and (b) (HL2)[Np(NO3)6] (4) at 50% probability level. Each one of disordering atoms of L1 and H atoms except for H+ involved in the hydrogen bond polymers are omitted for clarity. Both (R,R)- and (S,S)-enantiomers of L2 are present in the crystal structure of 4 to make it racemic, while only the (S,S)-isomer is displayed here. | ||
| 1a | 2a | 3b | 4b | |
|---|---|---|---|---|
| An= | U | U | Np | Np |
| L= | L1 | L2 | L1 | L2 |
| a Ref. 16.b This work.c Notations related to hydrogen bonds. D: hydrogen bond donor, A: hydrogen bond acceptor. | ||||
| Bond distance/Å | ||||
| An–ONO3 | 2.514(2) | 2.499(3) | 2.492(2) | 2.484(2) |
| 2.515(2) | 2.511(4) | 2.496(2) | 2.497(2) | |
| 2.518(2) | 2.516(4) | 2.500(2) | 2.508(2) | |
| 2.521(2) | 2.524(4) | 2.503(2) | 2.508(2) | |
| 2.523(2) | 2.525(4) | 2.505(2) | 2.511(2) | |
| 2.525(2) | 2.531(4) | 2.506(2) | 2.516(2) | |
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
1.255(5) | 1.268(6) | 1.255(4) | 1.271(3) |
| 1.261(5) | 1.270(6) | 1.260(4) | 1.271(3) | |
| C–N | 1.303(5) | 1.313(6) | 1.318(4) | 1.308(4) |
| 1.323(4) | 1.314(6) | 1.351(9) | 1.311(3) | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| H-bond param.c | ||||
| D–H/Å | 1.13 | 0.94 | 1.08 | 1.17 |
| H⋯A/Å | 1.28 | 1.48 | 1.34 | 1.24 |
| D⋯A/Å | 2.41 | 2.41 | 2.42 | 2.41 |
| D–H⋯A/Å | 178.3 | 167.9 | 178.0 | 177.1 |
Note that the coordination sphere of Np4+ is fully saturated by 12-coordination resulted from 6 bidentate NO3−, and that there are no direct interactions between Np4+ and L. Therefore, L does not play any roles as a ligand in both 3 and 4 despite its bridging nature that we found in 1-dimensional coordination polymers of UVIO22+, [UO2(NO3)2(L)]n.20,21 Nevertheless, L still plays another important role to construct the crystal structures of 3 and 4. Negative charge of [Np(NO3)6]2− has to be somehow compensated in crystal structures by incorporation of countercation(s). Based on the experimental conditions described above, no cations other than Np4+ and H+ were available in the current reaction systems. Therefore, the most plausible countercation in 3 and 4 should be H+. While usual status of H+ in aqueous solution is oxonium ion like H3O+, no isolated O atoms attributable to H3O+ have been found in the residual Fourier maps in both structures despite their deposition from 3 M HNO3 aq. Instead, a significant residual electron density was found between two carbonyl O atoms of neighbouring L molecules in both 3 and 4, and assigned to H+ with isotropic temperature factor. The final least-squares refinement of each structure was successfully converged to afford good agreement factors; R = 0.0192 (I > 2σ), wR = 0.0420 (all) for 3; R = 0.0188 (I > 2σ), wR = 0.0691 (all) for 4. Therefore, we conclude that anhydrous H+ is present as a countercation of [Np(NO3)6]2− in both 3 and 4.
During crystallization process, initial hydration around H+ in the reaction mixtures was fully removed. Instead, the dehydrated H+ interacts with the neighbouring L molecules to make a [H+⋯L]n hydrogen bond polymer as shown in Fig. 3. In 3, [H+⋯L1] units are extended through translational operation (1/2, −1/2, 0) to form the zigzag-type 1D hydrogen bond polymer. In 4, [H+⋯L2] units are expanded by 21 screw rotation along b axis to make a helical hydrogen bond polymer. As racemic L2 was used here, both z- and s-twisted helixes arising from the (S,S)- and (R,R)-isomers of L2, respectively, occur in this crystal structure. According to a comprehensive review by Steiner,32 all the hydrogen bond parameters listed in Table 1 indicate that these interactions in 3 and 4 are strongly covalent (63–170 kJ mol−1). However, its strength is still much weaker than that of an actual O–H bond (e.g., 436 kJ mol−1 for methanol).33 Indeed, the CO groups (mean 1.26 Å for 3, mean 1.27 Å for 4) are slightly longer than free L (1.23 Å for both L1 and L2),16 but still exhibit double bonding character on the basis of covalent radii (CO: 1.24 Å, C–O: 1.43 Å).34 This means that there is no direct protonation to make an actual covalent bond between the carbonyl O and H+.
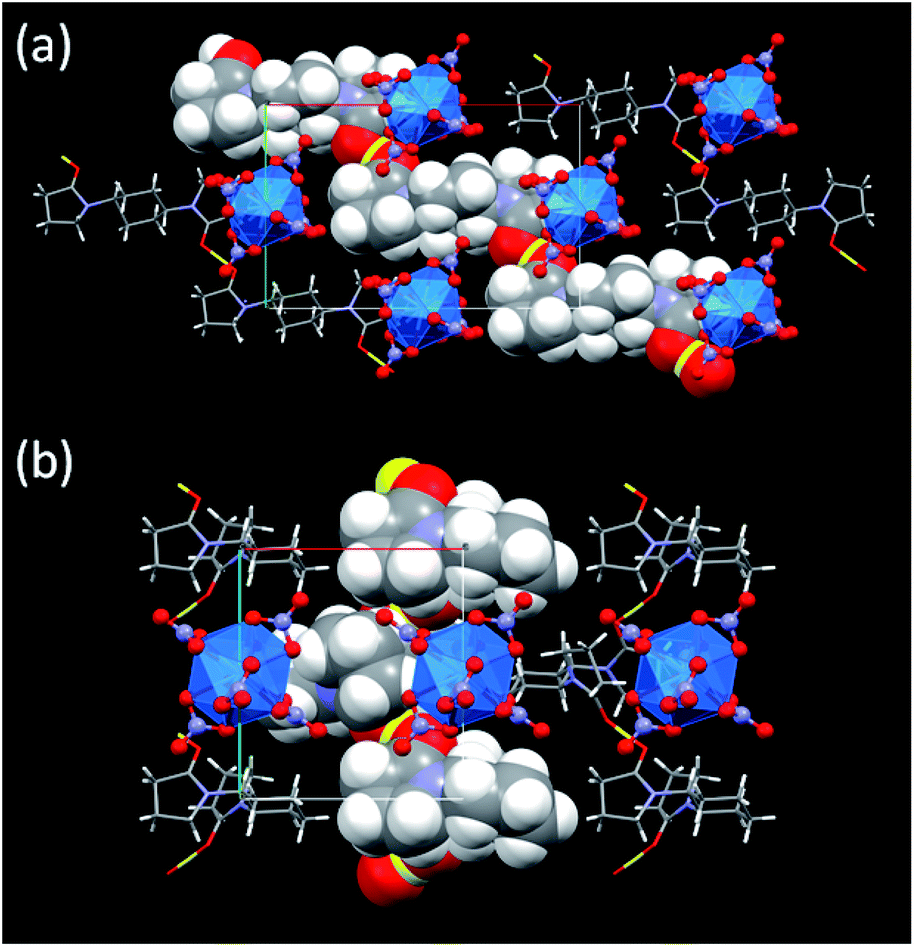 | ||
| Fig. 3 Crystal structures of (a) (HL1)2[Np(NO3)6] (3) and (b) (HL2)[Np(NO3)6] (4) along c axes. Yellow: H+, blue: Np, red: O, purple: N, grey: C, white: H. Blue transparent icosahedron shows coordination geometry around Np4+ centre surrounded by 6 bidentate NO3−. One of [H⋯L]n hydrogen bond polymers in each structure is drawn in van der Waals radius for its clarity. | ||
Unlike characteristic olive colour of ordinary Np(IV) species, [Np(NO3)6]2− in 3 and 4 do not exhibit any colour as shown in Fig. 1. At present, the solid-state UV-vis spectra of the Np(IV) compounds are not available due to strong radioactivity of Np samples. Quantitative discussion on light absorptivity at solid state would anyway be complicated due to difficulty in normalization of absorption intensity, as we previously experienced in a comparison between [U(NO3)6]2− crystalline compounds with different colours.16 However, the photomicrographs shown in Fig. 1 obviously contain some information about visual colour of 3 and 4. To make our discussion more quantitative, each pixel of these photographs was divided into colour appearance parameters, hue, saturation, and brightness by ImageJ.26 Here, hue defines intrinsic colour of a subject of interest. By comparing hue value of the crystals in Fig. 1 with that of the background, it is possible to quantify how these Np(IV) compounds are coloured. Fig. S1 and S2 (ESI†) show 8 bit grey scale colour components of photomicrographs of 3 and 4, respectively. While distribution is somewhat broad due to cracks, boundaries, and surface roughness, hue histograms of the crystal surfaces of 3 and 4 exhibit similar maxima to those of the background in both systems. These results clearly indicate that both Np(IV) compounds reported here are indeed colourless.
This fact can be explained by the Laporte selection rule. In a centrosymmetric system of the current [Np(NO3)6]2−, the electronic transitions between energy states with the same parity like f–f transitions in 5f3 electronic configuration of Np4+ are strictly forbidden. Decolouration of Np compounds due to the same reason have also been observed in 1:2 complexes of NpVO2+ with diglycolamide,35 oxidiacetate,36 and dipicolinate,37 where the inversion centre is present at the centre metal, Np5+, with 5f2 configuration.38
In order to theoretically confirm that Np compounds synthesized in this work are truly colourless, the intensities of 5f–5f electronic transitions in the [Np(NO3)6]2− entity of 3 has been estimated by CASSCF/sc-NEVPT2 approach using ORCA program.27 The calculated spectrum are given in Fig. S3 (ESI†) together with those of [Np(H2O)n]4+ (n = 8, 9)28 as references which exhibit typical olive green colour. The structures of [Np(H2O)n]4+ were optimized by DFT calculations29 at the B3LYP level in water. In CASSCF calculations, for all complexes, three electrons were distributed among seven active 5f orbitals (CAS(3,7)) and both quartet and doublet electronic configurations were considered for which 35 and 112 roots were included, respectively. Remind that only absorption stemming from 5f–5f transitions are included in these calculations and neither 5f–6d nor LMCT states are represented in the spectra. Furthermore, vibronic progression to the electronic transitions are not included here. As can be seen in Fig. S3,† typical absorption features of tetravalent Np are well-represented in the spectra of [Np(H2O)8]4+ and [Np(H2O)9]4+ which are characterized by two strong absorptions at around 500–600 nm and at around 900 nm. Transition energies are overall somewhat overestimated as we found previously in the case of U(IV) complexes. This is because of the use of isolated highly charged cation and thereby overestimating the effective charge on actinide centre.16 By contrast, these absorption features are totally diminished in the spectra of [Np(NO3)6]2− unit of 3. Closer look into the entire scale of the spectra (150–5000 nm) reveals that all 5f–5f transitions are strictly forbidden in this complex due to the perfect centrosymmetry of the complex. In reality, there are vibronic progressions to the electronic transitions,39 which are not included in our calculations. These effects may eventually give rise to absorption. However, as was previously demonstrated in the case of [U(NO3)6]2−,16 slight distortion to the Th symmetry of [AnIV(NO3)6]2− causes clear colorization of the complex. It suggests that symmetry plays by far the most crucial role to the colour of complexes compared to marginal contribution from vibrational progression. Therefore, we believe our calculations excluding vibrational progressions are still valid to discuss the colourless features of Np(IV) compounds.
The U(IV)-analogues 1 and 2 we reported previously do not exhibit typical green colour unlike ordinary U(IV) species, but are still pale colored.16 In contrast, the corresponding Np(IV) compounds 3 and 4 are more colourless as shown in Fig. 1 and as demonstrated by the colour component analysis. Interionic distances like H+⋯[An(NO3)6]2− and [An(NO3)6]2−⋯[An(NO3)6]2− and lattice parameters are almost identical between corresponding U(IV)- and Np(IV) compounds (Fig. S4, Tables S1 and S2, ESI†). Therefore, there are no large differences in electrostatic interactions that may cause perturbation of the centrosymmetric geometry of [An(NO3)6]2−. The only difference currently found is the decrease in the An–ONO3 bond distances by ∼0.02 Å, arising from the actinide contraction. Thus, the more compact structure of [Np(NO3)6]2− compared to [U(NO3)6]2− would suppress the static and/or dynamic structural perturbation from the centrosymmetry, presumably making 3 and 4 more colourless.
Conclusions
In conclusion, we have succeeded in crystallization of [Np(NO3)6]2− with anhydrous H+ and double-headed 2-pyrrolidone derivatives, L1 and L2. The isomorphic and isostructural features of the hexanitrato complexes of U4+ and Np4+ clearly indicate that the crystal structure of (HL)2[An(NO3)6] is common in coordination chemistry of An(IV) regardless of electronic configurations in 5f orbitals. On the other hand, some minor differences arising from the actinide contraction have also been observed in bond distances and colour of compounds. To further explore the systematic trend in An4+ coordination chemistry, our next target is Pu4+, which has 5f4 electronic configuration and has the highest relevance among An4+ in the nuclear fuel recycling. To our best knowledge, isolation of anhydrous H+ from aqueous systems without any direct covalent bonds and formation of hydrogen bond polymer [H+⋯L]n are also quite rare.17 We also intend to further explore these unique and interesting aspects of H+-involving chemical interactions constructed by linker molecules designed and optimized appropriately.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Mr Stephan Weiß (HZDR), Dr Atsushi Ikeda-Ohno (HZDR, currently JAEA), and Prof. Shinobu Takao (University of Electro-Communications) for their technical assistance related to γ-ray spectroscopy, single crystal XRD, and image analysis, respectively. This work was partially supported by JSPS KAKENHI Grant-in-Aid for Scientific Research (C) 19K05325 and World Research Hub Initiative (WRHI) Program of Institute of Innovative Research, Tokyo Institute of Technology. DFT calculations were performed on TSUBAME 3.0 at Tokyo Institute of Technology using Gaussian 16 program. CASSCF calculations were performed at the Center for Information Services and High-Performance Computing (ZIH) at the Technische Universität Dresden, Germany, using the library program ORCA.Notes and references
- M. Benedict, T. H. Pigford and H. W. Levi, Nuclear Chemical Engineering, McGraw-Hill, United States, 2nd edn, 1981 Search PubMed.
- L. R. Morss, N. M. Edelstein and J. Fuger, The Chemistry of the Actinide and Transactinide Elements, Springer, Dordrecht, The Netherlands, 4th edn, 2011 Search PubMed.
- G. B. Jin, J. Lin, S. L. Estes, S. Skanthakumar and L. Soderholm, J. Am. Chem. Soc., 2017, 139, 18003–18008 CrossRef CAS PubMed.
- M. Wang, B. Wang, P. Zheng, W. Wang and J. Lin, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 1913–1916 CrossRef.
- N. N. Rammo, K. R. Hamid and B. A. Khaleel, J. Less-Common Met., 1990, 162, 1–9 CrossRef CAS.
- M. R. Spirlet, J. Rebizant, C. Apostolidis, B. Kanellakopulos and E. Dornberger, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 1161–1164 CrossRef.
- G. E. Sigmon and P. C. Burns, J. Solid State Chem., 2010, 183, 1604–1608 CrossRef CAS.
- J. Rebizant, C. Apostolidis, M. R. Spirlet, G. D. Andreetti and B. Kanellakopulos, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 2098–2101 CrossRef.
- M. J. Crawford, A. Ellern, K. Karaghiosoff and P. Mayer, Inorg. Chem., 2009, 48, 10877–10879 CrossRef CAS PubMed.
- M. S. Grigor'ev, B. F. Gulev and N. N. Krot, Radiokhimiya, 1986, 28, 685–690 Search PubMed.
- M. S. Grigor'ev, A. I. Yanovskii, N. N. Krot and Y. Y. Struchkov, Radiokhimiya, 1987, 29, 574–579 Search PubMed.
- P. G. Allen, D. K. Veirs, S. D. Conradson, C. A. Smith and S. F. Marsh, Inorg. Chem., 1996, 35, 2841–2845 CrossRef CAS.
- S. D. Reilly, B. L. Scott and A. J. Gaunt, Inorg. Chem., 2012, 51, 9165–9167 CrossRef CAS PubMed.
- S. D. Conradson, K. D. Abney, B. D. Begg, E. D. Brady, D. L. Clark, C. den Auwer, M. Ding, P. K. Dorhout, F. J. Espinosa-Faller, P. L. Gordon, R. G. Haire, N. J. Hess, R. F. Hess, D. W. Keogh, G. H. Lander, A. J. Lupinetti, L. A. Morales, M. P. Neu, P. D. Palmer, P. Paviet-Hartmann, S. D. Reilly, W. H. Runde, C. D. Tait, D. K. Veirs and F. Wastin, Inorg. Chem., 2004, 43, 116–131 CrossRef CAS PubMed.
- U. Casellato, P. A. Vigato and M. Vidali, Coord. Chem. Rev., 1981, 36, 183–265 CrossRef CAS.
- K. Takao, H. Kazama, Y. Ikeda and S. Tsushima, Angew. Chem., Int. Ed., 2019, 58, 240–243 CrossRef CAS PubMed.
- H. Kazama, S. Tsushima and K. Takao, Cryst. Growth Des., 2019, 19, 6048–6052 CrossRef CAS.
- S. D. Reilly, J. L. Brown, B. L. Scott and A. J. Gaunt, Dalton Trans., 2014, 43, 1498–1501 RSC.
- K. Takao, K. Noda, Y. Morita, K. Nishimura and Y. Ikeda, Cryst. Growth Des., 2008, 8, 2364–2376 CrossRef CAS.
- H. Kazama, S. Tsushima, Y. Ikeda and K. Takao, Inorg. Chem., 2017, 56, 13530–13534 CrossRef CAS PubMed.
- K. Takao, Y. Ikeda and H. Kazama, Energy Procedia, 2017, 131, 157–162 CrossRef CAS.
- G. M. Sheldrick, SADABS, Program for Empirical Absorption Correction of Area Detector Data, 1996 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- S. Tsushima and T. Yang, Chem. Phys. Lett., 2005, 401, 68–71 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, A. P. B. Peng, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, 2016 Search PubMed.
- D. Aravena, M. Atanasov and F. Neese, Inorg. Chem., 2016, 55, 4457–4469 CrossRef CAS PubMed.
- J. Jung, M. Atanasov and F. Neese, Inorg. Chem., 2017, 56, 8802–8816 CrossRef CAS PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- J. S. Wright and K. U. Ingold, J. Chem. Educ., 2000, 77, 1062 CrossRef.
- P. Atkins, T. Overton, J. Rourke, M. Weller and F. Armstrong, Shriver & Atkins' Inorganic Chemistry, Oxford University Press, Oxford New York, 5th edn, 2010 Search PubMed.
- G. Tian, J. Xu and L. Rao, Angew. Chem., Int. Ed., 2005, 44, 6200–6203 CrossRef CAS PubMed.
- G. Tian, L. Rao and A. Oliver, Chem. Commun., 2007, 4119–4121 RSC.
- G. Tian, L. Rao and S. J. Teat, Inorg. Chem., 2009, 48, 10158–10164 CrossRef CAS PubMed.
- L. Rao and G. Tian, Symmetry, 2010, 2, 1–14 CrossRef CAS.
- C. D. Flint and P. A. Tanner, Mol. Phys., 1987, 61, 389–407 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Colour component analysis for (HL)2[Np(NO3)6], calculated absorption spectra of centrosymmetric [Np(NO3)6]2− and [Np(H2O)n]4+, schematic structures of [Np(NO3)6]2− and [H+⋯L]n hydrogen bond polymer, crystallographic data of (HL)2[An(NO3)6]. CCDC 1933983 and 1933984. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra10090c |
| This journal is © The Royal Society of Chemistry 2020 |