DOI:
10.1039/D0OB00350F
(Review Article)
Org. Biomol. Chem., 2020,
18, 3950-3964
Recent advances in the synthesis of imidazoles†
Received
17th February 2020
, Accepted 11th May 2020
First published on 18th May 2020
Abstract
The review highlights the recent advances (2018-present) in the regiocontrolled synthesis of substituted imidazoles. These heterocycles are key components to functional molecules that are used in a variety of everyday applications. An emphasis has been placed on the bonds constructed during the formation of the imidazole. The utility of these methodologies based around the functional group compatibility of the process and resultant substitution patterns around the ring are described, including discussion of scope and limitations, reaction mechanisms and future challenges.
Introduction
Imidazoles are an important heterocyclic structural motif in functional molecules and are utilized in a diverse range of applications.1,2 Despite recent advances, the development of novel methods for the regiocontrolled synthesis of substituted imidazoles is of strategic importance. This is due to the preponderance of applications to which this important heterocycle is being deployed, such as the traditional applications in pharmaceuticals and agrochemicals3,4 to emerging research into dyes for solar cells and other optical applications,5–16 functional materials,17 and catalysis18 (Fig. 1). It is due to their versatility and utility in a number of these areas that expedient methods for the synthesis of imidazoles are both highly topical and necessary. This review is focused on recent advances in the synthesis of imidazoles and is organized via the sorts of bond disconnections that were employed in order to construct the heterocycle. The bonds formed in the reaction are highlighted by being red colored throughout the review and the standard numbering of imidazoles is used in the description of disconnections (Fig. 1).
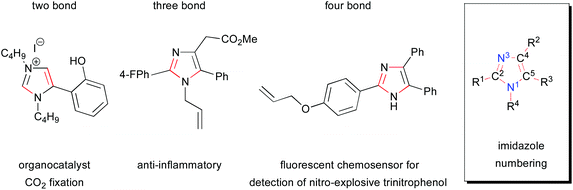 |
| | Fig. 1 Examples of two, three and four bond disconnections in the formation of functional imidazole as well as its generic core and substituent labels. | |
Synthesis
One-bond formed
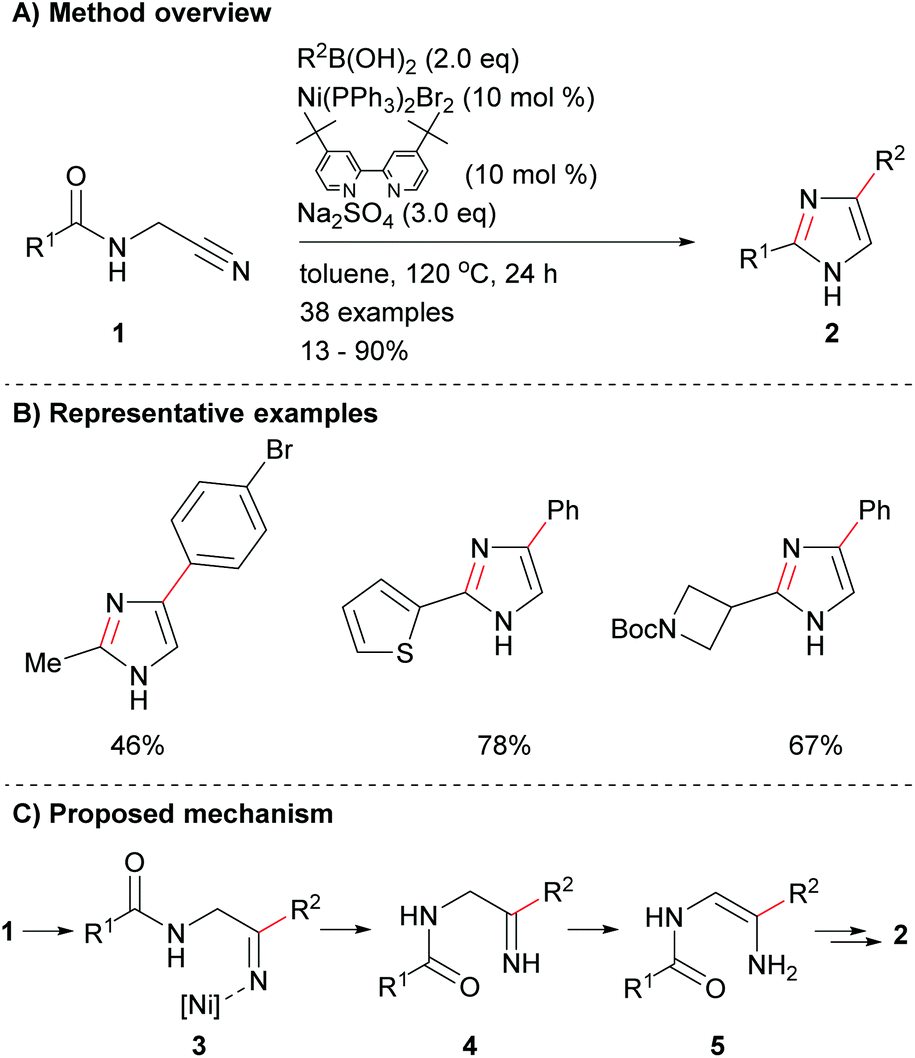
There has been limited recent research on the synthesis of imidazoles via methods that only form one of the heterocycle's core bonds. Fang et al. reported a novel protocol for the cyclization of amido-nitriles 1 to form disubstituted imidazoles 2 (Scheme 1a).19 The reaction conditions were mild enough for the inclusion of a variety of functional groups including, arylhalides as well as aromatic and saturated heterocycles (Scheme 1b). This reaction is reported to proceed via nickel-catalysed addition to nitrile 1, which followed by proto-demetallation, tautomerization and dehydrative cyclization afforded the desired 2,4-disubstituted NH-imidazoles 2 in poor to excellent yield depending on the coupling partners (Scheme 1c).
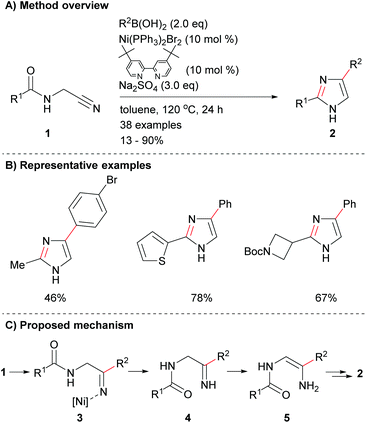 |
| | Scheme 1 Nickel-catalysed cyclization of amido-nitriles. | |
Two-bonds formed
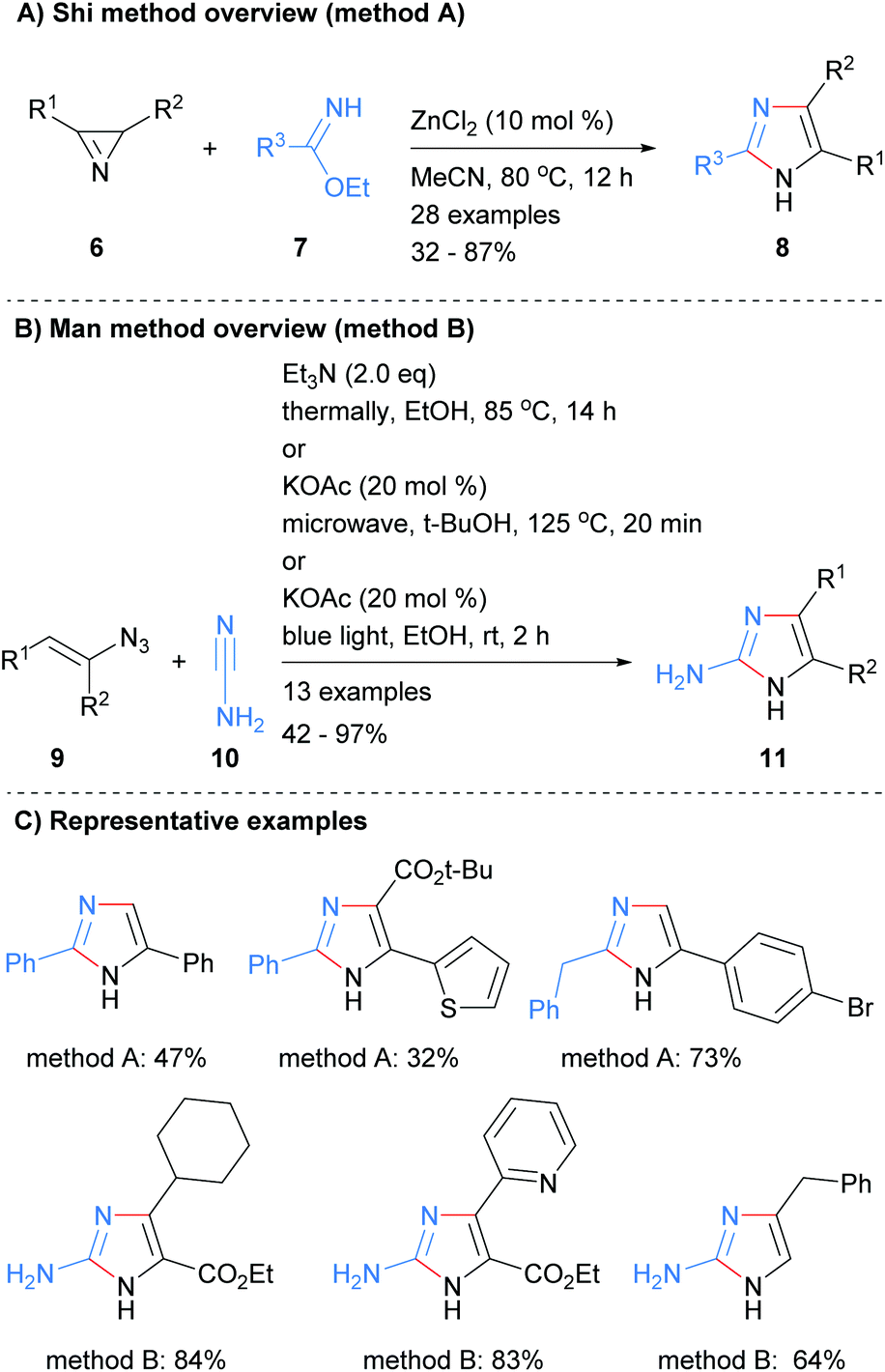
A two-bond disconnection for the synthesis of imidazoles that has been explored recently is to combine a C2–N3 fragment with a N1–C4–C5 unit. For example, Shi et al. used this disconnection to form trisubstituted NH-imidazoles 8 from the reaction of benzimidates 7 with 2H-azirines 6 in the presence of zinc(II) chloride (Scheme 2a).20 Man et al. used a related method for the synthesis of 2-aminoimidazoles 11 (Scheme 2b).21 Under a variety of condition, vinylazides 9 were converted in situ into 2H-azirines, which subsequently reacted with cyanamide (10) to form the desired 2-aminoimidazoles 11 in moderate to excellent yield. Both of these methods afford NH-imidazoles with control of substitution at the 2, 4 and 5 positions. An ester moiety could also be incorporated regioselective at either the C-4 or C-5 positions depending on which protocol was followed (Scheme 2c).
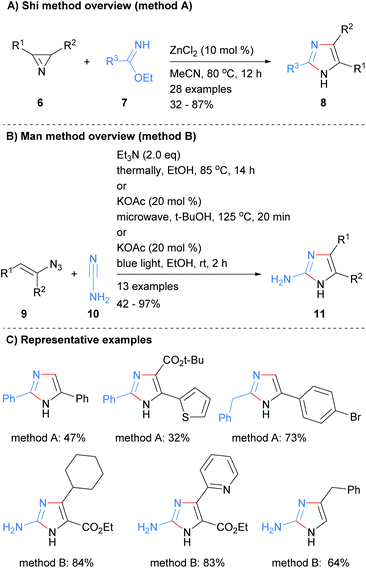 |
| | Scheme 2 Metal-catalysed 2H-azirines based protocols. | |
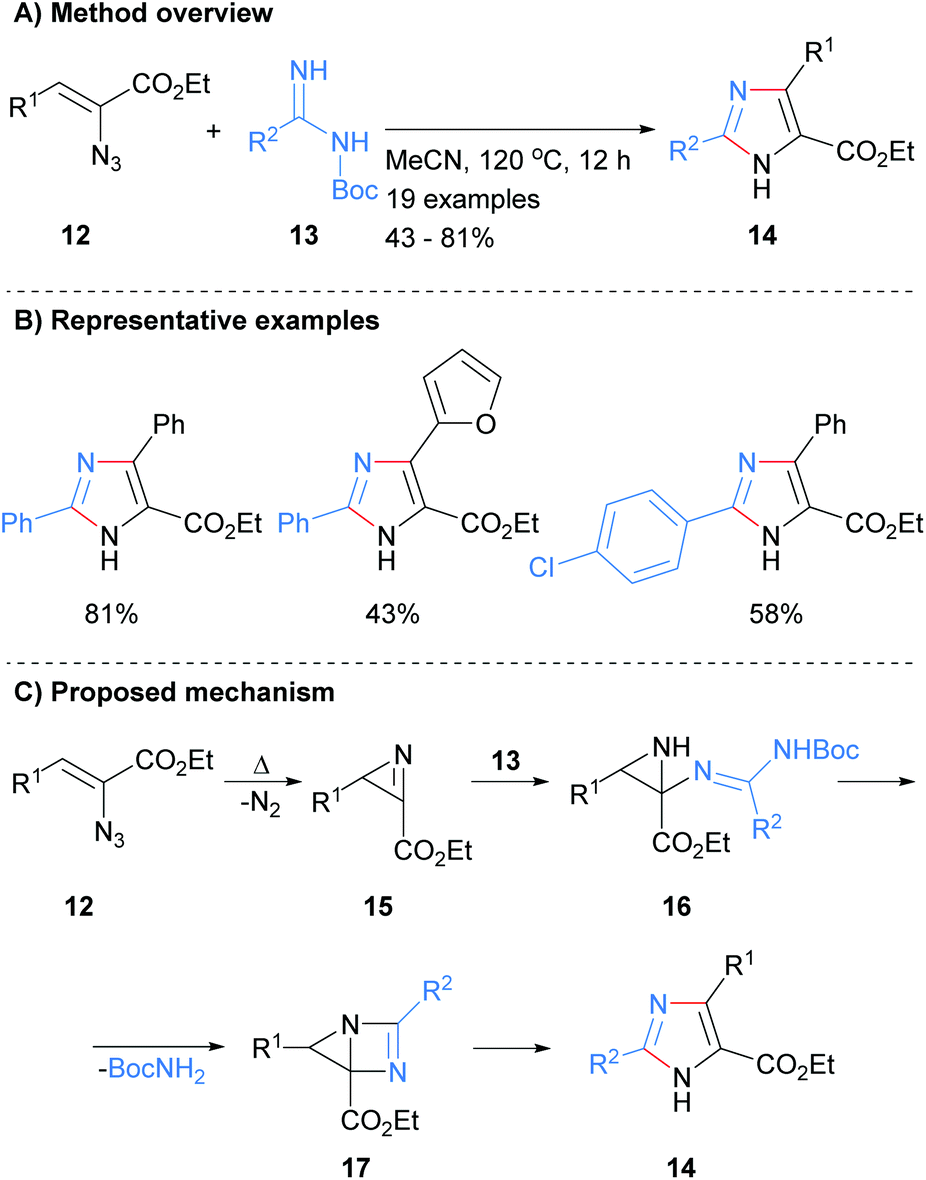
Related work by Tang et al. reported the synthesis of 2,4,5-trisubstited NH-imidazoles in moderate to good yield (Scheme 3).22 Thus, reaction of NBoc-imidamides 13 with α-azidoenones 12 at 120 °C in acetonitrile gave the desired imidazoles 14 without the need for the addition of a catalyst. The protocol allowed for the inclusion of aromatic and heteroaromatic substituents as well as the ester at the C-5 position. It was proposed that intramolecular cyclization with concomitant loss of nitrogen of azide 12 would afford 2H-azirines 15. Addition of NBoc-imidamides 13 to azirines 15 followed by intramolecular cyclization would afford bicyclic intermediate 17. Ring fragmentation would then lead to the desired imidazoles 14. In contrast to the methods outline in Scheme 2, this protocol require that the starting material contains an ester moiety.
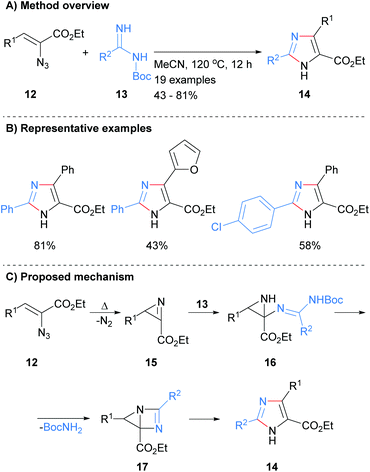 |
| | Scheme 3 Metal catalyst free 2H-azirines based protocol. | |
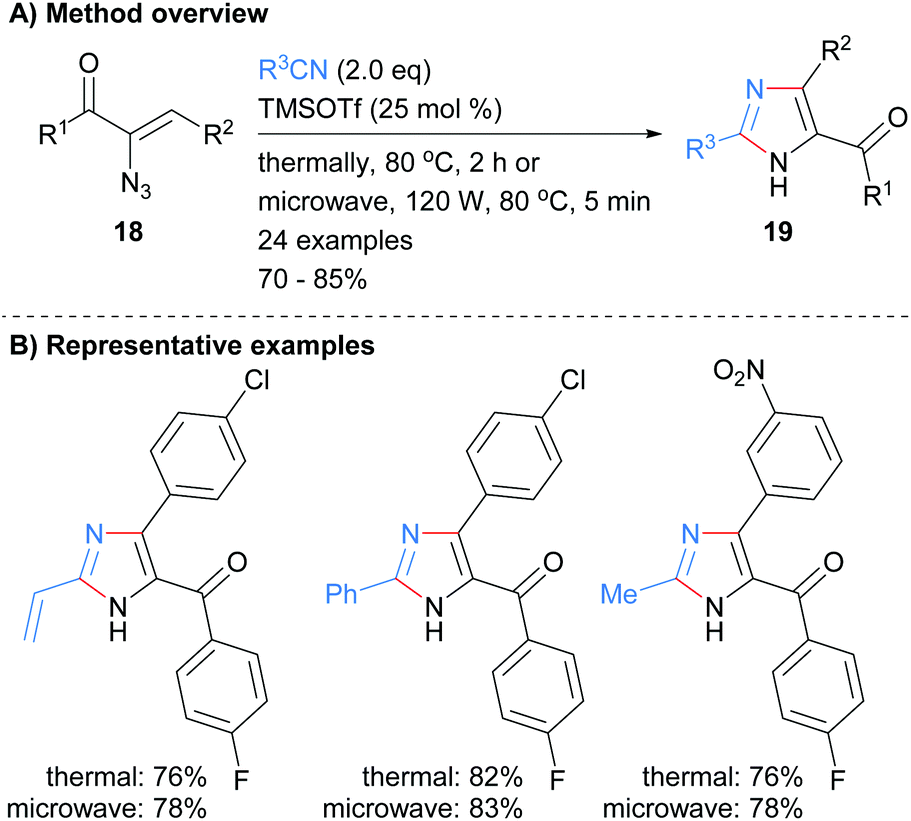
Nitriles have also been used as reagents in metal free reactions for the formation of substituted imidazoles in which two-bonds of the heterocycle are formed. For example Harisha et al. recently reported the reaction of α-azidoenones 18 with substituted nitriles to form tri-substituted NH-imidazoles 19 (Scheme 4).23 No significant difference was observed in isolated yield for reactions run under either thermal or microwave conditions. In addition to an arylketone moiety at the C-5 position, the protocol also allowed for the incorporation of vinyl and arylhalide functional groups.
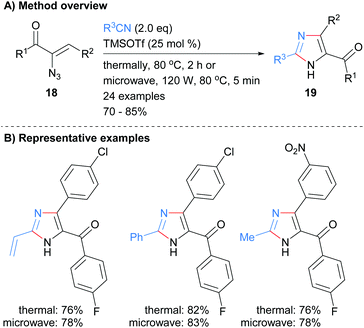 |
| | Scheme 4 TMSOTf-Catalysed [3 + 2] cycloaddition protocol. | |
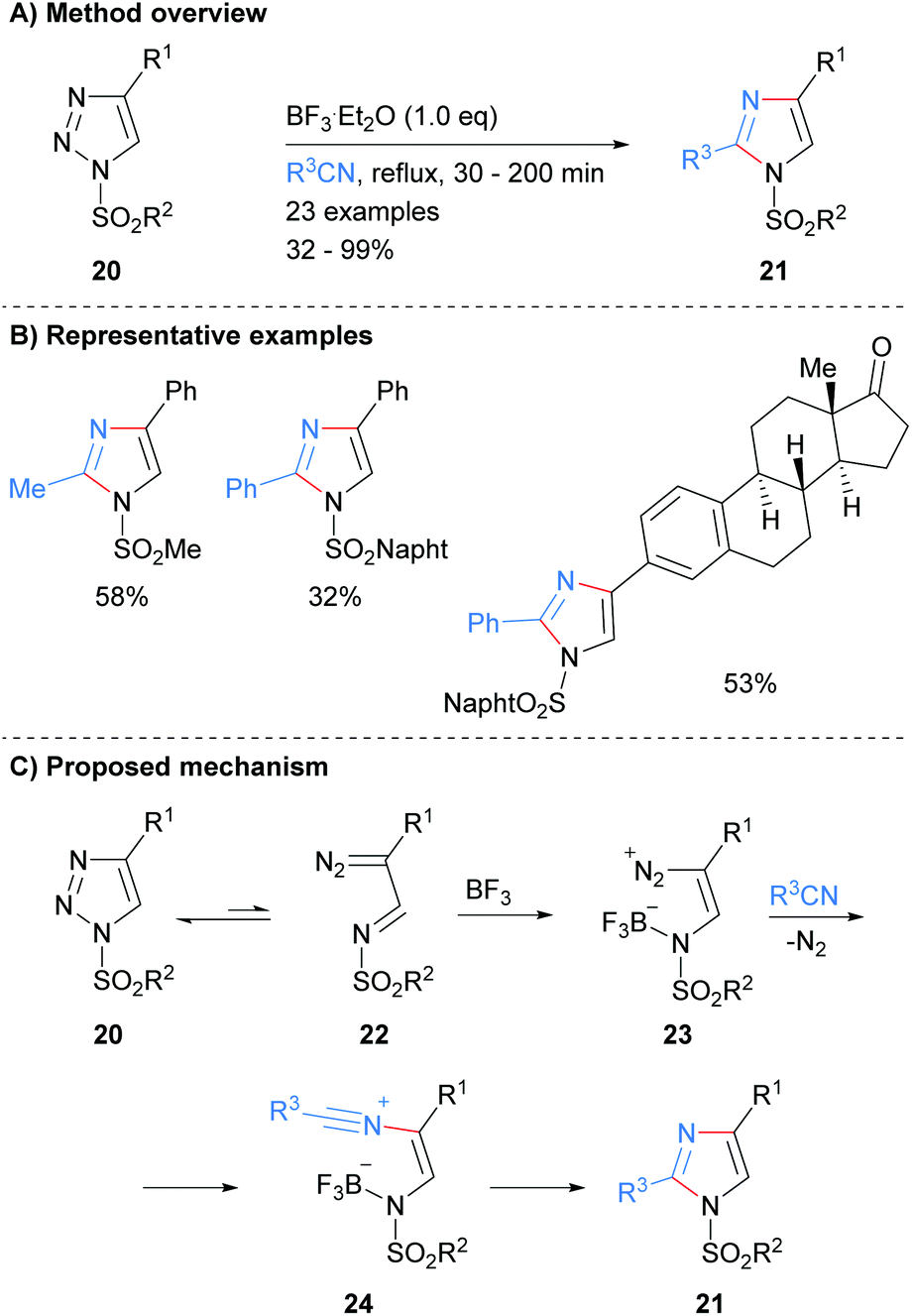
Recently, Yang and co-workers reported the synthesis of protected imidazoles 21via a BF3·Et2O promoted reaction of triazoles 20 and nitriles (Scheme 5).24 The products of the reaction are sulphone protected imidazoles that have substitution at the C-2 and C-4 positions. The reaction is proposed to proceed via initial ring opening of triazole 20 to form diazo-imine 22. BF3·Et2O promoted addition of the nitrile affords nitrilium intermediate 24, which subsequently cyclized to form substituted imidazoles 21 in average to excellent yield.
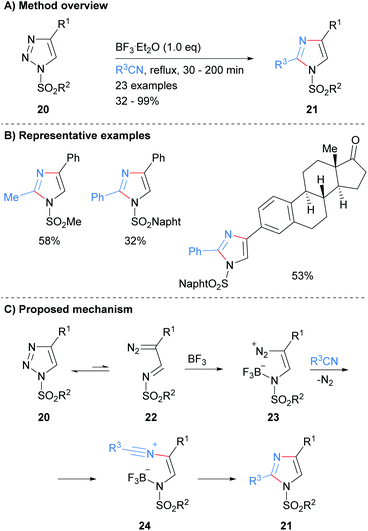 |
| | Scheme 5 BF3·Et2O-Catalysed recyclisation of triazoles. | |
In related work, Cai and co-workers showed that the addition of the anion derived from methylene isocyanides 26 to ketenimines 25 resulted in the formation of 1,4,5-trisubstitued imidazoles 27 (Scheme 6).25 Interestingly, this method allows for the synthesis of C-2 unsubstituted imidazoles whilst being functional group tolerant of esters, sulphones and arylhalides.
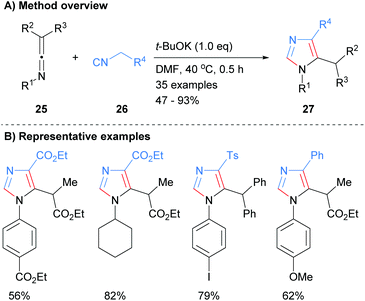 |
| | Scheme 6 Base-catalysed [3 + 2] cycloaddition protocol. | |
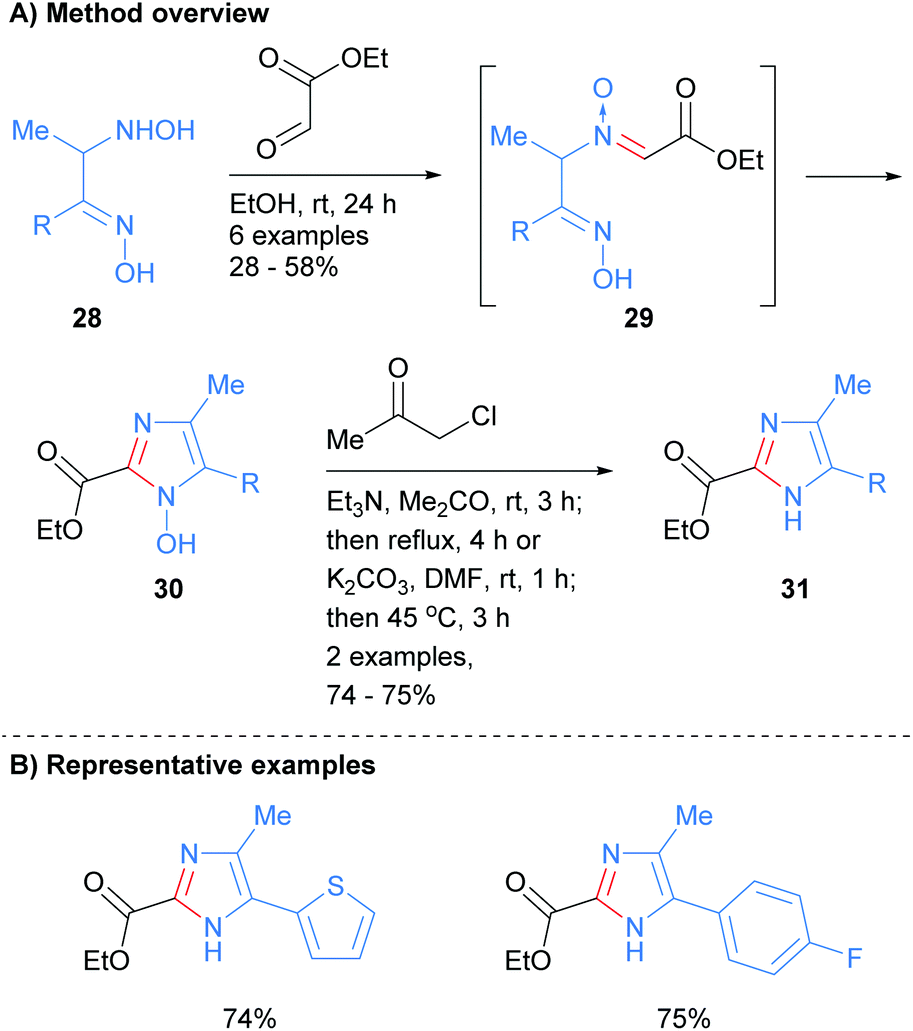
Recently, Nikolaenkova et al. reported the synthesis of 2-carboxylate substituted imidazoles from oxime-hydroxlyamines 28 (Scheme 7).26 The reaction is reported to proceed via initial condensation of hydroxylamine 28 with ethyl glyoxalate to form N-oxide 29in situ. Cyclization followed by dehydrative aromatization afforded 1-hydroxyimidazole 30, which was subsequently reacted with chloro-2-propanone to give NH-imidazole 31 in good yield. This method allowed for the synthesis of NH-imidazoles with an ester moiety at the C-2 position and was also tolerant of heterocycles and arylhalides.
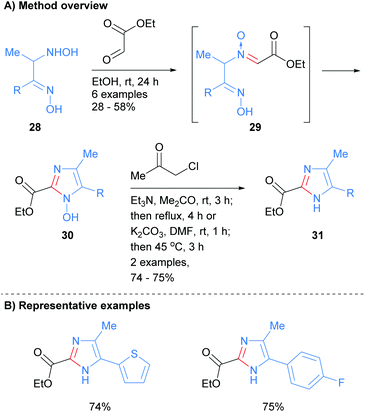 |
| | Scheme 7 Condensation of oxime-hydroxlyamine with ethyl glyoxalate. | |
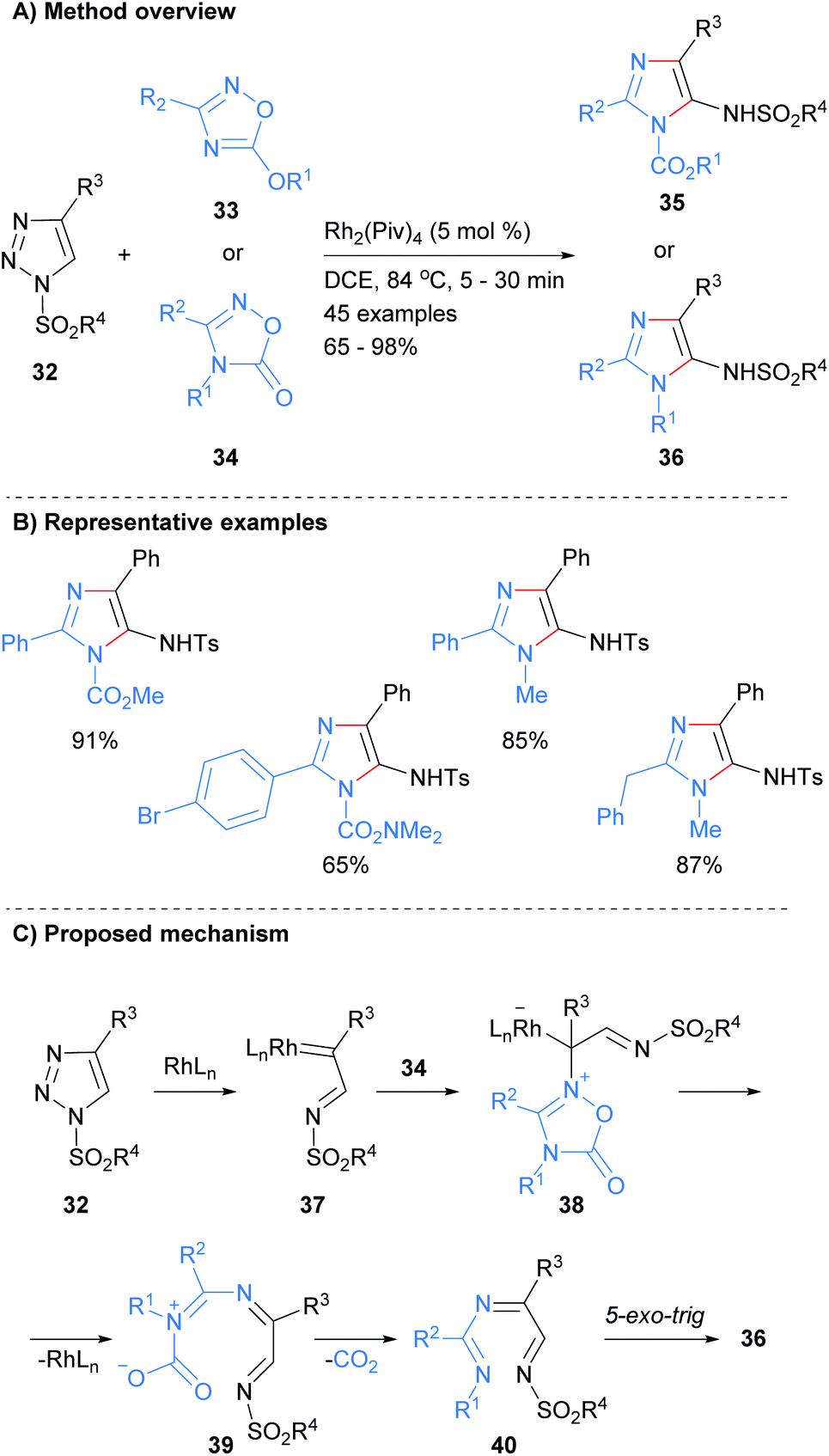
There has been a significant amount of recent works on the formation of both the N1–C5 and N3–C4 bonds in a single operation. For example, Strelnikova et al. reported the synthesis of 5-sulphonamidoimidazoles 35 or 36 from the reaction of two different heterocyclic starting materials in the presence of a rhodium catalyst (Scheme 8).27 Thus, reaction of 1,2,4-oxadiazole derivatives 33 and 34 with 1-sulphonyl-1,2,3-triazoles 32 at 84 °C in DCE afforded the desired imidazoles 35 and 36 in good to excellent yield. In addition to a suphonolated amine at the C-5 position, this protocol produced imidazoles substituted at the N-1 position with either an ester or alkyl moieties. The mechanism is proposed to proceed via insertion of the rhodium catalyst into triazole 32 to afford carbenoid 37. Attack of 1,2,4-oxadiazole derivatives 34 onto carbenoid 37 followed by ring-opening, loss of the metal catalyst and carbon dioxide afforded tri-imine 40. A 5-exo-trig cyclization of imine 40 then gave the desired imidazole 36.
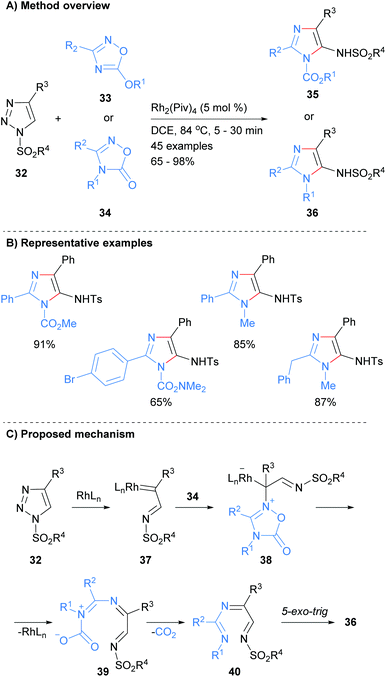 |
| | Scheme 8 Rhodium-catalysed transannulation of 1,2,4-oxadiazoles with 1-sulphonyl-1,2,3-triazoles. | |
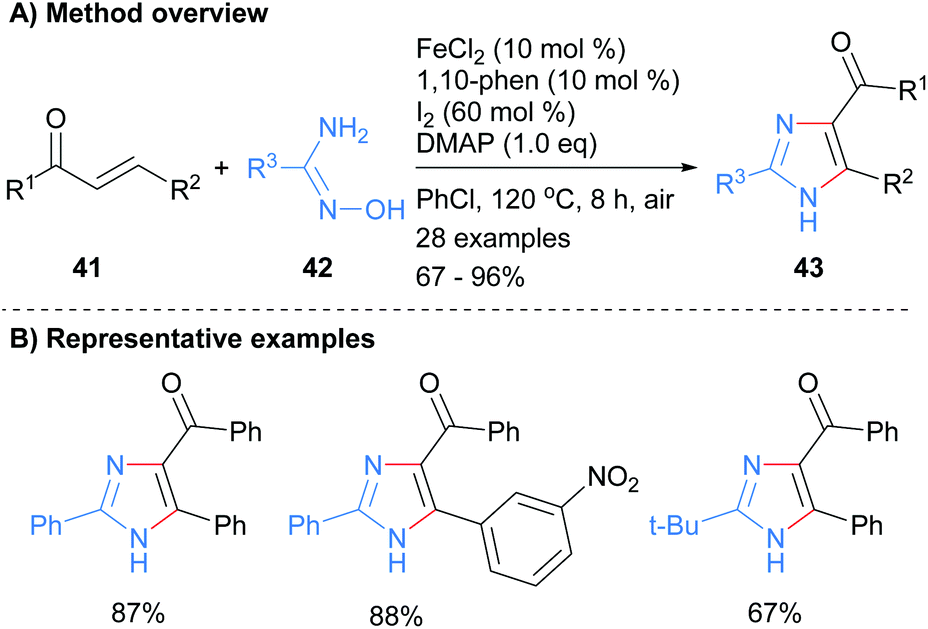
The 1,3-diamine component of an imidazole has also been introduced using either amidoximes or N-iminylsulphilimine. For example, Wu and co-workers used an iron catalyst to promote the reaction between amidoximes 42 with enones 41 (Scheme 9).28 Heating the reagents at 120 °C in the presence of the metal catalyst and iodine afforded imidazole 43 in good to excellent yields. The product NH-imidazoles were substituted with a ketone moiety at the C-4 position and the reaction protocol was tolerant to nitroarenes.
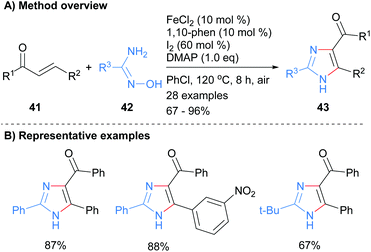 |
| | Scheme 9 Iron-catalysed [3 + 2] addition protocol. | |
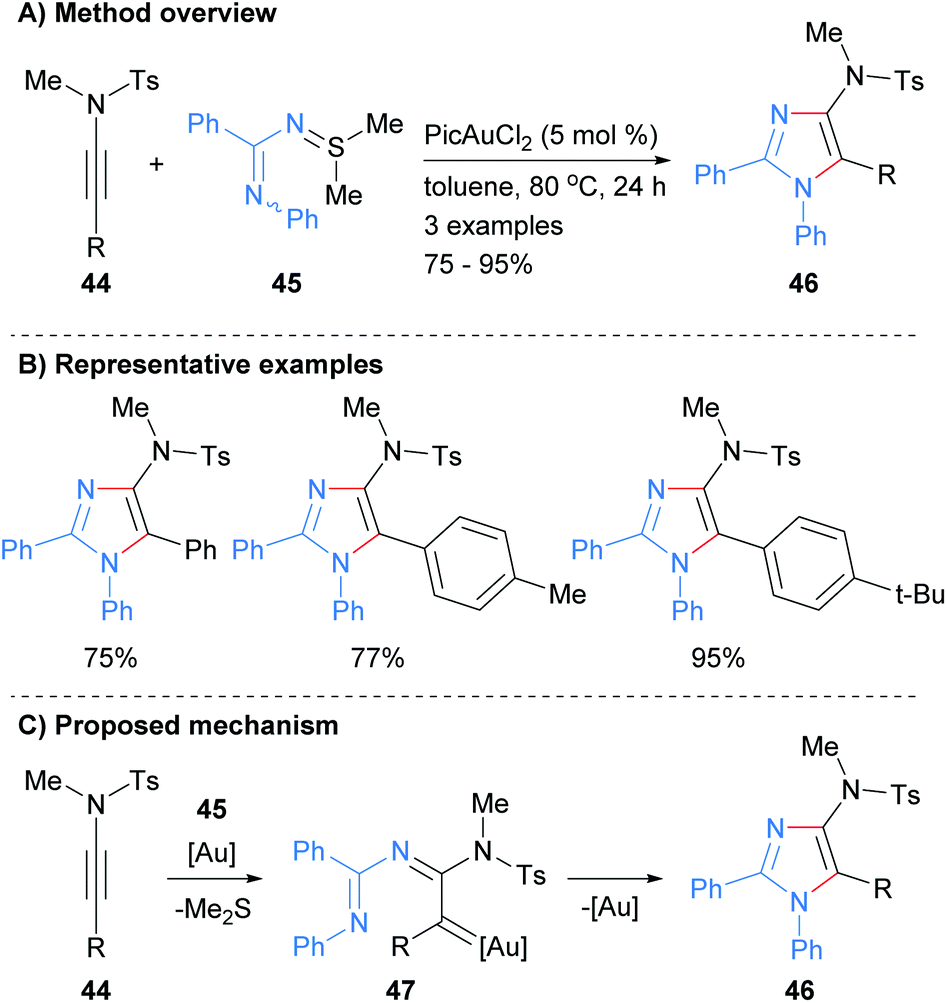
In a related protocol, the reaction of N-iminylsulphilimine 44 with ynamides 45 was recently reported to afford 4-amino substituted imidazoles 46 in good to excellent yields (Scheme 10).29 In this gold-catalyzed process the sulphilimines are used as nitrene transfer reagents. The products of the reaction are N-1 phenyl substituted imidazoles that have sulphonolated amines at the C-4 position as well as aryl substitution at the C-2 and C-5 positions.
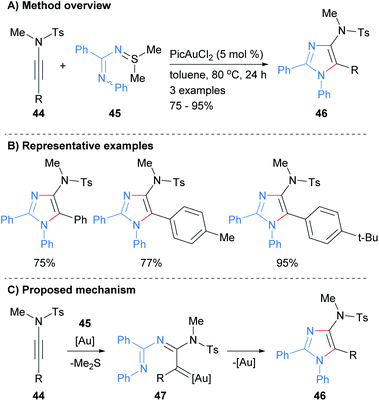 |
| | Scheme 10 Gold-catalysed [3 + 2] addition protocol. | |
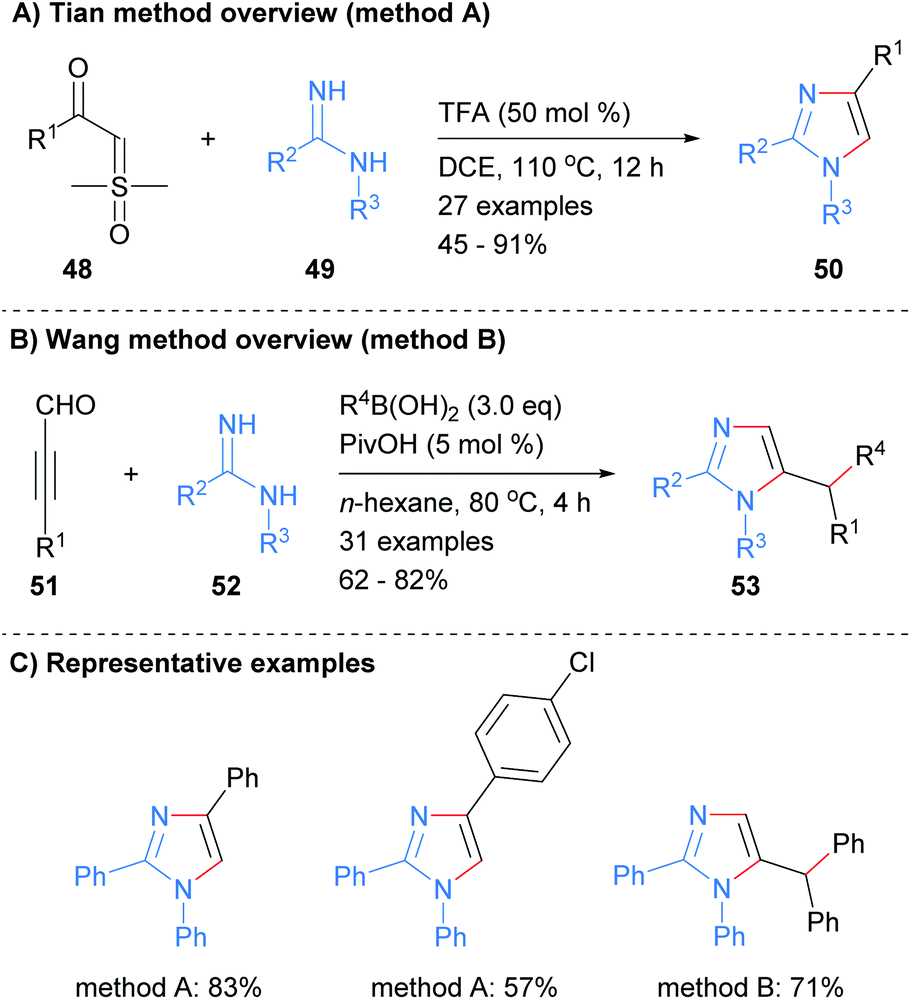
Imidamides can also be used as starting materials for the synthesis of imidazoles without the need for a metal catalyst. For example, Tian et al. reported the synthesis of substituted imidazole 50 from the reaction of imidamides 49 with sulphoxonium ylides 48 in the presence of trifluoroacetic acid (Scheme 11a).30 The resultant imidazoles were substituted at the 1, 2 and 4-positions. In contrast, Wang and co-workers reported that the reaction of imidamides 52 with propargyl aldehydes in the presence of a boronic acid afforded 1,2,5-substituted imidazoles in which the boronic acid has transferred its substituent to the C-5′ position (Scheme 11b).31
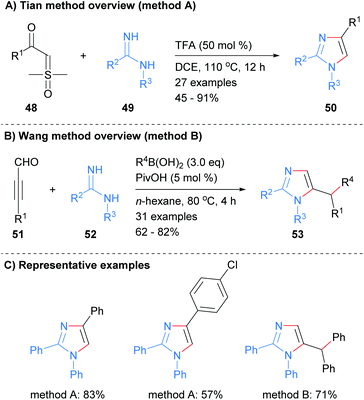 |
| | Scheme 11 Acid-catalysed [3 + 2] addition protocols. | |
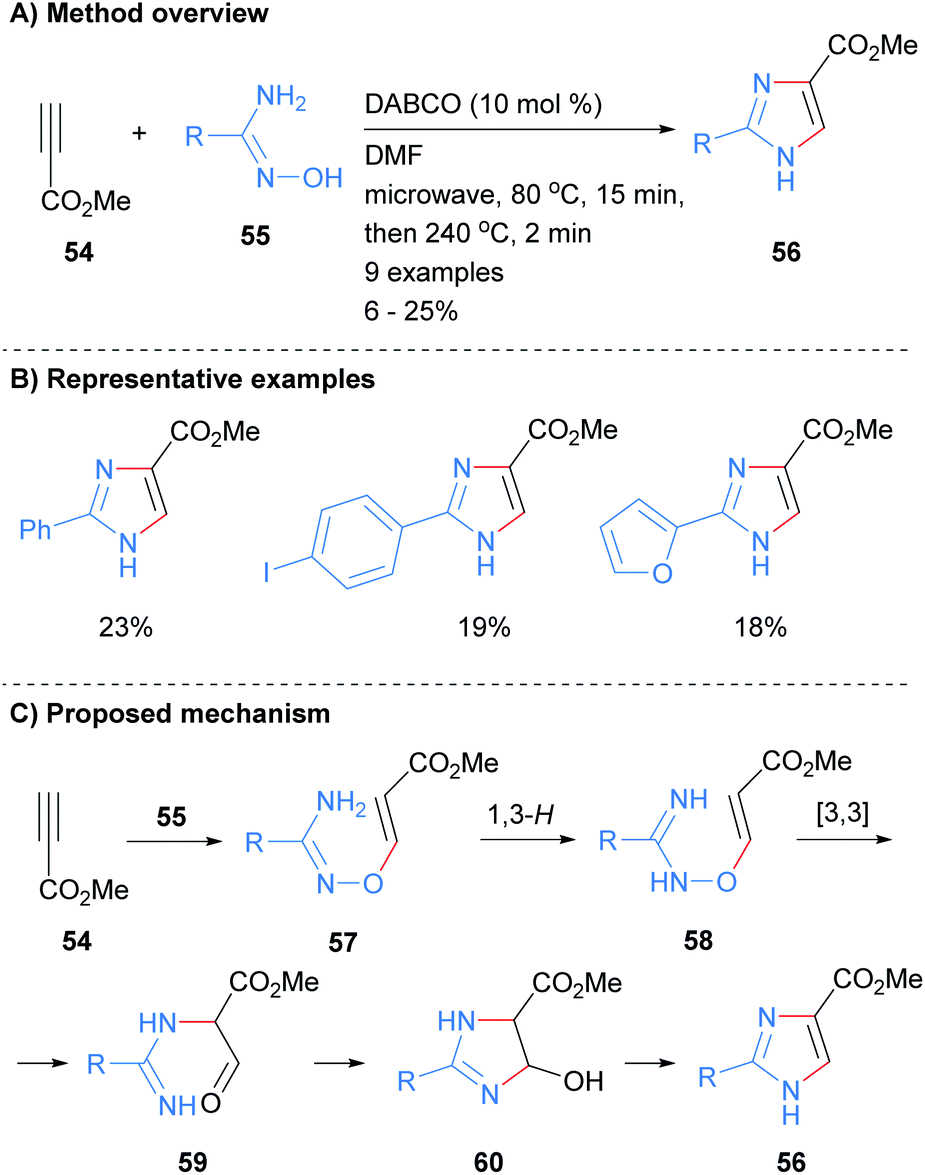
Recently we reported the synthesis of NH-imidazoles from the reaction of methyl propiolate (54) and substituted amidoximes 55 (Scheme 12).32 Thus, subjection of activated alkyne 54 with a variety of amidoximes 55 to a two stage microwave protocol in the presence of a catalytic amount of 1,4-diazabicyclo[2.2.2]octane (DABCO) rapidly afford the desired NH-imidazoles in moderate yield. The C-4 position was substituted by an ester moiety and the reaction conditions were also tolerant to arylhalides and heterocycles.
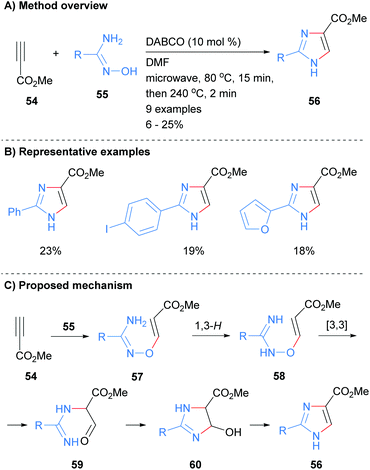 |
| | Scheme 12 Nucleophilic catalyst-based protocol. | |
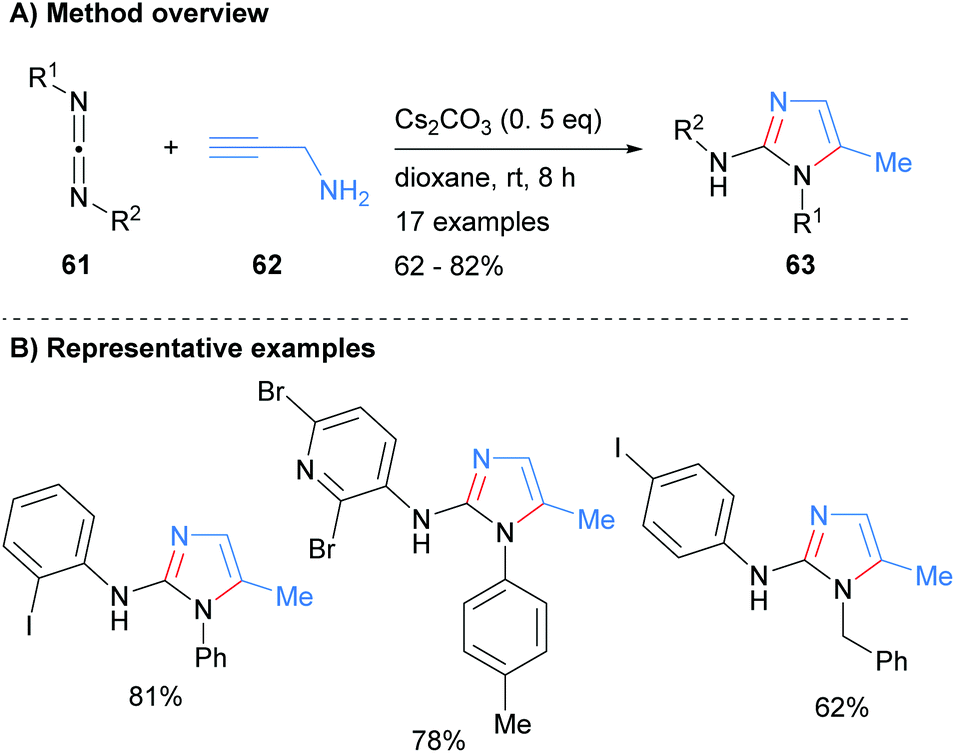
The N3–C4–C5 unit can also be introduced in the form of a carbodiimide. For example, Li et al. recently reported the synthesis of 2-aminoimidazoles 63 from the reaction of carbodiimides 61 with propargyl amine (62) in moderate to good isolated yield (Scheme 13).33 The reaction conditions were tolerant of both arylhalides and heteroarylhalides. In addition, the N-1 nitrogen could be substituted by either aryl or benzyl groups depending on the starting material that was employed.
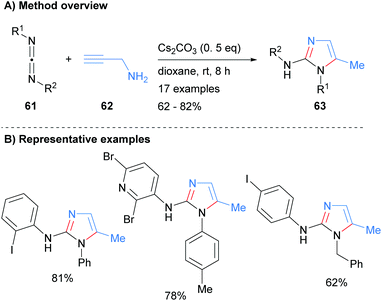 |
| | Scheme 13 Base-catalysed addition of propargylamines to carbodiimides. | |
Three-bonds formed
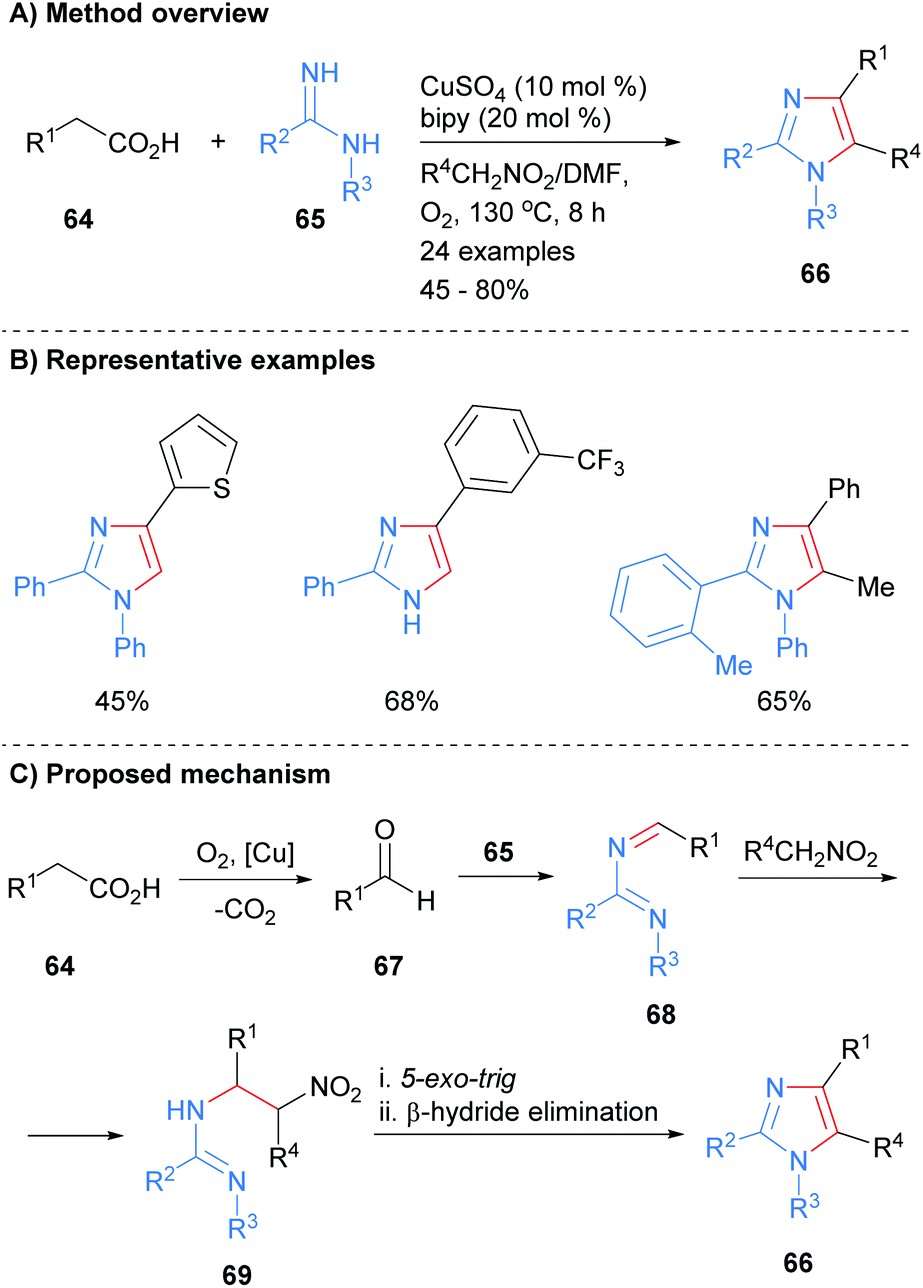
A number of recent methods for the synthesis of imidazoles in which three of the heterocycle bonds are formed have been reported. For example, imidamides 65 were reacted with carboxylic acids 64 in the presence of a copper catalyst to form imidazoles 66 (Scheme 14).34 In addition to substrate control over the substitution at the N-1 position (H or aryl), the product imidazoles were regioselectively substituted at the C-2 and C-4 positions. In addition, functionality could be introduced at the C-5 position via inclusion of a nitroalkane in the reaction mixture. Mechanistically, the reaction is believed to proceed via oxidative decarboxylation of carboxylic acid 64 to afford aldehyde 67. Condensation of imidamide 65 with aldehyde 67 followed by addition of the anion derived from the nitroalkane to the in situ formed imine 68 gave substituted imidamide 69. A 5-exo-trig cyclization of imidamide 69 followed by β-hydride elimination afforded imidazole 66.
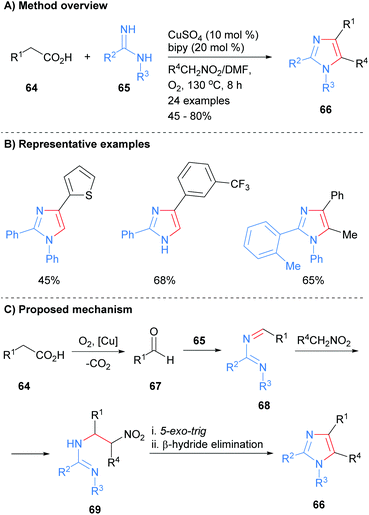 |
| | Scheme 14 Copper-catalysed decarboxylative coupling of carboxylic acids, imidamides and nitroalkanes. | |
The reaction of excess amine with enones has been shown to afford trisubstitued imidazoles. For examples, Salfeena et al. recently reported the copper-catalyzed reaction of amines 71 with enones 70 gave imidazoles 72 with substitution at the 1, 2 and 4 positions in poor to moderate yield (Scheme 15).35 The resultant imidazoles contained a benzyl group at the N-1 position and the reaction conditions were tolerant of aromatic and heteroaromatic functional groups. Mechanistically the reaction was reported to proceed via copper promoted imine and iodonium formation on enone 70 to give iodonium 73. Addition of a second equivalent of amine 71 followed by intramolecular cyclization afforded iminium salt 75. Finally, addition of a third equivalent of amine 71 to alkyl iodide 75 followed by aromatization via fragmentation gave the desired imidazole 72.
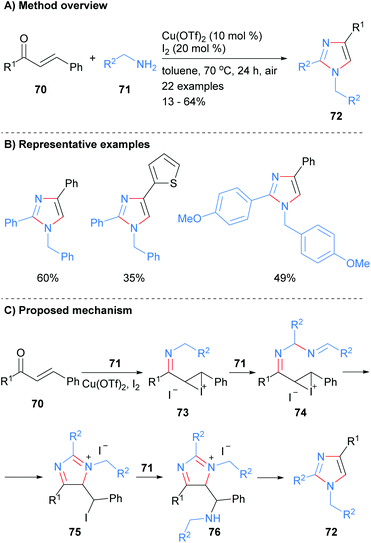 |
| | Scheme 15 Copper-catalysed enones C–C bond cleavage protocol. | |
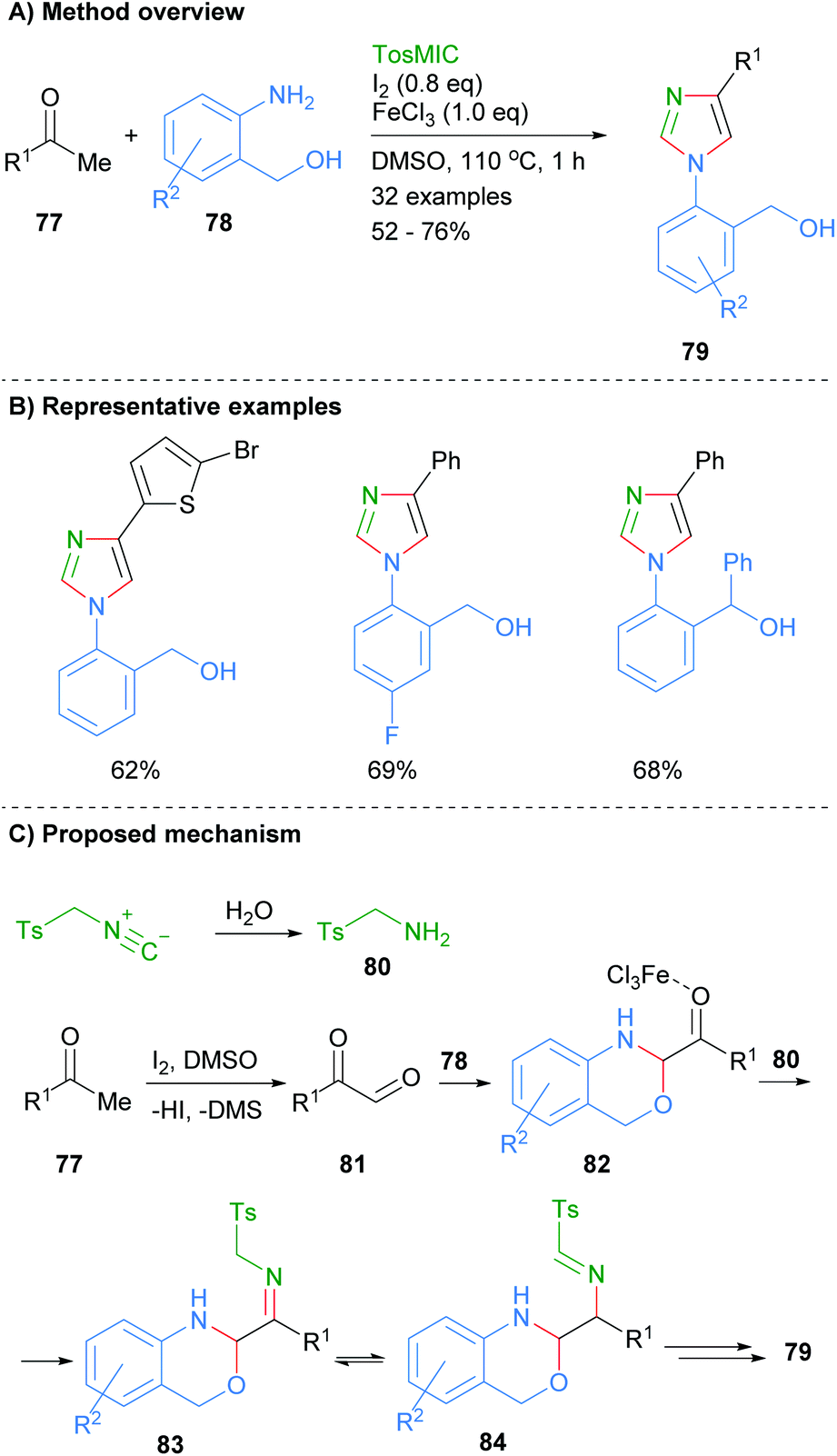
In related work, Geng et al. found that ketones 77 and 2-aminobenzylalcohols 78 could be reacted together in the presence of iodine, ferric chloride and toluenesulphonylmethyl isocyanide to afford 1,4-disubstitued imidazoles 79 (Scheme 16).36 Interestingly, this is one of the few recent examples in which the product imidazole is regioselectively substituted at only the N-1 and C-4 positions with an aromatic group. The reaction is reported to proceed via initial oxidation of ketone 77 to keto-aldehyde 81. Subsequent condensation of aldehyde 81 with 2-aminobenzylalcohol 78 and reaction with the in situ formed tosylamine (80), afforded imine 83. Intramolecular cyclization of amine 83 followed by elimination of TsH and aromatization via C–O bond cleavage gave 1,4-disubstitued imidazole 79.
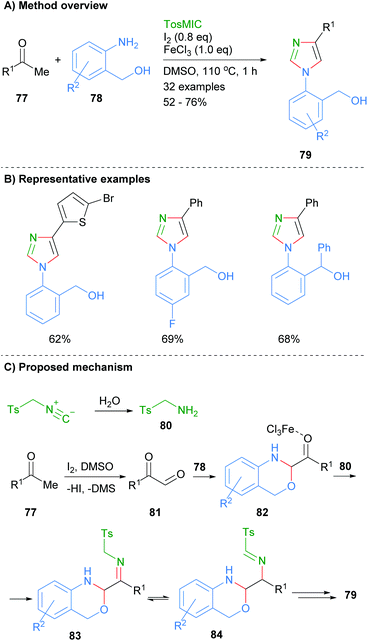 |
| | Scheme 16 Ferric chloride/iodine-catalysed [2 + 2 + 1] addition protocol. | |
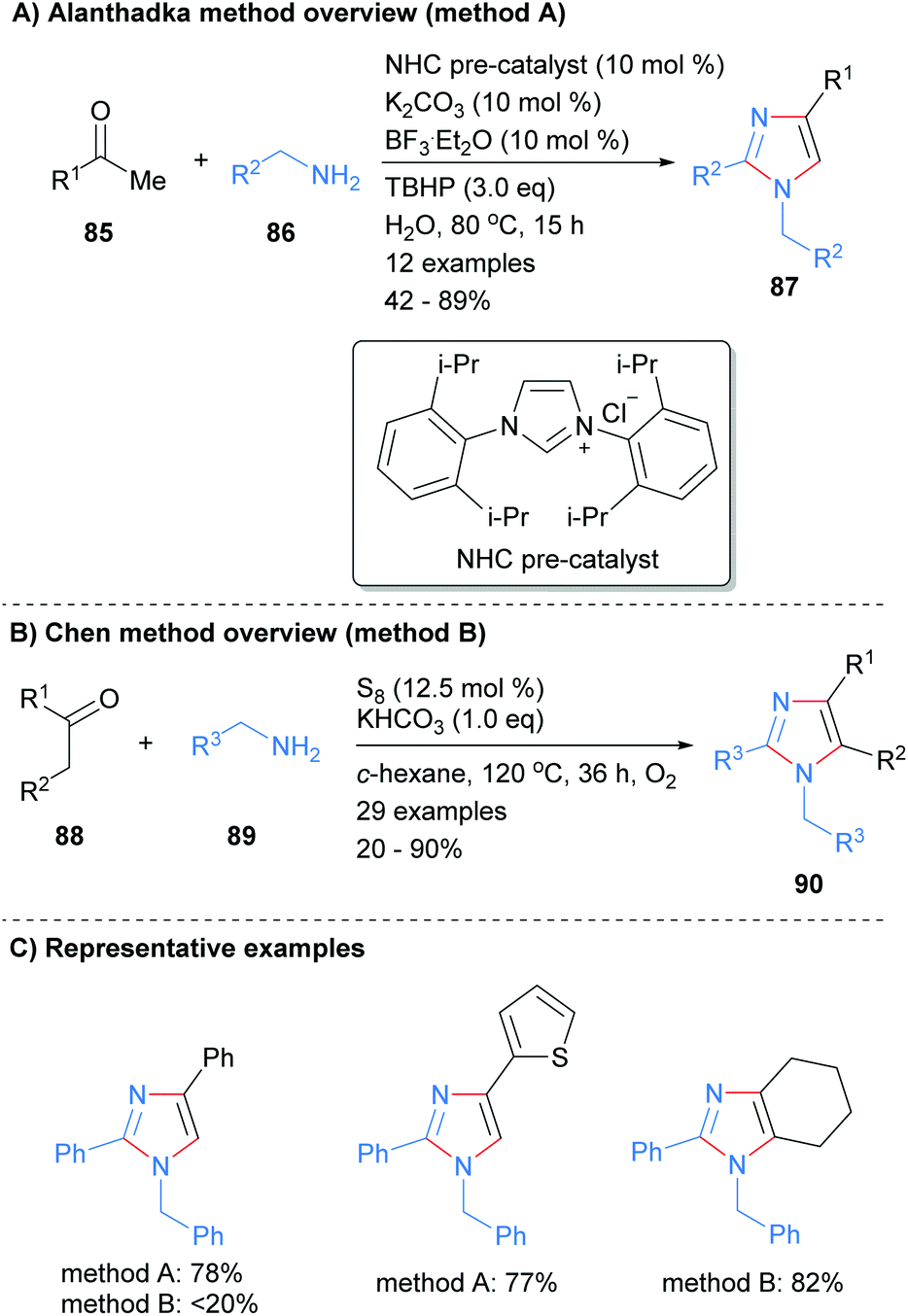
The use of N-heterocyclic carbenes (NHC) as both ligand and organocatalysts have become commonplace in modern organic synthesis since they were characterized by Arduengo et al.37 Alanthadka and co-workers recently built on this extensive research to develop a NHC-catalyzed protocol for the synthesis of 1,2,4-trisubstituted imidazoles 87 from the reaction of acetophenones 85 and benzylic amines 86 (Scheme 17a).38 The reaction was conducted in the presence of an excess of an oxidant, tert-butylhydroperoxide (TBHP), which serves to oxidize an in situ formed enamine to an α-aminoaldehyde. Importantly, this process can be run under solvent free conditions. In related work, Chen et al. reported the synthesis of tetrasubstituted imidazoles 90 from the reaction of ketones and amines in the presence of elemental sulphur (Scheme 17b).39 Both methods reported the synthesis of the N-1 benzyl, C-2/C-4 phenyl substituted imidazoles, though the isolated yields were markedly different.
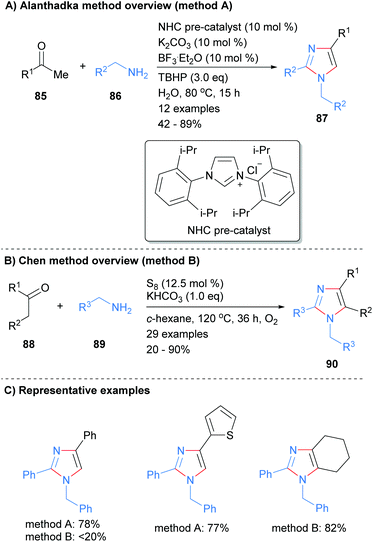 |
| | Scheme 17 Metal catalyst free ketones/benzylamines coupling protocols. | |
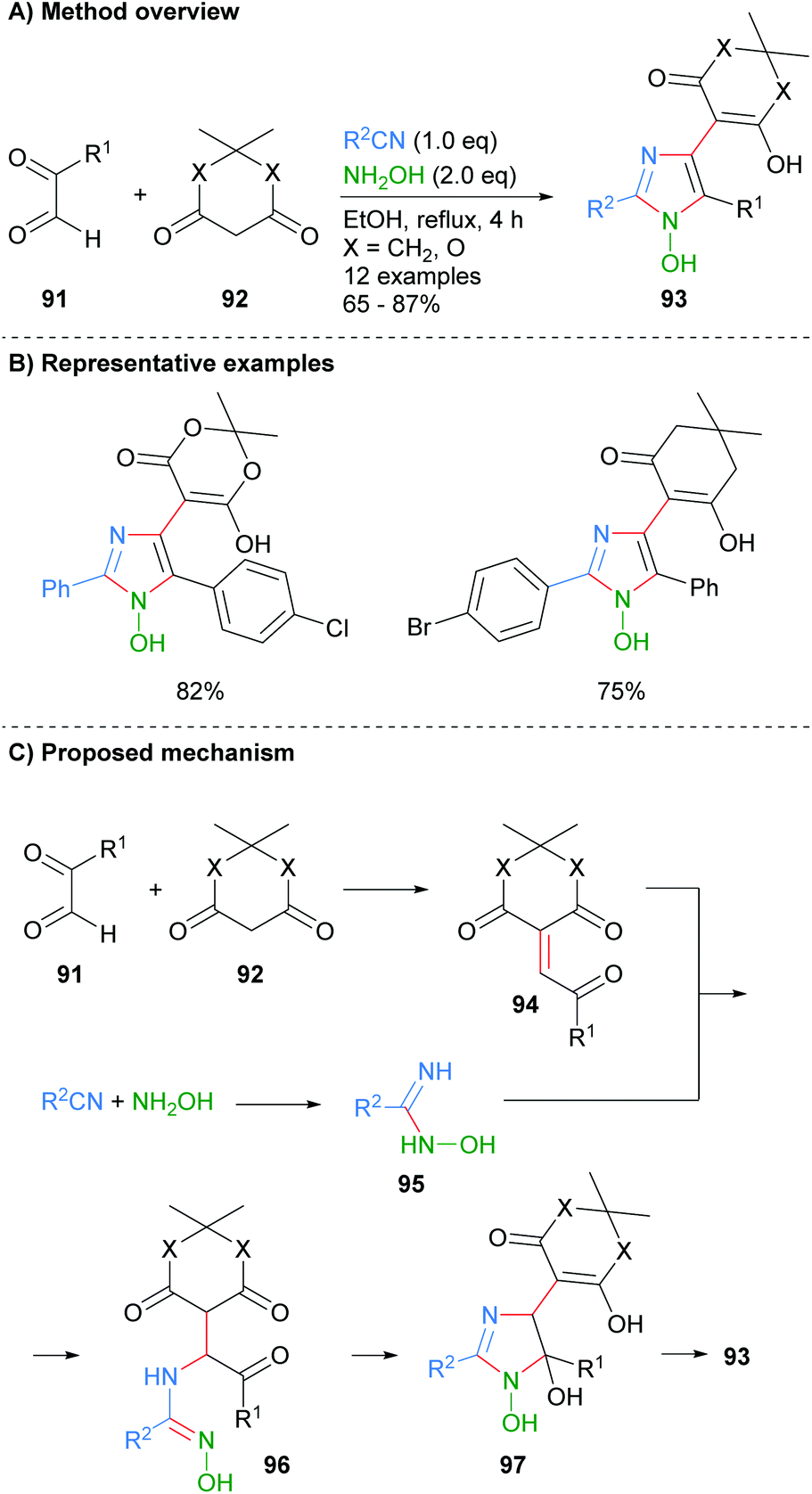
Alizadeh-Bami et al. reported a similar disconnection for the synthesis of substituted 1-hydroxyimidazole 93 (Scheme 18).40 This protocol allowed for the introduction of an additional carbonyl nucleophile, derived from 92, which resulted in additional substitution at the C-4 position. The product imidazoles were substituted with a hydroxide moiety at the N-1 position (cf.Scheme 7 for the conversion of this functional group to an NH-imidazole). Mechanistically the reaction is reported to proceed via an initial condensation reaction between aldehyde 91 and dicarbonyl 92 to afford enone 94. Nucleophilic addition of the in situ formed N-hydroxy-imidamide 95 to enone 94 gave α-aminoketone 96, which after a cyclodehydration process afforded 1-hydroxyimidazole 93.
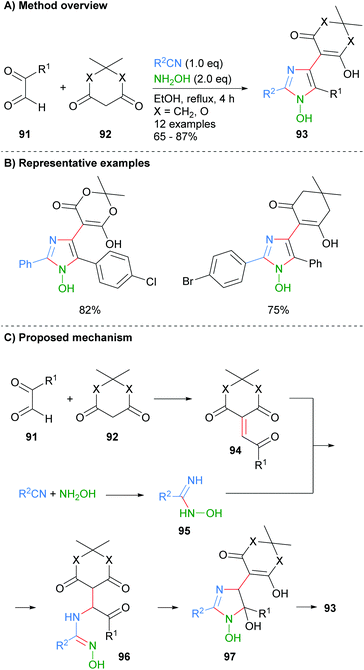 |
| | Scheme 18 Metal catalyst free four-component protocol. | |
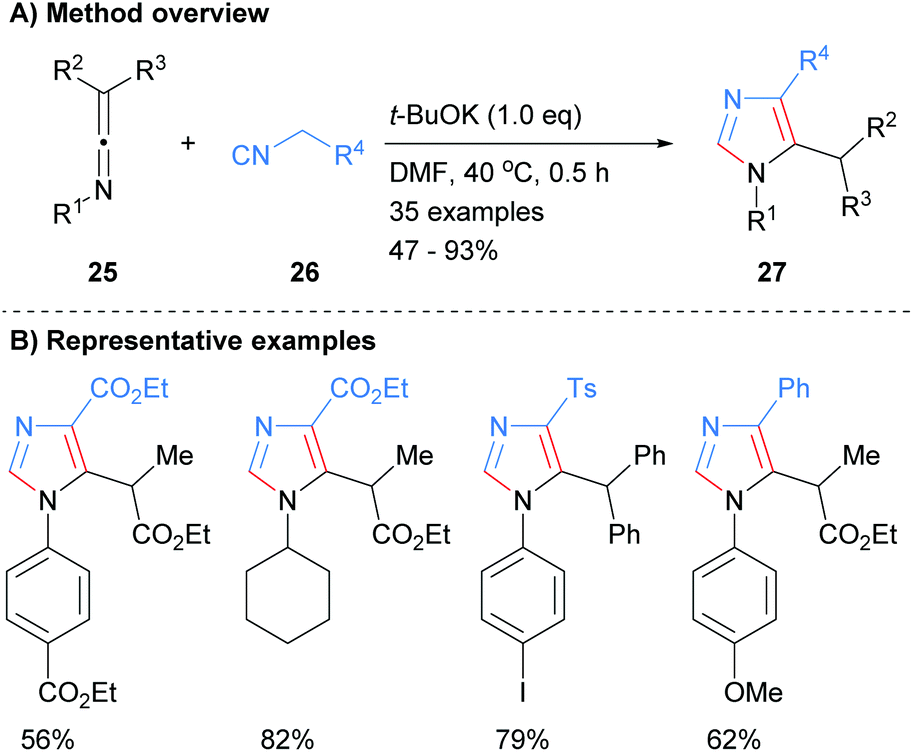
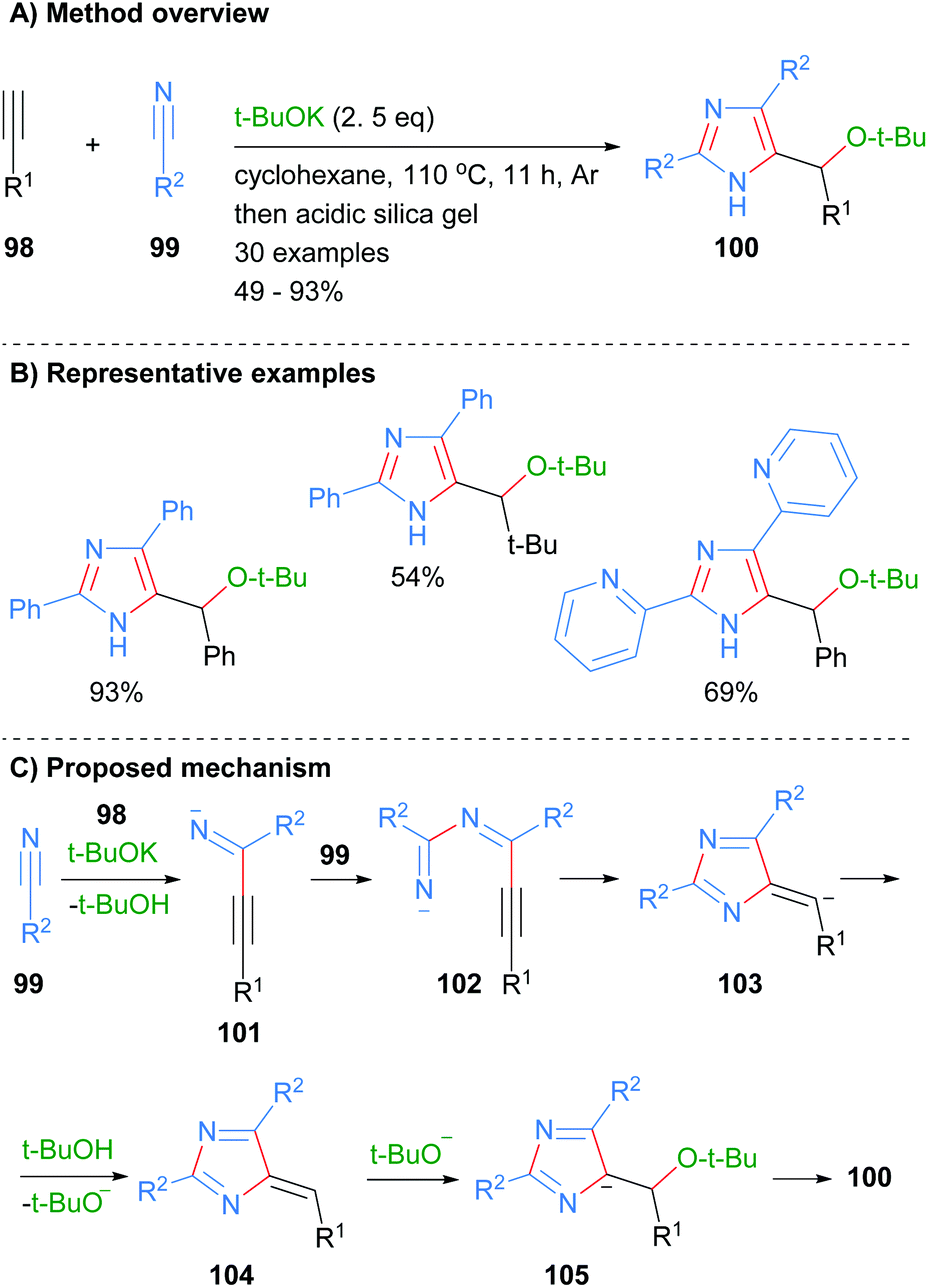
Wang and co-workers also constructed imidazoles 100 using the less common three component disconnection of the heterocycle (Scheme 19).41 Two equivalents of nitrile 99 were reacted with the acetylide generated in situ from alkynes 98 to afford the desired substituted imidazoles in average to excellent yield. The t-BuOK acts as both a base and a nucleophile under the reaction conditions. In addition to the alkyl substituent at the C-5 position, reaction protocol afforded NH-imidazoles with aryl or heteroaryl substituents. The proposed mechanism for the reaction starts with the reaction of in situ formed acetylide of alkyne 98 with first equivalent of nitrile 99 to form imine 101. Addition of the second equivalent of nitrile 99 to imine 101 followed by 5-exo-dig cyclization and protonation gave heterocycle 104. Addition of tert-butoxide to exocyclic methylene 104 followed by aromatization and protonation when purifying on silica afforded imidazoles 100.
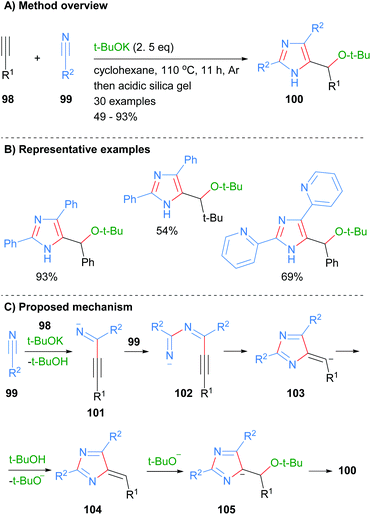 |
| | Scheme 19 Base-catalysed [2 + 2 + 2] addition protocol. | |
Four-bonds formed
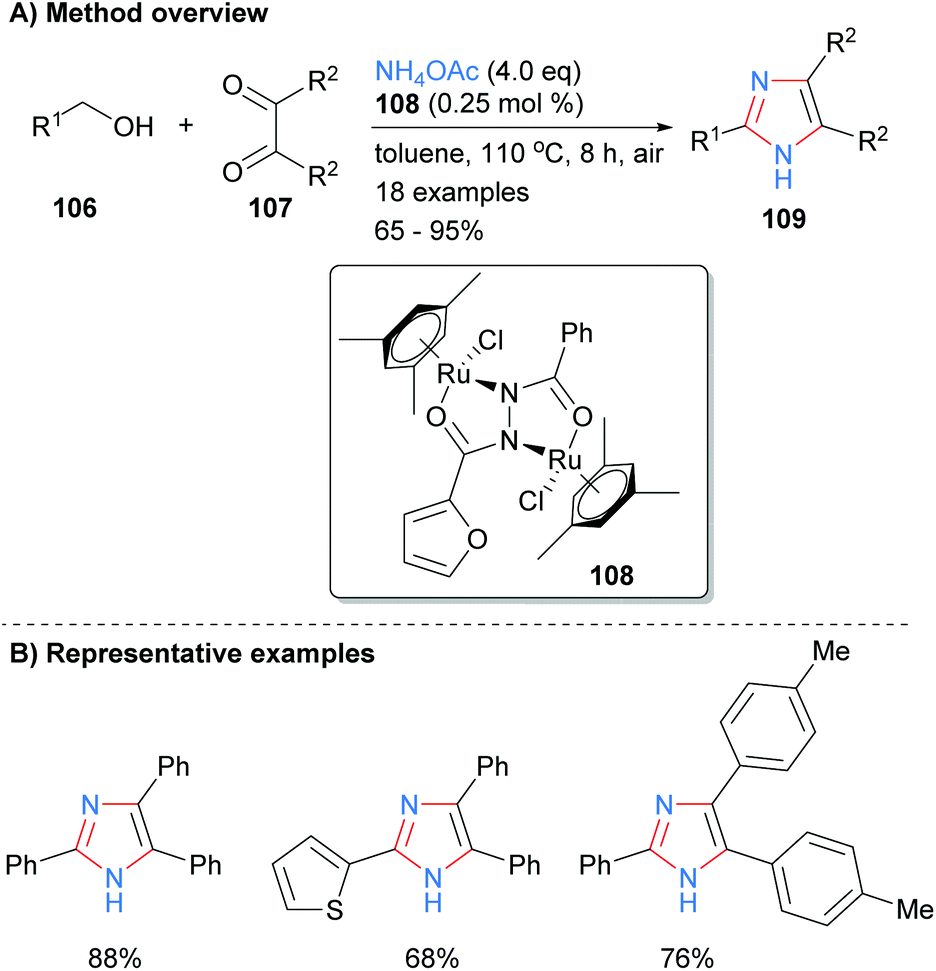
One of the most popular disconnections for the synthesis of imidazoles is the concurrent formation of four bonds of the heterocycles core with both metal-catalyzed and metal-free processes being reported recently. For example, Sundar and Rengan synthesized imidazoles 109 from the three component reaction between benzylic alcohol 106, 1,2-diketone 107 and excess ammonium acetate (Scheme 20).42 This borrowing hydrogen process was catalyzed by diruthenium(II) catalyst 108 under aerobic conditions. The method allows for the synthesis of NH-imidazoles with regioselective substitution at the C-2, C-4 and C-5 positions and is tolerant of aryl and heteroaryl functional groups.
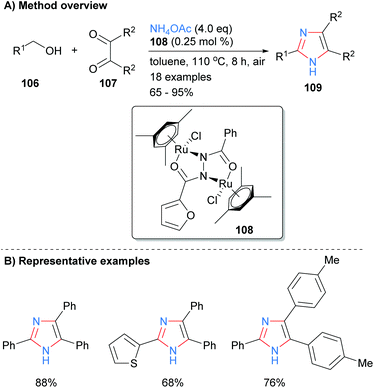 |
| | Scheme 20 Ruthenium-catalysed borrowing hydrogen protocol. | |
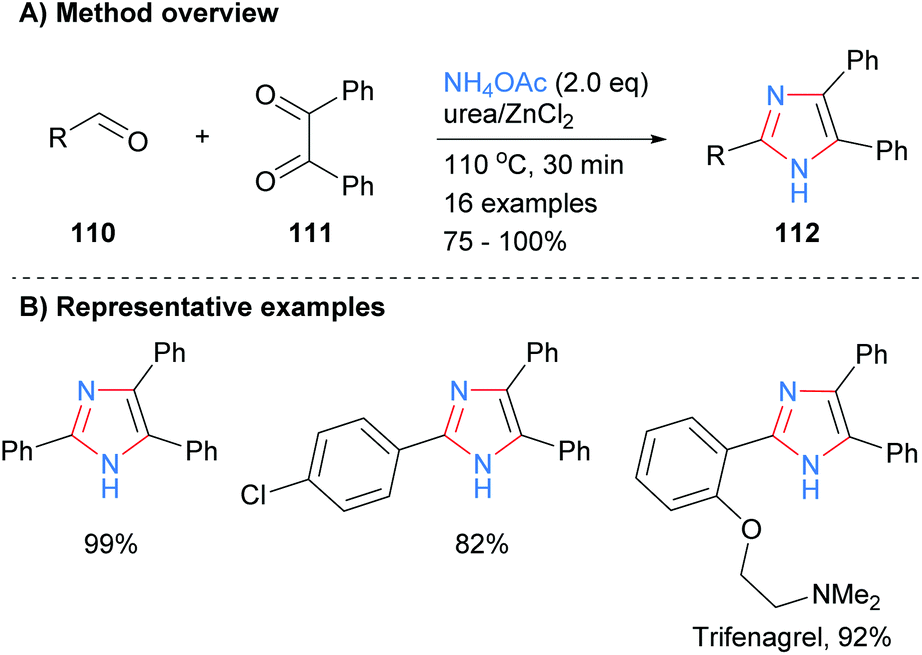
Related work by Higuera et al. demonstrated that the use of the deep eutectic solvent (DES) urea/zinc(II) dichloride could catalyze the synthesis of 4,5-diphenyl-2-substituted imidazoles 112 from the reaction of aldehydes 110 with benzyl (111) and excess ammonium acetate (Scheme 21).43 This methodology was applied to the synthesis of the drug trifenagrel, which was isolated in 92% yield. In addition to the arylether and alkylamine functionalities found in trifenagrel, the method also tolerated arylhalides as the aromatic aldehyde partner.
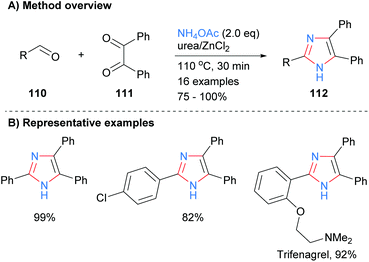 |
| | Scheme 21 Urea/zinc(II) dichloride-catalysed aldehyde/diketone/ammonia coupling protocol. | |
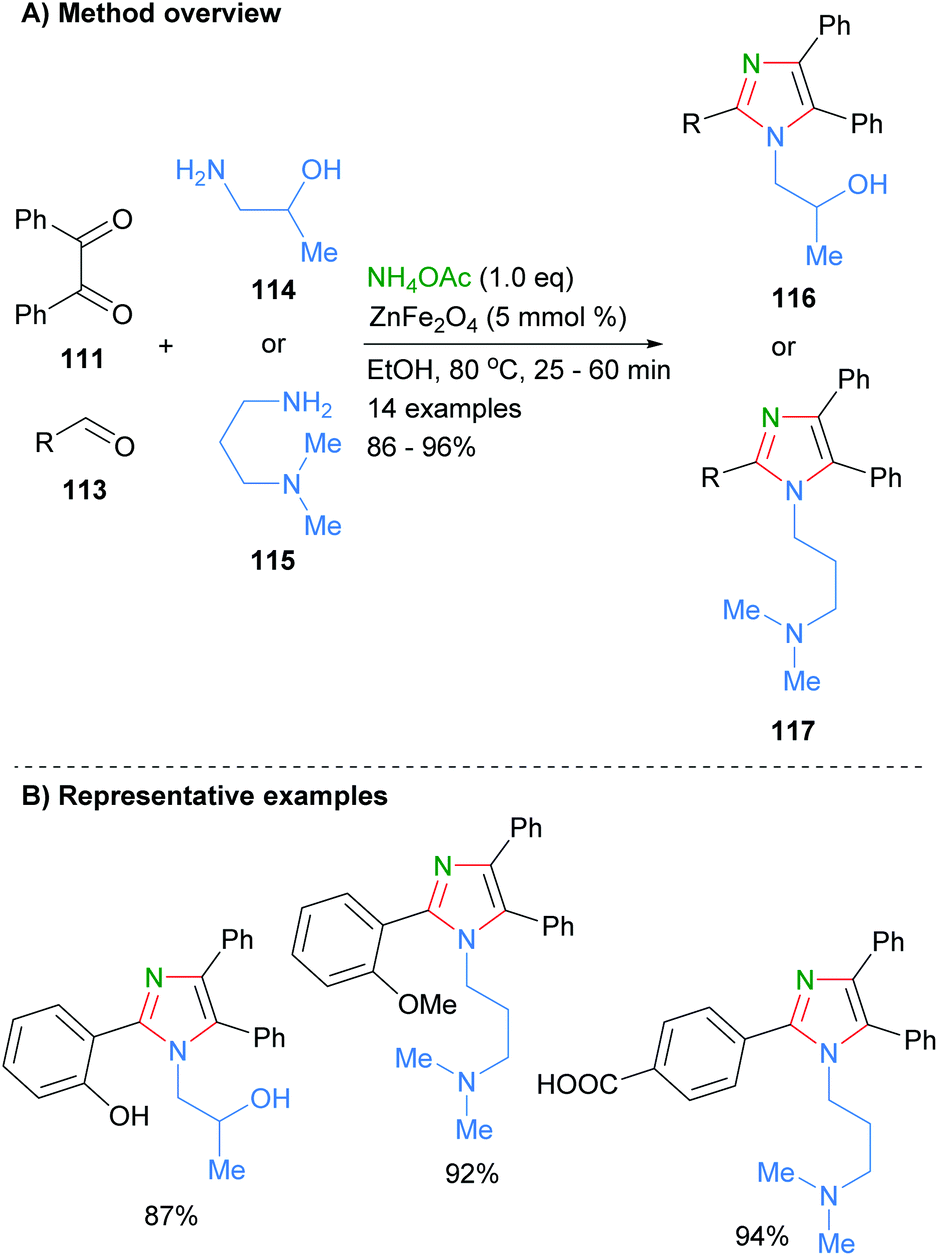
Marzouk et al. recently reported the preparation of ZnFe2O4 nanoparticles as well as their use as catalysts for the synthesis of substituted imidazoles (Scheme 22).44 Reaction of aldehyde 113 and benzil (111) and primary amines in the presence of the heterogenous catalyst afford the desired tetra-substituted imidazoles 116 and 117 in good isolated yields. The authors proposed that the iron acts as a Lewis acid to activate the carbonyl towards nucleophilic addition of amines and imines. Importantly, the catalyst can be easily isolated and reused up to four times without a significant decrease in product yield. A wide range of functional groups are tolerated under the reaction conditions including phenols, alcohols, carboxylic acids and amines.
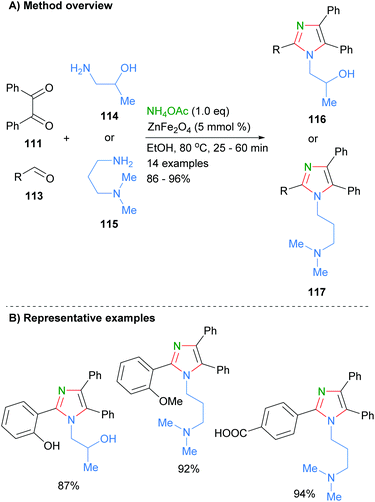 |
| | Scheme 22 ZnFe2O4-catalysed aldehyde/diketone/amine coupling protocol. | |
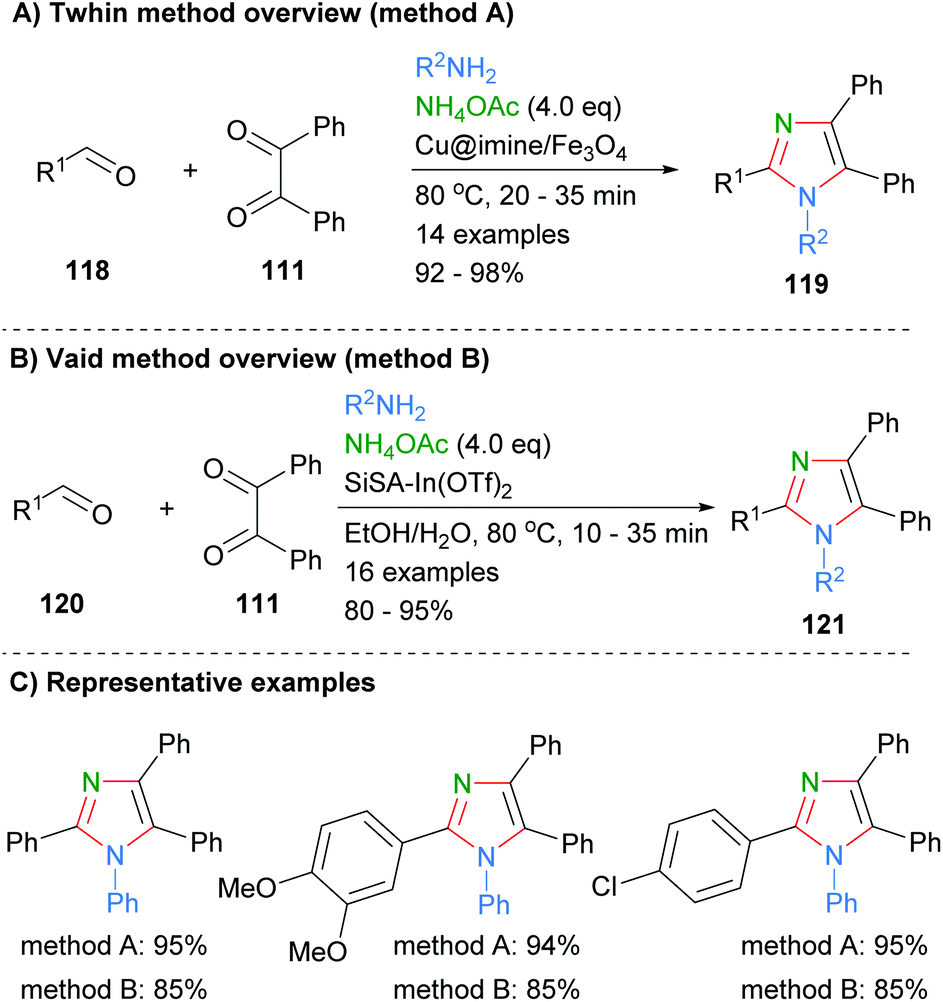
Similar approaches were recently reported by Thwin et al. who used a copper catalyst to facilitate formation of the imidazoles 119 (Scheme 23a).45 In addition, a heterogeneous indium catalyst supported on SiSA was used by Vaid and co-workers to promote the formation of imidazoles 121via the same disconnection in good to excellent yields (Scheme 23b).46 Both of these methods had a similar substrate scope and resulted in the formation of tetrasubstituted imidazoles. On average the use of the cheaper metal, copper, gave approximately a 10% increase in yield when the same substrates were directly compared (Scheme 23c).
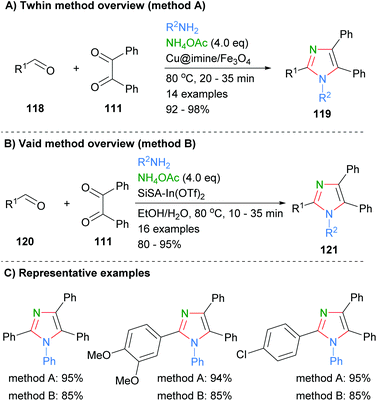 |
| | Scheme 23 Other examples of aldehyde/diketone/amine coupling protocols. | |
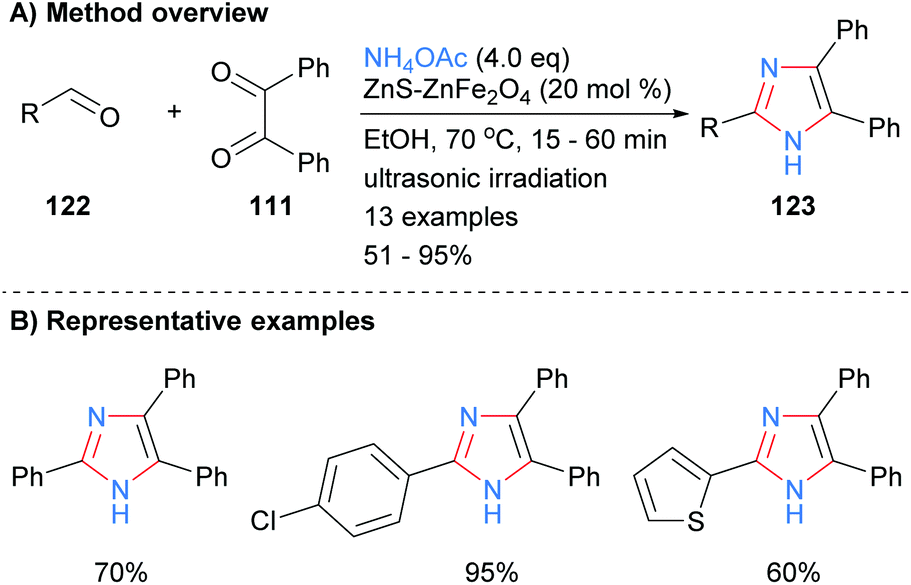
A similar disconnection was undertaken by Varzi and Maleki in which a ZnS-ZnFe2O4 nanocatalyst was used to synthesize 2,4,5-trisubsttituted-NH-imidazoles (Scheme 24).47 In contrast to the related heterogeneous catalysis methods outlined above (cf.Schemes 22 and 23), the resultant imidazoles were unsubstituted at the N-1 position. Aryl and heteroaryl aldehydes were well tolerated under the reaction conditions.
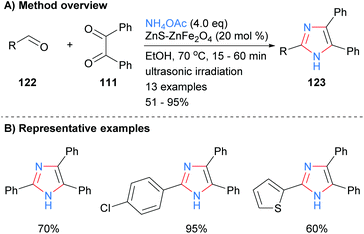 |
| | Scheme 24 ZnS-ZnFe2O4-catalysed aldehyde/diketone/ammonia coupling protocol. | |
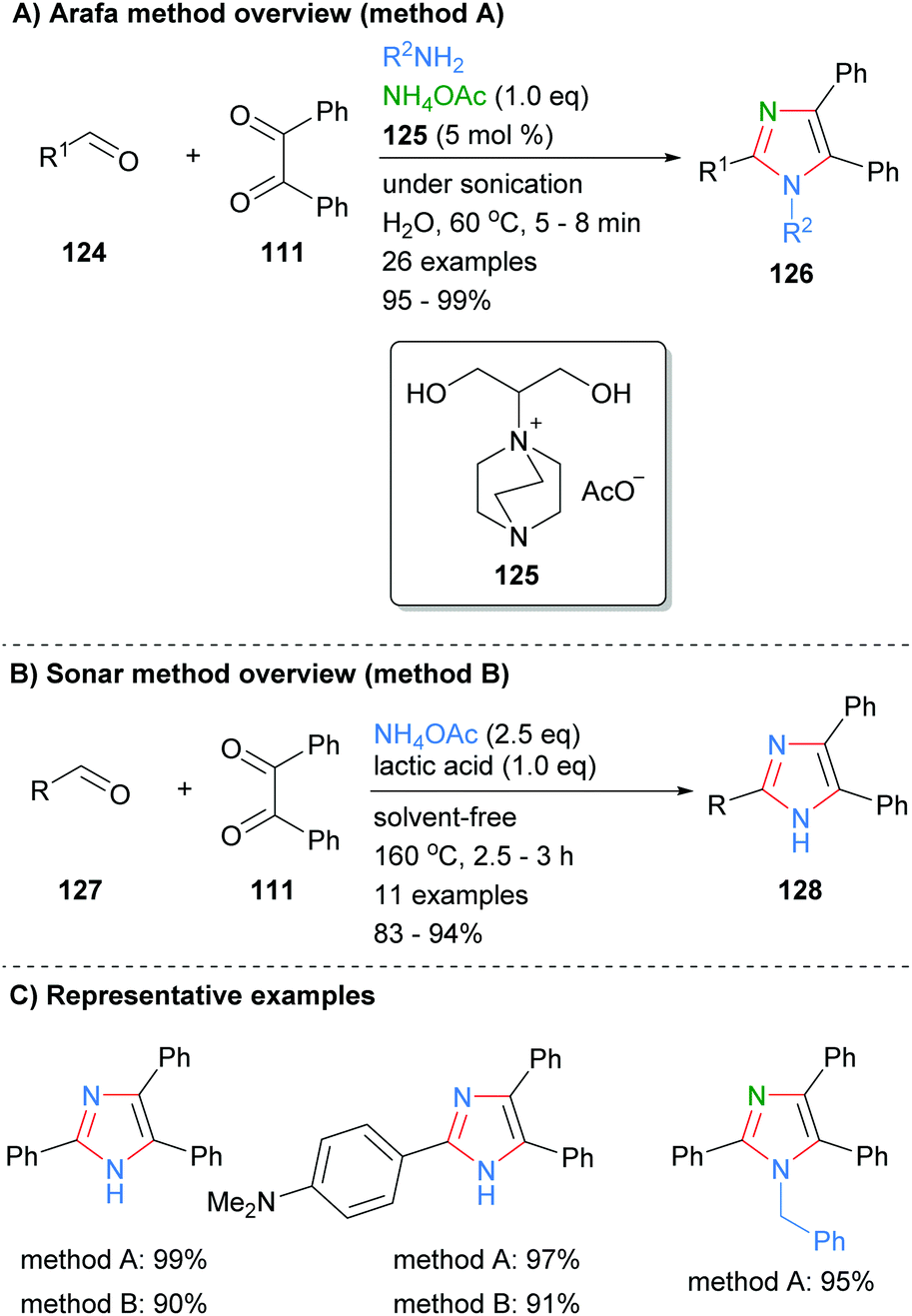
Condensation reactions to form the core four bonds of imidazoles can also be run under a variety of metal-free conditions. For example, Arafa recently reported the synthesis of tri- and tetra-substituted imidazoles 126 over an aqueous 1,4-diazabicyclo[2.2.2]octane (DABCO)48 based ionic liquid (Scheme 25a).49 Thus, sonication of aldehyde 124, 1,2-diketone 111 and an amine donor (ammonium acetate or primary amine) afford the desired imidazoles in excellent yields. The methodology was tolerant of a wide variety of functional groups and the catalyst could be used up to seven times without significant loss of activity. In contrast, Sonar et al. used lactic acid to promote the formation of trisubstitued imidazoles from the reaction of aldehydes 127, benzyl (111) and ammonium acetate (Scheme 25b).50 The high temperature, solvent free conditions afford the desired imidazoles in good to excellent yields. Both of these methods gave the desired imidazoles in good isolated yields, though the high temperatures invoked by Sonar, 160 °C, may limit the ability to incorporate certain functionality.
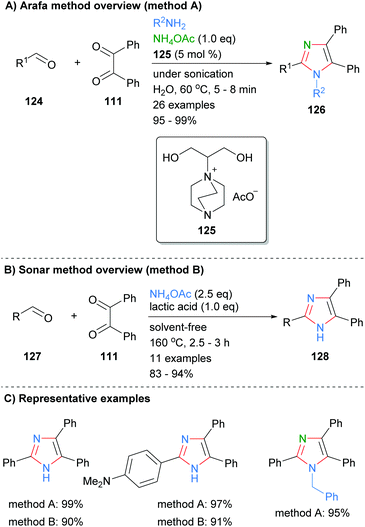 |
| | Scheme 25 Metal catalyst free aldehyde/diketone/amine(ammonia) coupling protocols. | |
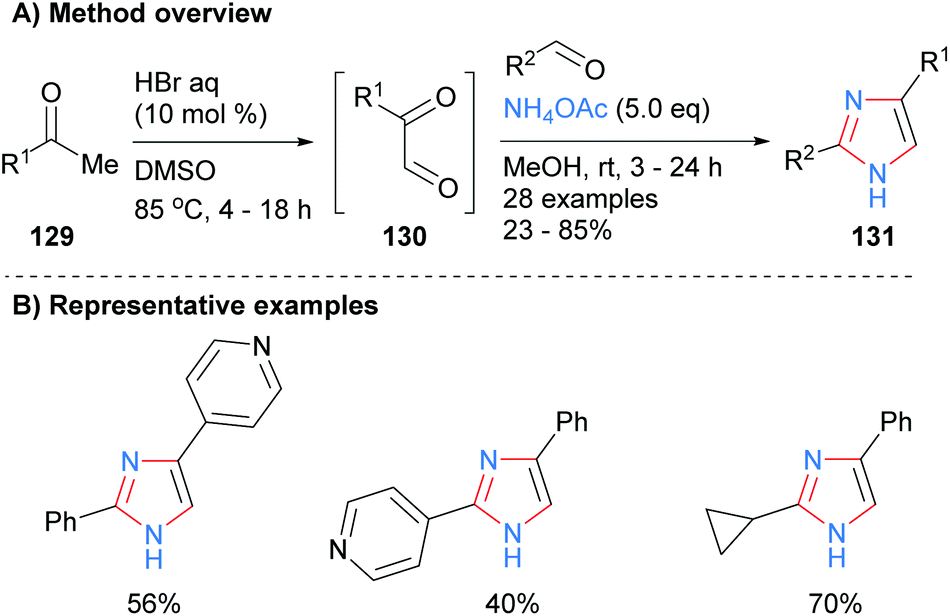
Toledo and co-workers recently reported that imidazoles 131 could also be synthesized by a metal-free, one-pot process from ketones 129via oxidation and subsequent dehydrative coupling with aldehydes and ammonium acetate (Scheme 26).51 This method had a reasonable level of functional group tolerance including basic pyridines and cyclopropanes.
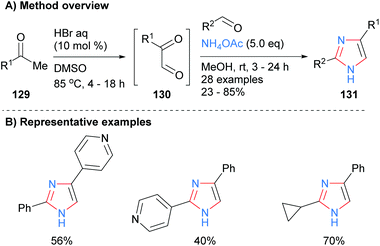 |
| | Scheme 26
In situ generation of ketoaldehydes via acid-catalysed oxidation of ketones. | |
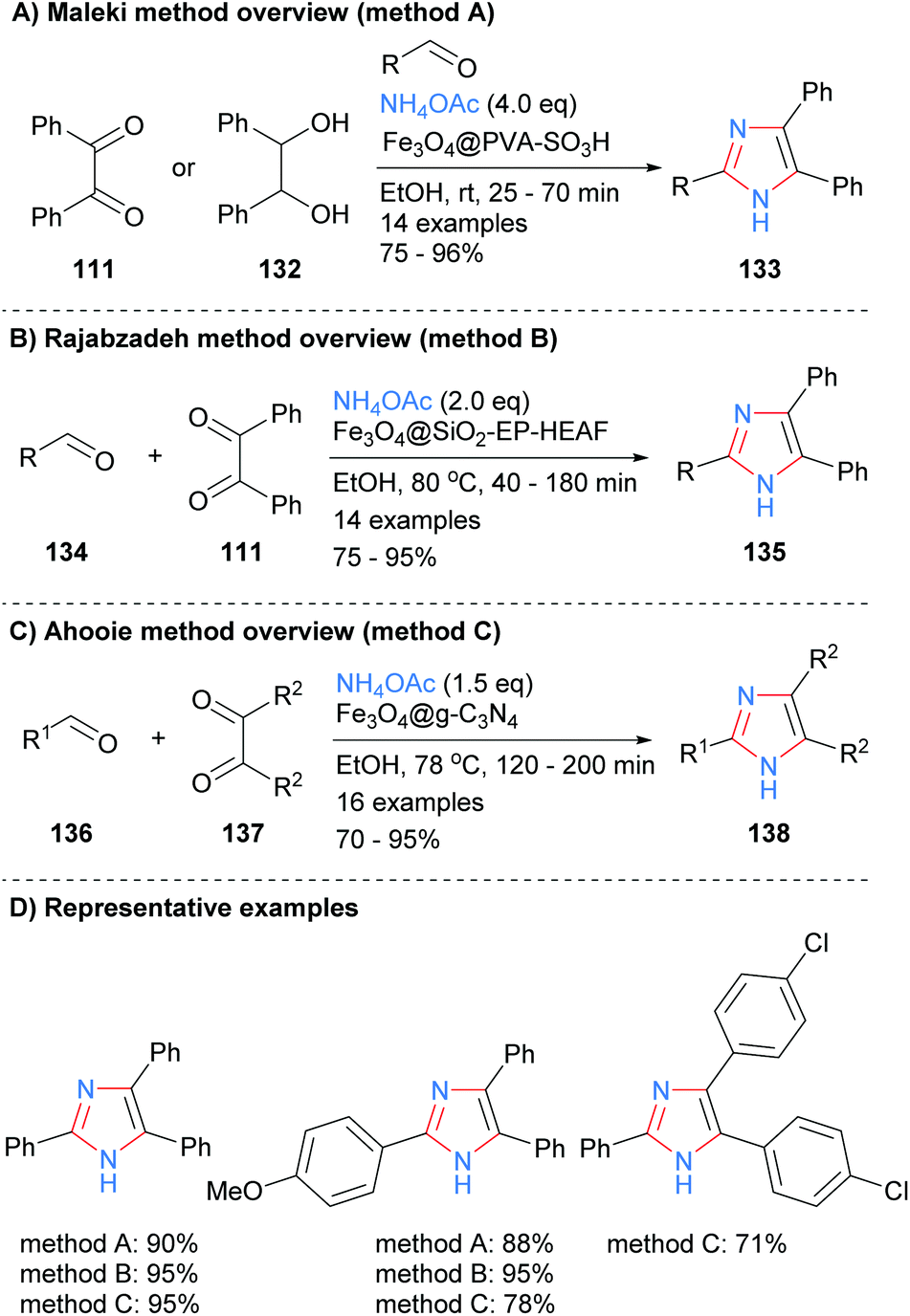
Derivatized magnetic nano-catalysts have been used in a wide range of synthetic applications due to the fact that they are easily recoverable.52 For example, a sulphonated Fe3O4@PVA superparamagnetic nano-catalyst was employed by Maleki et al. for the synthesis of trisubstitued-NH-imidazoles from the condensation reaction of benzyl (111), an aldehyde and ammonium acetate (Scheme 27a).53 The catalyst acts as an efficient organic–inorganic Brønsted acid and can be recycled up to ten times without a significant loss of reactivity. In contrast to the work above (cf.Scheme 22), the authors propose that the iron core plays no role in the catalytic cycle and that its major role is to provide an easy way to recycle the catalysts via its magnetic properties. Similar protocols were developed by both Rajabzadeh et al.54 and Ahooie et al.55 who used Fe3O4@SiO2-EP-HEAF and Fe3O4@g-C3N4 magnetic nanoparticles, respectively, to catalyse the three-component condensation reaction in ethanol to form 2,4,5-trisubstituted imidazoles (Scheme 27b and c). When the yields of the same product imidazoles were compared it was found that all three methods were relatively high yields, with only method C having one slightly lower for one of the substrates.
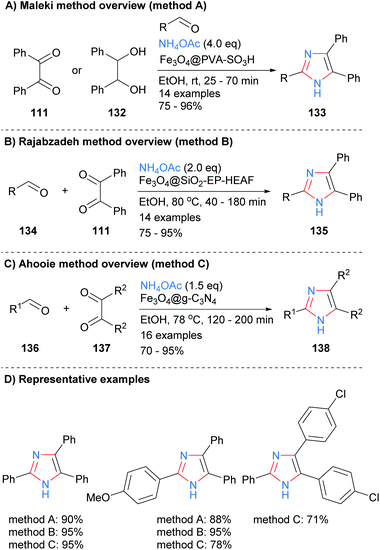 |
| | Scheme 27 Derivatized magnetic nano-catalysts in aldehyde/diketone/ammonia coupling protocols. | |
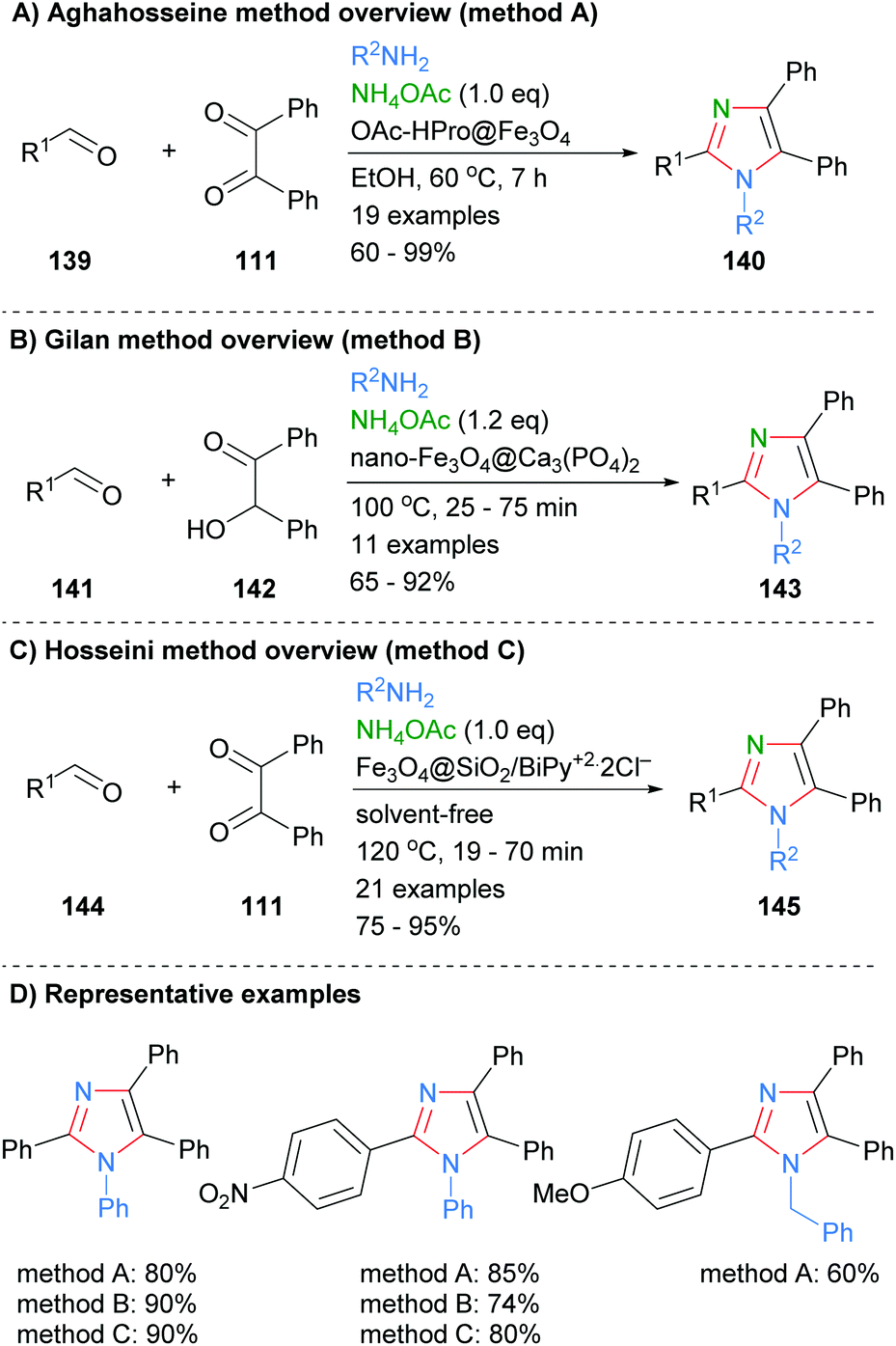
Similar condensation protocols for the synthesis of nitrogen substituted imidazoles using magnetic nano-catalysts have also been developed. For example, Aghahosseini et al. developed a method using L-proline derived magnetic core nanoparticles as organocatalysts (Scheme 28a).56 These catalysts were used to form tri- and tetra-substituted imidazoles in moderate to good yield. In a related example, Gilan and co-workers were also able to synthesis 2-substituted imidazoles via a three-component condensations approach (Scheme 28b).57 The reaction was catalyzed by a nano-magnetic catalyst Fe3O4@Ca3(PO4)2, which was synthesized using eggshell waste. The catalyst gave a good yield of 1,2,4,5-tetra-substitued imidazoles 143 and could be recycled up to four time without significant decrease in isolated product yield. Recently, Hosseini et al. also reported that the synthesis of imidazoles 145 could be catalyzed by a supported pyridinium catalyst (Scheme 28c).58 On average the yields for the synthesis of nitrogen substituted imidazoles using nano-magnetic catalyst were approximately 10% lower than for the NH-imidazoles described above (cf.Scheme 27).
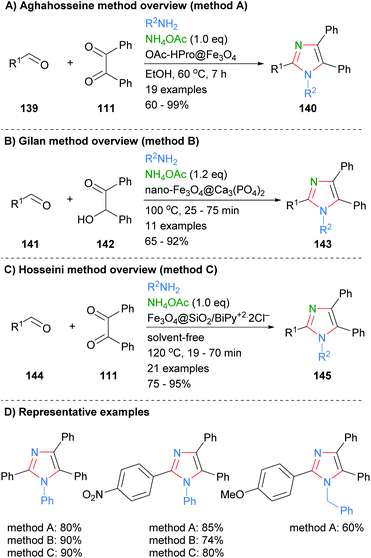 |
| | Scheme 28 Derivatized magnetic nano-catalysts in aldehyde/diketone/amine coupling protocols. | |
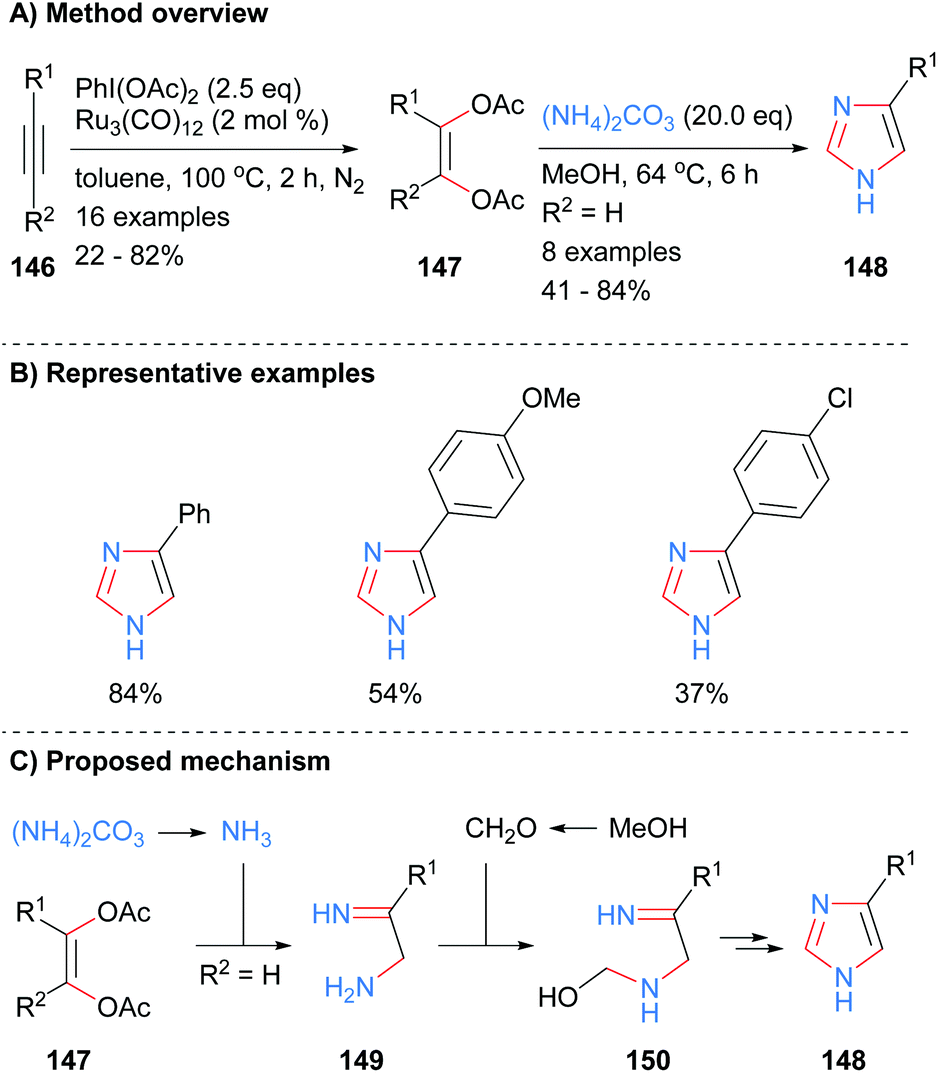
The four core bonds of monosubstitued imidazole 148 were constructed via bisfunctionalization of 1,2-disubstituted acetylenes 146 by ruthenium carbonyl to form cis-enediol diacetates 147 followed by subsequent reaction with ammonium carbonate (Scheme 29).59 The difunctionalization protocol obviates the substrate limitations and use of toxic reagents required of previous methodologies. Interestingly, this is one of the few recent examples in the literature in which mono-substituted NH-imidazoles were synthesized. Both electron rich and electron deficient aromatics were tolerated under the reaction conditions. Mechanistically, the reaction is proposed to proceed via in situ formation of ammonia, which reacts with cis-enediol diacetates 147 to give amino imine 149. Reaction of amine 149 with formaldehyde followed by intramolecular cyclization and aromatization afforded imidazole 148.
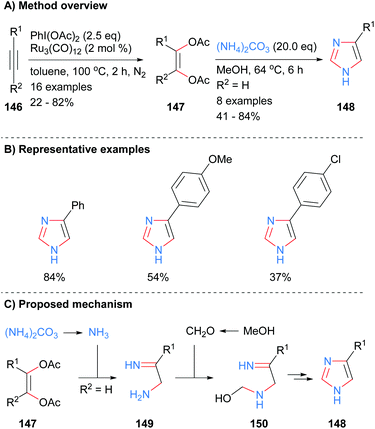 |
| | Scheme 29
In situ generation of diketone equivalents via ruthenium-catalysed oxidation of alkynes. | |
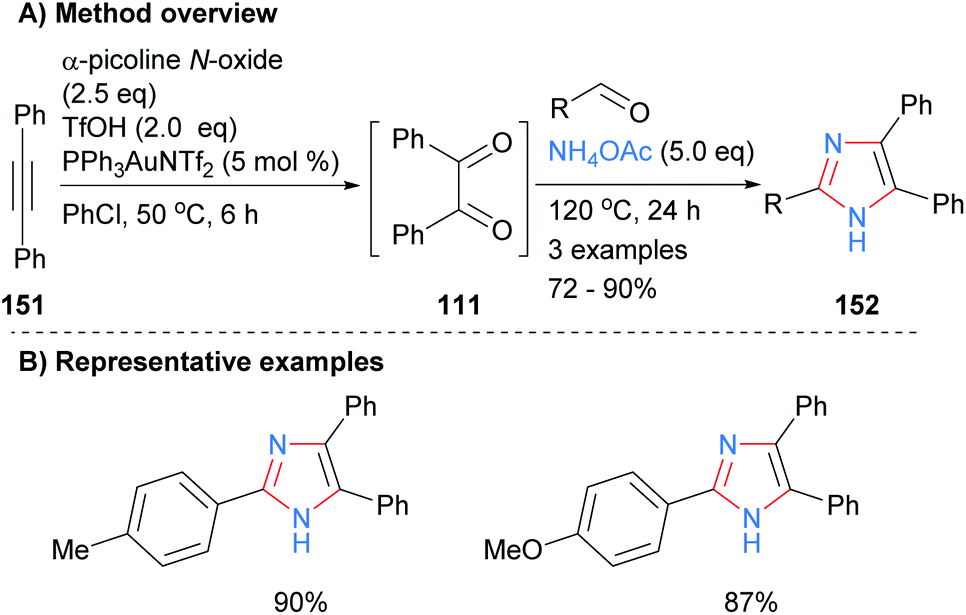
In a related approach Dubovtsev et al. also started from a disubstitued acetylene 151, which when subjected to a one-pot gold-catalyzed oxidation formed benzil (111). Subsequent reaction of the in situ generated diketone 111 with an aldehyde and ammonium acetate formed 2,4,5-trisubstituted imidazoles (Scheme 30).60 Both electron neutral and rich arylaldehydes gave the desired NH-imidazoles 152 in good isolated yields. Benzil generated in a similar manner was also converted into quinoxaline, 1,2,4-triazines, pyrazines and 1,4-diazepine in one-pot reaction sequences.
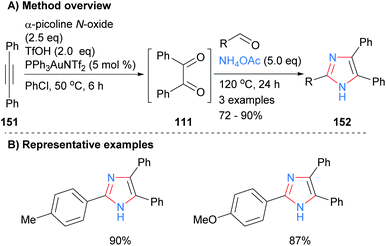 |
| | Scheme 30
In situ generation of diketone via gold-catalysed oxidation of alkynes. | |
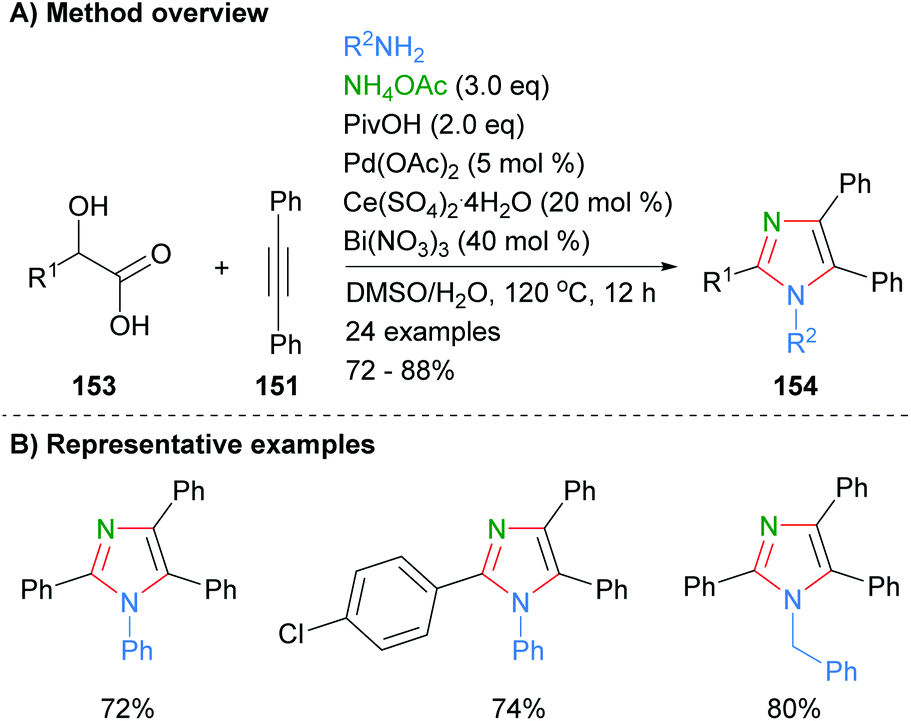
Internal alkynes were also employed by Sun et al. to from tetrasubstituted imidazoles (Scheme 31).61 Thus, reaction of diphenylacetylene (151) with α-hydroxy carboxylic acids 153 in the presence of excess ammonium acetate and a combination of palladium, cerium and bismuth reagents afforded the desired imidazoles in good yields. In contrast to the methods above (cf.Schemes 29 and 30), the method resulted in the formation of tetrasubstitued rather than NH-imidazoles. Both phenyl and benzyl functionalities were tolerated at N-1.
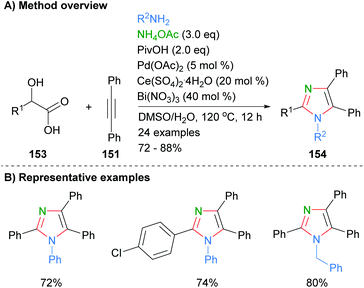 |
| | Scheme 31 Palladium/cerium/bismuth salts-catalysed condensation of α-hydroxy carboxylic acids with alkynes and amines. | |
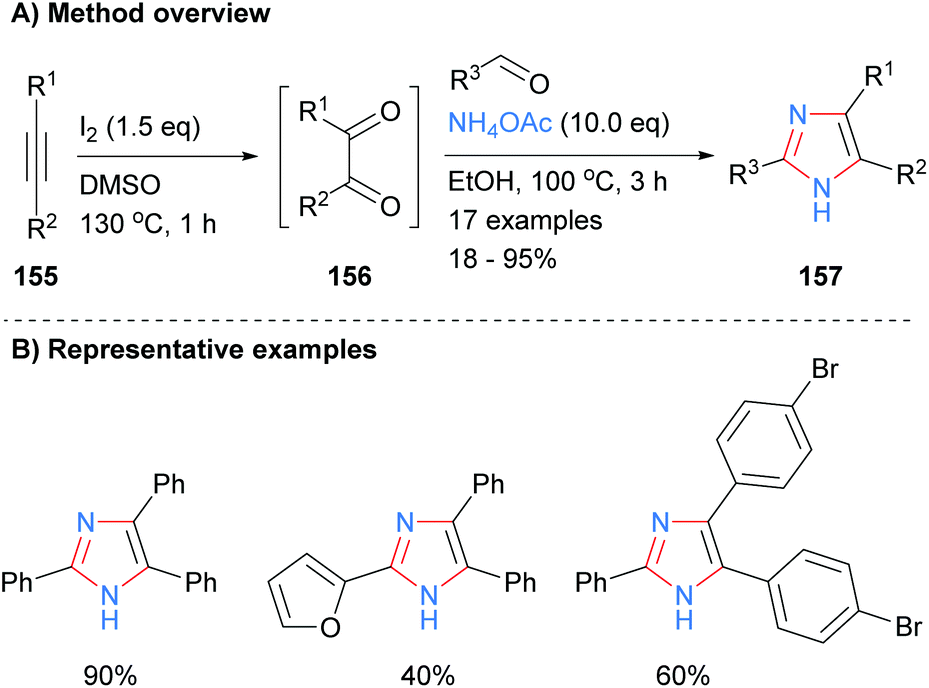
A one-pot metal and acid free synthesis of 2,4,5-trisubstituted imidazoles was recently reported by Naidoo and Jeena (Scheme 32).62 Reaction of internal alkynes 155 with iodine in dimethyl sulphoxide (DMSO) afford benzils, which were subsequently reacted with an aldehyde and ammonium acetate in situ. Both of the solvents involved, DMSO and ethanol, can be obtained from renewable resources63 and thus the overall methodology provides a greener alternative to many existing protocols. Both heterocycles and halogenated aromatics were compatible with the reaction conditions.
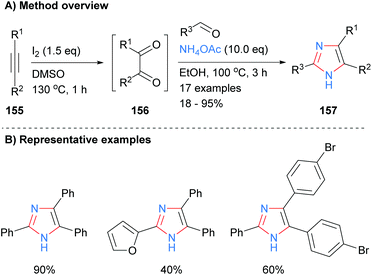 |
| | Scheme 32
In situ generation of diketone via iodine-catalysed oxidation of alkynes. | |
Outlook and conclusion
This review highlights that there has been a significant amount of recent research (2018-present) on the regioncontrolled synthesis of imidazoles. Table 1 and Table S1† provide an overview of the bond disconnections discussed as well as the resultant substitution pattern obtained in the study and can be used as starting point to determine which method(s) would be best suited for a particular application. Due to the importance of this structural motif in existing and emergent technologies, such as pharmaceuticals and agrochemicals as well as dyes for solar cells and other optical applications, functional materials, and catalysis, the development of additional methods is likely to increase. Despite these recent advances, the development of novel methods for the regiocontrolled synthesis of substituted imidazole is of strategic importance. An important consideration in new methods will be to provide regiochemically flexible access to the myriad of imidazole substitution patterns whilst increasing the functional group tolerance of the process. In addition, the ability to rationally and predicatively synthesise imidazoles from renewable starting materials under mild conditions that have a minimal impact on the environment, but which are economically viable will also be key to realise of the applications outlined above. Increased mechanistic understanding as well as reaction design based on the Twelve Principles of Green Chemistry64 should help deliver clean, concise and high yielding methods for the synthesis of imidazoles. Due to the wide range of potential permutations and the growing importance sustainability, it is clear that there is still significant scope for further discovery.
Table 1 Substitution pattern of imidazoles covered in this reviewa
| Entry |
Bonds |
NH |
NR |
C2 |
C4 |
C5 |
Yield range (%) |
Author |
Ref. |
|
Examples we chosen based on the following criteria: (a) number of bonds formed (b) substitution pattern (c) reaction conditions with priority given to methods using lower temperatures and green solvents (d) yield range.
|
| 1 |
One |
✓ |
|
✓ |
✓ |
|
13–90 |
Fang |
19
|
| 2 |
Two |
✓ |
|
✓ |
✓ |
|
6–25 |
Shabalin |
32
|
| 3 |
Two |
✓ |
|
✓ |
✓ |
✓ |
67–96 |
Wu |
28
|
| 4 |
Two |
|
✓ |
|
✓ |
✓ |
47–93 |
Cai |
25
|
| 5 |
Two |
|
✓ |
✓ |
|
✓ |
62–82 |
Wang |
31
|
| 6 |
Two |
|
✓ |
✓ |
✓ |
|
45–91 |
Tian |
30
|
| 7 |
Two |
|
✓ |
✓ |
✓ |
✓ |
65–98 |
Strelnikova |
27
|
| 8 |
Three |
✓ |
|
✓ |
✓ |
✓ |
49–93 |
Wang |
41
|
| 9 |
Three |
|
✓ |
|
✓ |
|
52–76 |
Geng |
36
|
| 10 |
Three |
|
✓ |
✓ |
✓ |
|
42–89 |
Alanthadka |
38
|
| 11 |
Three |
|
✓ |
✓ |
✓ |
✓ |
65–87 |
Alizadeh-Bami |
40
|
| 12 |
Four |
✓ |
|
|
✓ |
|
41–84 |
Ruan |
59
|
| 13 |
Four |
✓ |
|
✓ |
✓ |
|
23–85 |
Toledo |
51
|
| 14 |
Four |
✓ |
|
✓ |
✓ |
✓ |
75–96 |
Hosseini |
58
|
| 15 |
Four |
|
✓ |
✓ |
✓ |
✓ |
60–99 |
Aghahosseini |
56
|
Conflicts of interest
There are no conflicts to declare.
Notes and references
- For recent example of natural product synthesis, see:
(a) M. S. Roy, X. Meg, K. Koda, S. Rasapalli, D. Gout and C. J. Lovely, Tetrahedron Lett., 2019, 60, 979–982 CrossRef;
(b) Z. Jin, Nat. Prod. Rep., 2011, 28, 1143–1191 RSC;
(c) D. P. O'Malley, K. Li, M. Maue, A. L. Zografos and P. S. Baran, J. Am. Chem. Soc., 2007, 129, 4762–4775 CrossRef PubMed.
- For recent reviews on the synthesis and applications of imidazoles, see:
(a) N. Kerru, S. V. H. S. Bhaskaruni, L. Gummidi, S. N. Maddila, S. Maddila and S. B. Jonnalagadda, Synth. Commun., 2019, 49, 2437–2459 CrossRef CAS;
(b) R. Rossi, G. Angelici, G. Casotti, C. Manzini and M. Lessi, Adv. Synth. Catal., 2019, 361, 2737–2803 CrossRef CAS;
(c) M. Hossain and A. K. Nanda, Sci. J. Chem., 2018, 6, 83–94 CrossRef CAS;
(d) M. M. Heravi, M. Daraie and V. Zadsirjan, Mol. Diversity, 2015, 19, 577–623 CrossRef CAS PubMed;
(e) K. D. Kreuer, J. Membr. Sci., 2001, 185, 29–39 CrossRef CAS;
(f) J. Wang and C. Chen, Biotechnol. Adv., 2009, 27, 195–226 CrossRef CAS PubMed;
(g) B. H. Lipshultz, Chem. Rev., 1986, 86, 795–820 CrossRef;
(h) S. Steenken, Chem. Rev., 1989, 89, 503–520 CrossRef CAS;
(i) M. Xue, Y. Yang, Z. Xiadong, Z. Zhang and F. Hunag, Acc. Chem. Res., 2012, 45, 1294–1308 CrossRef CAS PubMed;
(j) J. Yoon, S. K. Kim, N. J. Singh and K. S. Kim, Chem. Soc. Rev., 2006, 35, 355–360 RSC;
(k) R. J. Sundberg and R. B. Martin, Chem. Rev., 1974, 74, 471–517 CrossRef CAS.
- S. Naureen, F. Chaudhry, M. A. Munawar, M. Ashraf, S. Hamid and M. A. Khan, Bioorg. Chem., 2018, 76, 365–369 CrossRef CAS PubMed.
- M. V. P. dos Santos Nascimento, A. C. M. Munhoz, B. M. De Campos Facchin, E. Fratoni, T. A. Rossa, M. M. Sá, C. C. Campa, E. Ciraolo, E. Hirsch and E. M. Dalmarco, Biomed. Pharmacother., 2019, 111, 1399–1407 CrossRef CAS PubMed.
- Z. Mi, Y. Chen, X. Chen, L. Yan, Q. Gu, H. Zhang, C. Chen and Y. Zhang, Chem. Res. Chin. Univ., 2018, 34, 369–374 CrossRef CAS.
- M. Mao, X.-L. Zhang and G.-H. Wu, Int. J. Photoenergy, 2018, 2061472, DOI:10.1155/2018/2061472.
- K. Dhanunjayarao, V. Mukundam, R. V. R. N. Chinta and K. Venkatasubbaiah, J. Organomet. Chem., 2018, 865, 234–238 CrossRef CAS.
- S. A. Khan, A. M. Asiri, A.-A. M. Al-Dies, O. I. Osman, M. Asad and M. E. M. Zayed, J. Photochem. Photobiol., A, 2018, 364, 390–399 CrossRef CAS.
- A. A. Bhagwat, K. C. Avhad, D. S. Patil and N. Sekar, Photochem. Photobiol., 2019, 95, 740–754 CrossRef CAS PubMed.
- A. Hariharan, S. Kumar, M. Alagar, K. Dinakaran and K. Subramanian, Polym. Bull., 2018, 75, 93–107 CrossRef CAS.
- Jigyasa and J. K. Rajput, Sens. Actuators, B, 2018, 259, 990–1005 CrossRef CAS.
- J. Tagare, D. K. Dubey, J.-H. Jou and S. Vaidyanathan, Dyes Pigm., 2019, 160, 944–956 CrossRef CAS.
- S. Sambathkumar, S. Priyadharshini, M. Fleisch, D. W. Bahnemann, G. Gnana Kumar, S. Senthilarasu and R. Renganathan, Mater. Lett., 2019, 242, 28–31 CrossRef CAS.
- Y. Dong, J. Qian, Y. Liu, N. Zhu, B. Xu, C.-L. Ho, W. Tian and W.-Y. Wong, New J. Chem., 2019, 43, 1844–1850 RSC.
- H.-L. Ding, L.-D. Chen, N. Wang, K. Li, Y. An and C.-W. Lü, Talanta, 2019, 195, 345–353 CrossRef CAS PubMed.
- J. Jang, Y. Sim, S. Kang, D. Shin, M. Park, K.-Y. Kay and J. Park, Mol. Cryst. Liq. Cryst., 2019, 687, 14–20 CrossRef CAS.
- M. Mohagheghnezhad and Z. Rafiee, Polym. Bull., 2019 DOI:10.1007/s00289-019-02992-9.
- J. A. Castro-Osma, J. Martínez, F. de la Cruz-Martínez, M. P. Caballero, J. Fernández-Baeza, J. Rodríguez-López, A. Otero, A. Lara-Sánchez and J. Tejeda, Catal. Sci. Technol., 2018, 8, 1981–1987 RSC.
- S. Fang, H. Yu, X. Yang, J. Li and L. Shao, Adv. Synth. Catal., 2019, 361, 3312–3317 CrossRef CAS.
- S. Shi, K. Xu, C. Jiang and Z. Ding, J. Org. Chem., 2018, 83, 14791–14796 CrossRef CAS PubMed.
- L. Man, R. C. B. Copley and A. L. Handlon, Org. Biomol. Chem., 2019, 17, 6566–6569 RSC.
- P. Tang, D. Ke, J. Shao, W. Chen and Y. Yu, Tetrahedron, 2019, 75, 4419–4424 CrossRef CAS.
- M. B. Harisha, P. Dhanalakshmi, R. Suresh, R. R. Kumar, S. Muthusubramanian and N. Bhuvanesh, ChemistrySelect, 2019, 4, 2954–2958 CrossRef CAS.
- D. Yang, L. Shan, Z.-F. Xu and C.-Y. Li, Org. Biomol. Chem., 2018, 16, 1461–1464 RSC.
- J. Cai, H. Bai, Y. Wang, X. Xu, H. Xie and J. Liu, Chem. Commun., 2019, 55, 3821–3824 RSC.
- E. B. Nikolaenkova, A. Ya. Tikhonov and S. Yu. Grishchenko, Chem. Heterocycl. Compd., 2019, 55, 142–146 CrossRef CAS.
- J. O. Strelnikova, N. V. Rostovskii, G. L. Starova, A. F. Khlebnikov and M. S. Novikov, J. Org. Chem., 2018, 83, 11232–11244 CrossRef CAS PubMed.
- P. Wu, X. Zhang and B. Chen, Tetrahedron Lett., 2019, 60, 1103–1107 CrossRef CAS.
- X. Tian, L. Song, M. Rudolph, F. Rominger, T. Oeser and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2019, 58, 3589–3593 CrossRef CAS PubMed.
- Y. Tian, M. Qin, X. Yang, X. Zhang, Y. Liu, X. Guo and B. Chen, Tetrahedron, 2019, 75, 2817–2823 CrossRef CAS.
- C. Wang, Y. Yu, Z. Su, X. Li and H. Cao, Org. Lett., 2019, 21, 4420–4423 CrossRef CAS PubMed.
- D. A. Shabalin, J. J. Dunsford, S. Ngwerume, A. R. Saunders, D. M. Gill and J. E. Camp, Synlett, 2020, 31, 797–800 CrossRef CAS.
- E. Li, Y. Lin, X. Wu, X. Mao, H. Kang, Y. Wen, Y. Bai, X. Wang and X. Lv, Tetrahedron Lett., 2019, 60, 151122 CrossRef.
- S. D. Pardeshi, P. A. Sathe, K. S. Vadagaonkar, L. Melone and A. C. Chaskar, Synthesis, 2018, 50, 361–370 CrossRef CAS.
- C. T. F. Salfeena, R. Jalaja, R. Davis, E. Suresh and S. B. Somappa, ACS Omega, 2018, 3, 8074–8082 CrossRef CAS PubMed.
- X. Geng, C. Wang, C. Huang, Y. Bao, P. Zhao, Y. Zhou, Y.-D. Wu, L. Feng and A.-X. Wu, Org. Lett., 2020, 22, 140–144 CrossRef CAS PubMed.
-
(a) A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef;
(b) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed.
- A. Alanthadka, S. D. Elango, P. Thangavel, N. Subbiah, S. Vellaisamy and U. M. Chockalingam, Catal. Commun., 2019, 125, 26–31 CrossRef CAS.
- X. Chen, Z. Wang, H. Huang and G.-J. Deng, Adv. Synth. Catal., 2018, 360, 4017–4022 CrossRef CAS.
- F. Alizadeh-Bami, M. Salehzadeh, H. Mehrabi and R. Ranjbar-Karimi, ARKIVOC, 2019,(vi), 55–63 Search PubMed.
- Q. Wang, X. Chen, X.-G. Wang, H.-C. Liu and Y.-M. Liang, Org. Lett., 2019, 21, 9874–9877 CrossRef CAS PubMed.
- S. Sundar and R. Rengan, Org. Biomol. Chem., 2019, 17, 1402–1409 RSC.
- N. L. Higuera, D. Peña-Solórzano and C. Ochoa-Puentes, Synlett, 2019, 30, 225–229 CrossRef CAS.
- A. A. Marzouk, A. M. Abu-Dief and A. A. Abdelhamid, Appl. Organomet. Chem., 2018, 32, e3794 CrossRef.
- M. Thwin, B. Mahmoudi, O. A. Ivaschuk and Q. A. Yousif, RSC Adv., 2019, 9, 15966–15975 RSC.
- R. Vaid, M. Gupta, G. Kour and V. K. Gupta, ChemistrySelect, 2019, 4, 9179–9184 CrossRef CAS.
- Z. Varzi and A. Maleki, Appl. Organomet. Chem., 2019, e5008 Search PubMed.
- For recent examples of nucleophilic catalysis, see:
(a) J. P. Tellam, G. Kociok-Köhn and D. R. Carbery, Org. Lett., 2008, 10, 5199–5202 CrossRef CAS;
(b) G. Sabitha, D. V. Reddy, A. S. Rao and J. S. Yadav, Tetrahedron Lett., 2010, 51, 4195–4198 CrossRef CAS;
(c) S. Ngwerume and J. E. Camp, J. Org. Chem., 2010, 75, 6271–6274 CrossRef CAS PubMed;
(d) W. Chung, P. Lindovská and J. E. Camp, Tetrahedron Lett., 2011, 52, 6785–6787 CrossRef CAS.
- W. A. A. Arafa, RSC Adv., 2018, 8, 16392–16399 RSC.
- J. Sonar, S. Pardeshi, S. Dokhe, R. Pawar, K. Kharat, A. Zine, B. Matsagar, K. Wu and S. Thore, SN Appl. Sci., 2019, 1, 1045 CrossRef.
- I. de Toledo, T. A. Grigolo, J. M. Bennett, J. M. Elkins and R. A. Pilli, J. Org. Chem., 2019, 84, 14187–14201 CrossRef CAS PubMed.
-
(a) B. Liu and Z. Zhang, ACS Catal., 2016, 6, 326–338 CrossRef CAS;
(b) R. G. Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef PubMed.
- A. Maleki, J. Rahimi and K. Valadi, Nano-Struct. Nano-Objects, 2019, 18, 100264 CrossRef.
- M. Rajabzadeh, H. Eshghi, R. Khalifeh and M. Bakavoli, Appl. Organomet. Chem., 2018, 32, e4052 CrossRef.
- T. S. Ahooie, N. Azizi, I. Yavari and M. M. Hashemi, J. Iran. Chem. Soc., 2018, 15, 855–862 CrossRef CAS.
- H. Aghahosseini, A. Ramazani, K. Ślepokura and T. Lis, J. Colloid Interface Sci., 2018, 511, 222–232 CrossRef CAS PubMed.
- M. M. Gilan, A. Khazaei and N. Sarmasti, Res. Chem. Intermed., 2019 DOI:10.1007/s11164-018-03724-w.
- S. Hosseini, A. R. Kiasat and A. Farhadi, Polycyclic Aromat. Compd., 2019 DOI:10.1080/10406638.2019.1616306.
- Y. Ruan, Y. Chen, L. Gu, Y. Luo, Z. Yang and L. He, Synthesis, 2019, 51, 3520–3528 CrossRef CAS.
- A. Yu. Dubovtsev, D. V. Dar'in, M. Krasavin and V. Yu. Kukushkin, Eur. J. Org. Chem., 2019, 1856–1864 CrossRef CAS.
- W. Sun, M. Zhang, P. Li and Y. Li, Synthesis, 2019, 51, 3221–3230 CrossRef CAS.
- S. Naidoo and V. Jeena, Eur. J. Org. Chem., 2019, 1107–1113 CrossRef CAS.
- For recent work on bio-renewable solvents from our group, see:
(a) T. W. Bousfield, K. P. R. Pearce, S. B. Nyamini, A. Angelis-Dimakis and J. E. Camp, Green Chem., 2019, 21, 3675–3681 RSC;
(b) J. E. Camp, ChemSusChem, 2018, 11, 3048–3055 CrossRef CAS PubMed;
(c) L. Mistry, K. Mapesa, T. W. Bousfield and J. E. Camp, Green Chem., 2017, 19, 2123–2128 RSC;
(d) J. E. Camp, S. B. Nyamini and F. J. Scott, RSC Med. Chem., 2020, 11, 111–117 RSC.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob00350f |
|
| This journal is © The Royal Society of Chemistry 2020 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a and
Jason E.
Camp
a and
Jason E.
Camp