
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAll-in-one microfluidic assembly of insulin-loaded pH-responsive nano-in-microparticles for oral insulin delivery†
Clarinda
Costa‡
 abc,
Zehua
Liu‡
a,
João P.
Martins
a,
Alexandra
Correia
a,
Patrícia
Figueiredo
a,
Antti
Rahikkala
a,
Wei
Li
a,
Jani
Seitsonen
d,
Janne
Ruokolainen
d,
Sami-Pekka
Hirvonen
e,
Ana
Aguiar-Ricardo
b,
M. Luísa
Corvo
*c and
Hélder A.
Santos
*af
abc,
Zehua
Liu‡
a,
João P.
Martins
a,
Alexandra
Correia
a,
Patrícia
Figueiredo
a,
Antti
Rahikkala
a,
Wei
Li
a,
Jani
Seitsonen
d,
Janne
Ruokolainen
d,
Sami-Pekka
Hirvonen
e,
Ana
Aguiar-Ricardo
b,
M. Luísa
Corvo
*c and
Hélder A.
Santos
*af
aDrug Research Program, Division of Pharmaceutical Chemistry and Technology, Faculty of Pharmacy, University of Helsinki, FI-00014, Helsinki, Finland. E-mail: helder.santos@helsinki.pt
bLAQV-REQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, 2829-516 Caparica, Portugal
cInstituto de Investigação do Medicamento (iMed.ULisboa), Faculdade de Farmácia, Universidade de Lisboa, Avenida Professor Gama Pinto, 1649-003 Lisboa, Portugal. E-mail: lcorvo@ff.ulisboa.pt
dNanomicroscopy center, Aalto University, FI-00076, Aalto, Finland
eDepartment of Chemistry, Faculty of Science, University of Helsinki, FI-00014, Helsinki, Finland
fHelsinki Institute of Life Science (HiLIFE), University of Helsinki, FI-00014, Helsinki, Finland
First published on 27th May 2020
Abstract
Here, a continuous two-step glass-capillary microfluidic technique to produce a multistage oral delivery system is reported. Insulin is successfully encapsulated into liposomes, which are coated with chitosan to improve their mucoadhesion. The encapsulation in an enteric polymer offers protection from the harsh gastric conditions. Insulin permeability is enhanced across an intestinal monolayer.
The oral route of administration represents the most convenient way to administer drugs in terms of high patient compliance,1,2 promoting a fast and safe route, overcoming several main disadvantages from parenteral administration namely intravenous administration.3–6 However, the oral delivery of therapeutic proteins and peptides represents a big challenge for the pharmaceutical technology field. In fact, the degradation of proteins and peptides under acidic conditions in the stomach concomitantly with digestive enzyme action, as well as the poor oral bioavailability, characterized by a poor protein permeability in the intestinal epithelium7–9 has led to the need for designing and developing alternative approaches to enhance the oral protein/peptide administration.10–12 Over the last decade, the research for different strategies for the oral delivery of proteins and peptides, such as microemulsions,13,14 polymeric nanoparticles,8,9,16 silicon nanoparticles15,17,18 and liposomes,19,20 has been significantly increasing. Liposomes, introduced for the first time by Bangham et al.21 at the beginning of the 1960s, have been described as potential protein and peptide drug carriers able to enhance their blood residence time and targeting.22–24 However, even with high potential to deliver proteins, conventional liposomes are still likely to disintegrate under gastric conditions25,26 and can show poor permeation across the intestinal epithelial cell membrane,27 thus resorting to coatings with other molecules, such as polymers28,29 or protein corona.30,31 Yet, liposomes are usually prepared by bulk methods such as thin-film hydration,32,33 ethanol injection,34 and reverse phase evaporation,35,36 in which some of them require a post-processing step to obtain a better control of size and polydispersity index (PDI), such as high pressure extrusion.37
Microfluidics, a sophisticated technique for particle production, has gained substantial attention to prepare liposomes. High batch-to-batch reproducibility, reduced time and cost, and increased control on the production of nanoparticles are some of the advantages of microfluidics.38–40 Polydimethylsiloxane (PDMS)-based microfluidic devices are widely used in liposome preparation. PDMS is compatible with some pharmaceutical grade solvents, such as ethanol.41 However, PDMS microfluidic devices have low resistance to high flow rates and pressures and, upon contact with common organic solvents, tend to swell. In addition, due to the hydrophobicity of the polymer, post-surface functionalization of the polymer is required.42–44 Attending to these drawbacks, glass-capillary microfluidic devices are a good alternative when there is the need to use organic solvents. Besides, they can tolerate high flow rates and pressures.45 Glass-capillary microfluidic devices enable the production of liposomes through a nanoprecipitation method.46,47 If a central flow of a phospholipid-containing alcohol solution merges into a main channel with the aqueous solution in adequate concentrations, the lipids spontaneously self-assemble, producing liposomes,48 with diameters in the range of 73–131 nm, as described by Vladisavljevic et al.49 Moreover, these devices enable the generation of monodisperse droplets via co-flow or coaxial flow-focusing geometry or via the combination of both geometries,45,50 offering an excellent control of the size of the droplets by adjusting the flows of immiscible fluids inside the microchannels and the dimensions of the orifices of the channels.51,52 By the solidification of the monodisperse droplets, microparticles with an uniform size distribution can be produced.53 In this way, using glass-capillary microfluidic devices both nano- and nano-in-microparticles can be obtained, which represents a huge advantage for oral administration since microparticles have been described as preferable for this kind of administration compared to nano-sized systems.54 Besides, microparticles are more attractive for oral delivery over the conventional larger capsules or pill-type vehicles due to the patient compliance and convenience, especially in children that have some difficulties in swallowing larger pills or capsules.55,56
In this work, we present an optimized and continuous “all-in-one” two-step microfluidic production of a nano-in-micro composite system, as shown in Fig. 1A. Firstly, the microfluidic parameters, such as flow rate ratio (FRR), initial lipid concentration and total flow rate, were optimized in order to prepare liposomes with a size between 100 and 150 nm and an acceptable narrow PDI (<0.2). To assess the influence of the FRR (the ratio between the aqueous and the organic phases) on the size and PDI of the liposomes, the microfluidic parameters outer flow rate and initial lipid concentration were kept constant (outer flow rate = 20 mL h−1 and initial lipid concentration = 43.2 μmol mL−1). The FRR was varied between 1 and 10 (Fig. 1B(a)). As expected, increasing the FRR reduces the size of the liposomes.41,57 This phenomenon can be related to the non-equilibrium kinetic model described by Zook and Vreeland.57 This kinetic model applied to liposomes suggests that the size of the liposomes is closely related to the growth rate of the lipid planar discs (bilayered phospholipid fragments (BPFs)) and to the closing rate of the discs into spherical vesicles. In this way, the model predicts that at higher FRRs, the alcohol concentration decreases faster and the lipid disc growth rate decreases. In addition, since BPFs grow with less stabilization, the lipid discs close faster, producing smaller liposomes. In contrast, at lower FRRs, the BPFs take more time to grow, and due to the stabilization from the alcohol stream, they take more time to close, making the liposomes larger.37,57 The optimization was pursued towards the study of the effect of the total flow rate (TFR) on the morphological properties of the liposome. The TFR was varied between 10 and 100 mL h−1, while the FRR was kept at 1 and the initial lipid concentration at 43.2 μmol mL−1. Fig. 1B(b) shows that the TFR has no significant effect (p > 0.05) on the size of the liposomes, which is in accordance with previous studies.58,59 This represents an advantage of the scalable microfluidics – increasing the throughput does not affect significantly the liposome size. Likewise, it does not affect the PDI, which remained below 0.2. Thus, since the desired size and acceptable PDI were obtained with a total flow rate of 20 mL h−1, the studies were pursued with a lipid concentration of 43.2 μmol mL−1 and a total flow rate of 20 mL h−1. As the lipid concentration plays an important role in the morphology of the liposomes, the initial lipid concentration was varied from 20.8 to 54.0 μmol mL−1 (the total flow rate was kept at 20 mL h−1 and the FRR was kept at 1). Fig. 1B(c) shows an increase of the size of the liposomes with the increase of lipid concentration. Upon contact with the aqueous solution, the alcohol, in which the lipids were initially solubilized, diffuses into the water, leading to a lipid supersaturation.57 Similar to the FRR, the intermediate lipid discs, described as BPFs, are formed in the ethanol/water interface and are thermodynamically less stable. When the BPFs grow, they tend to fuse and enclose into a vesicle;60 thus, higher lipid concentrations can produce more BPFs, enabling the generation of larger vesicles. An initial lipid concentration of 43.2 μmol mL−1 was selected to pursue the experiments. Cryo-TEM was performed in order to confirm the morphology/structure of the InsLip nanoparticles. For the optimized parameters, the cryo-TEM images show a thin single-wall structure enclosing a vesicle, confirming that the structure of the nanoparticles corresponds to that of liposomes, with a mean size of 110 ± 20 nm, similarly to the values obtained by DLS (Fig. 1C(a)). In addition, using Gatan Microscopy Suite software, it was possible to measure the thickness of the bilayer of the liposomes, which was around 6 ± 1 nm (Fig. 1C(b)).
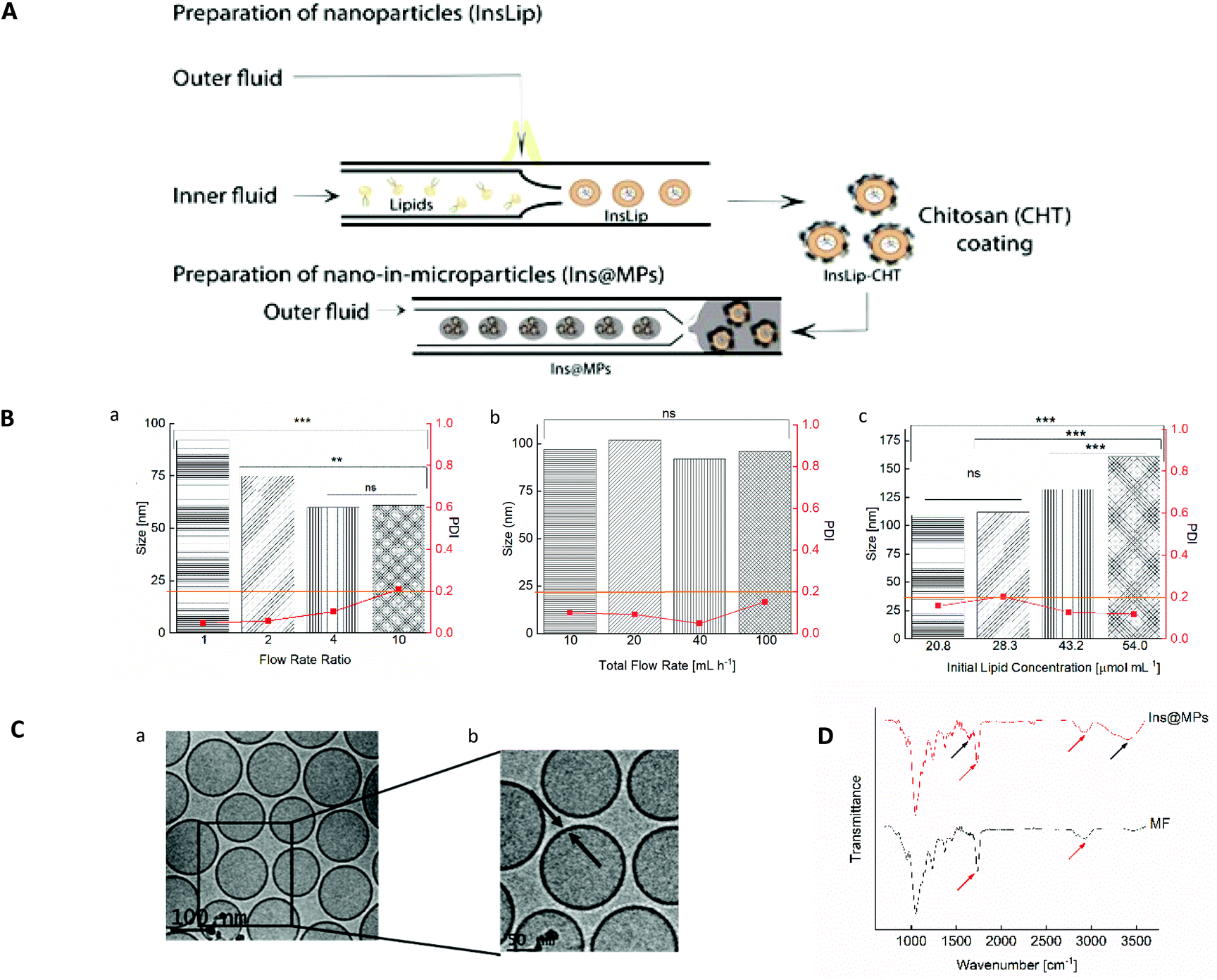 | ||
| Fig. 1 Preparation and characterization of Lip and Ins@MPs. (A) Schematic representation of the two-step microfluidic process of insulin-loaded nano-in-microparticles (not to scale), in which insulin is encapsulated in PEGylated liposomes (InsLip) through a nanoprecipitation technique, using a glass-capillary microfluidic device. The InsLip-CHT were further encapsulated in an enteric polymer (MF) using a double-emulsion microfluidic process, forming the final microcarrier system (Ins@MPs). (B) Size (bar) and PDI (red squared dots) of liposomes on the optimization of the microfluidic parameters for liposome production: a. flow rate ratio, keeping constant the total flow rate (TFR) of 20 mL h−1 and initial lipid composition (ILC) of 43.2 μmol mL−1; b. TFR, keeping fixed the FRR of 1 and ILC of 43.2 μmol mL−1 and c. initial lipid concentration (μmol mL−1), keeping fixed the FRR of 1 and TRF of 20 mL h−1. The orange line represents the maximum PDI value acceptable for liposomes (0.2). The level of significance was set at the probabilities of **p < 0.01 and ***p < 0.001. (C) Cryo-TEM images of liposomes, using the optimized microfluidic parameters described above. (D) ATR-FTIR spectra of MF and insulin-loaded nano-in-microparticles (Ins@MPs). Black arrows: bands from chitosan chemical groups; red arrows: bands from MF chemical groups. | ||
The encapsulation of insulin in liposomes was then carried out with the previous optimized parameters. Recombinant human insulin (Ins) was first dissolved in a citrate solution, at pH 2.0, at a concentration of 100 μg mL−1 (outer phase), whereas lipids (egg-phosphatidylcholine (E-PC) and distearoylphosphatidylethanolamine poly(ethyleneglycol)2000 (DSPE-PEG2000)) and cholesterol were dissolved in ethanol at a concentration of 43.2 μmol mL−1 (inner phase). Through a nanoprecipitation phenomenon,47 lipids upon contact with the citrate solution self-assembled, forming protein-loaded PEGylated liposomes (InsLip). After being collected from the glass capillary, InsLip were characterized in terms of hydrodynamic size, PDI, and encapsulation efficiency (E.E.). InsLip were obtained with an average size of 144 ± 23 nm, a mean PDI of 0.130 ± 0.003 and an E.E. of 91 ± 4%. Considering that this process is continuous, the E.E. was calculated from the total concentration of insulin in the liposomes and the insulin present in the supernatant after liposome washing. During the assays no precipitated insulin was observed. To the best of our knowledge, the E.E. obtained represents an improvement compared to previous studies, involving liposomes with the same morphological properties (Niu et al. presented insulin-loaded liposomes with a size of 150 nm and an E.E. of 30%,20,27 whereas Muramatsu et al. reported liposomes with a size between 106 and 108 nm with an E.E. of 21.5%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 61).
61).
The physical coating of chitosan on the InsLip (InsLip-CHT) was achieved by adding the InsLip suspension dropwise to the chitosan solution (10 mg mL−1 in 1% of acetic acid solution (v/v), pH 5.5), at the same volume ratio, and stirring for 6 h. Then, the dispersion containing chitosan-coated liposomes (InsLip-CHT) was centrifuged and washed twice. The coating was observed by the change in the InsLip-CHT average size and PDI, up to a mean size of 363 ± 54 nm and a PDI of 0.315 ± 0.057. In addition, the increase of the zeta-potential from a value near to zero (−0.5 ± 0.1 mV) to a positive value (+23 ± 1 mV) suggests the successful chitosan coating of the liposomal surface. Since the zeta-potential of InsLip was near zero, it is expectable that the interaction between the liposomes and chitosan might have occurred by van der Waals and hydrogen bonding forces.62
Nano-in-microparticles are mostly used for their ability to effectively deliver higher local doses in a specific tissue.63 In this way, through a double emulsion microfluidics method, the nanoparticles were encapsulated in an FDA approved excipient for oral administration, hydroxypropyl methylcellulose acetate succinate (HPMCAS-MF; M grade fine powders, abbreviated as MF), in order to protect the liposomes against the harsh stomach acidic conditions (Ins@MPs). MF is a synthetic enteric polymer with an average molecular weight of around 17000 Da and a polydispersity of approximately 1.364 and since it presents a pKa ∼ 5, this polymer is widely used for oral drug delivery applications.65 The encapsulation of the InsLip-CHT in the MF matrix resulted in a batch of 15 mL of insulin-loaded nano-in-microparticles (Ins@MPs) with a size of 19 ± 1 μm. The ATR-FTIR spectra (Fig. 1D) of Ins@MPs show a band at 1710 cm−1, which corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the carboxylic group from MF, as well as a band at 2900 cm−1, which belongs to the C–H stretching of the alkyl group. Nevertheless, the broad band at 3388 cm−1 and the band at 1630 cm−1 correspond to the N–H stretching of the amine and CO stretching of the amides groups, respectively, suggesting the presence of chitosan.66,67 Apart from these bands, the spectrum of Ins@MPs is very similar to the MF spectra, suggesting an efficient encapsulation of InsLip-CHT by MF. The microparticles were characterized in terms of loading degree, which was 0.39 ± 0.02%.
O stretching of the carboxylic group from MF, as well as a band at 2900 cm−1, which belongs to the C–H stretching of the alkyl group. Nevertheless, the broad band at 3388 cm−1 and the band at 1630 cm−1 correspond to the N–H stretching of the amine and CO stretching of the amides groups, respectively, suggesting the presence of chitosan.66,67 Apart from these bands, the spectrum of Ins@MPs is very similar to the MF spectra, suggesting an efficient encapsulation of InsLip-CHT by MF. The microparticles were characterized in terms of loading degree, which was 0.39 ± 0.02%.
Then, the release profile of the insulin was evaluated. To mimic the gastric conditions, the Ins@MPs were dispersed for 2 h in simulated gastric fluid (SGF) at pH 1.2. Afterwards, the microparticles were dispersed in fasted state simulated intestinal fluid (FaSSIF) at pH 6.8, in order to test their behavior after the transition from the stomach to the small intestine conditions. Regarding the pH-responsiveness of MF-based microparticles, TEM imaging and the size measurement over the time have shown that MF microparticles remain intact at pH 1.2, but upon pH increase to 6.8, the enteric polymer starts to dissolve, releasing the CHT-based NPs, and after 2 h, the polymer is completely dissolved.67–69 With respect to the pH of the small intestine in the fed or fasted state, McConnell et al. reported that the fed state of the animal (rats or mice) had no significant effect on the pH.70 Thus, it is hypothesized that the insulin release might not be affected by food intake, although this needs to be still confirmed in the future for our particular system. The pH-responsiveness of MF was observed in the release studies. Fig. 2A shows that there was no release in SGF during the first 2 h, due to the successful encapsulation in the enteric polymer (MF) that is not soluble at a pH below 6.0.71 However, MF dissolved when the pH was changed to 6.8, exposing InsLip-CHT to the solution, and a burst release of 7.5% of insulin was observed in the first minutes, whereas after 24 h an insulin release up to 25% was achieved. These results corroborate that, in this work, MF plays a relevant role in the nanoparticle protection, since previous works have demonstrated that in the absence of MF, at pH 1.2, insulin would be released from CHT-based nanoparticles.67,68
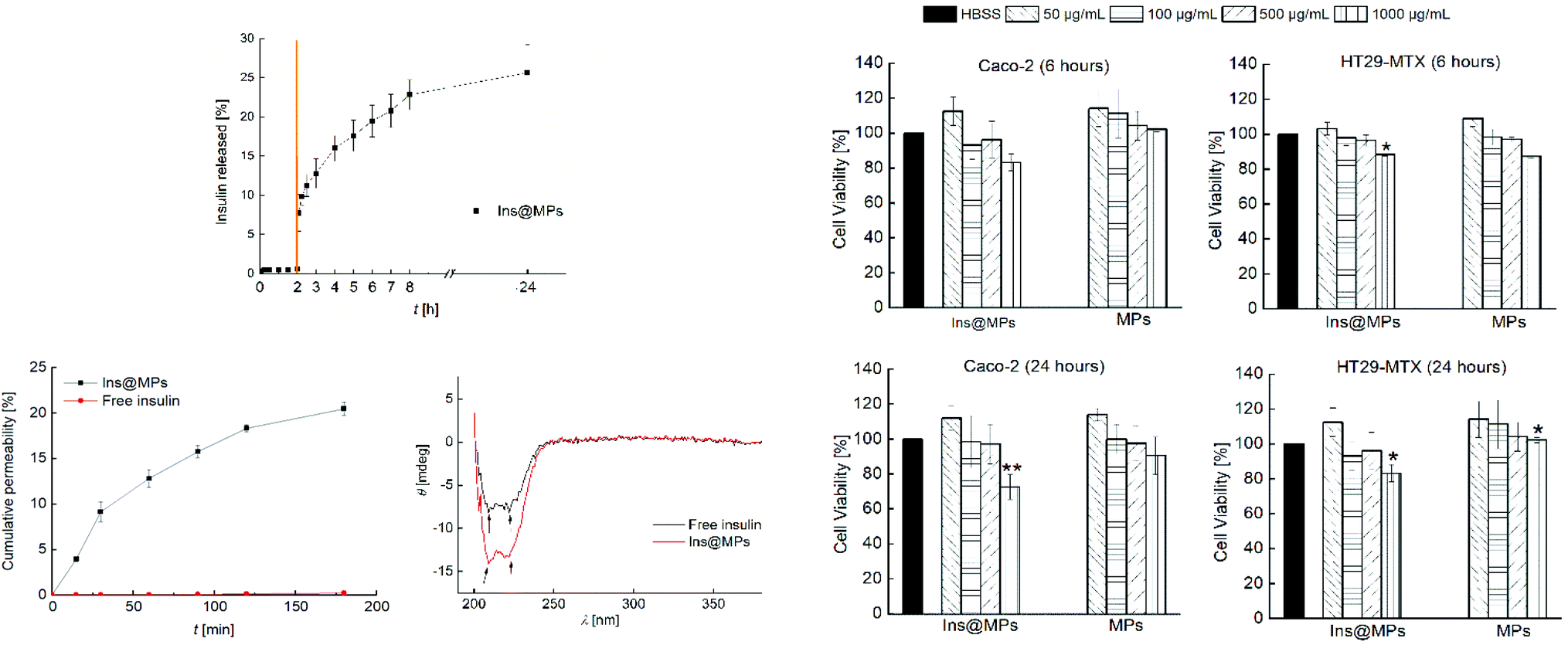 | ||
| Fig. 2 (A) Insulin release profile from insulin-loaded nano-in-microparticles (Ins@MPs), in the first 2 h in SGF (pH 1.2) and then in FaSSIF (pH 6.8) for 24 h at 37 °C. The transition from SGF to FaSSIF is represented by an orange line. (B) Caco-2 and HT29-MTX cell line viability when exposed to different concentrations of nano-in-microparticles with and without insulin, after 6 h and 24 h of incubation at 37 °C. All the data were compared to the negative control (HBSS–HEPES buffer, pH 7.4). (C) a. Insulin permeation profile across Caco-2 and HT29-MTX co-cultured (ratio of 9:1) cell monolayers. The Ins@MPs were incubated with FaSSIF (pH 6.8) in the apical compartment and HBSS–HEPES (pH 7.4) in the basolateral compartment. All the experiments were carried out for 3 h at 37 °C, using an orbital shaker at 100 rpm. b. Circular dichroism of permeated free insulin at 2.3 × 10−2 μg mL−1 (black line) and insulin from Ins@MPs at 1.1 μg mL−1 (red line), in HBSS–HEPES (pH 7.4). The level of significance was set at the probabilities of *p < 0.05 and **p < 0.01. The results are expressed as mean ± S.D. (n = 3). | ||
Next, cell viability assays were carried out in order to evaluate the toxicity of the nano-in-microparticles with and without insulin. The intestinal cell lines Caco-2 and HT29-MTX were used in this study. Human colon carcinoma Caco-2 cells have been used over the years as a model of the intestinal barrier. Upon differentiation, Caco-2 cells express similar characteristics to the small enterocytes.72 The HT29 cells (colon adenocarcinoma) are frequently used as a model of epithelial cells of the intestinal mucosa. When they are treated with methotrexate (MTX), they become resistant to cytostatic drugs and an adhesive mucus layer is formed on the apical side of a monolayer cell culture.73 The in vitro cytotoxicity assay was conducted by exposing both cell lines to the nano-in-microparticles with and without insulin for 6 and 24 h, in concentrations ranging from 50 μg mL−1 to 1000 μg mL−1 of microparticles (corresponding to 0.195 μg mL−1 to 0.390 μg mL−1 of insulin, respectively) (Fig. 2B). After the first 6 h, for the Caco-2 cells, the nano-in-microparticles without insulin did not show the signs of cytotoxicity, with a cell viability above 80%, for all concentrations tested. For Ins@MPs, a cell viability decrease was observed for the higher concentration used (1000 μg mL−1); however, it was not statistically significant (p < 0.05). Furthermore, after 24 h, a slight decrease in the viability of Caco-2 cells (13% for Ins@MPs and 12% for the nano-in-microparticles) was observed at a concentration of 1000 μg mL−1. Since at pH 6.8, the cells were exposed to the chitosan from InsLip-CHT, this decrease in their viability can be related to the Caco-2 cytotoxicity dependence on the chitosan concentration, as reported in previous studies.74,75 Regarding the HT29-MTX cell line, when the nano-in-microparticles were in contact with cells, even at higher concentrations, their viability remained higher than 80%, for both time points tested. This low cytotoxicity is in accordance with other studies reported in the literature and can be explained by the presence of mucus on the HT29-MTX surface, which acts as a barrier.8
Once verified the cell viability, the insulin permeability across a monolayer was evaluated. In order to reach the bloodstream, which is mandatory for a successful insulin drug delivery, insulin must cross from the apical to the basolateral side of the intestinal epithelium. To closely mimic the in vivo physiological intestinal environment, Caco-2 and HT29-MTX were co-cultured for 21 days to form a monolayer.8 The Ins@MPs were incubated with the cells for 3 h at pH 6.8 (apical compartment), whereas the pH in the basolateral compartment was kept at 7.4. The experiment was carried out using free insulin as a control. The permeated insulin dissolved in the basolateral compartment was quantified by HPLC. As observed in Fig. 2C(a), a higher permeation was observed for the Ins@MPs than for free insulin (Papp of 2.27 × 10−5 cm s−1 and 1.19 × 10−7 cm s−1, respectively). Since the pH in the apical compartment was 6.8, the existing enteric polymer (MF) was dissolved, exposing the chitosan-coated liposomes. These results are in accordance with those of the release studies, where insulin release occurred only at pH 6.8. At each time-point, transepithelial electrical resistance (TEER) was measured (Fig. S1, ESI†) in order to monitor the ion conductance across the monolayers. This phenomenon is characterized by the opening of the monolayer tight junctions. Once opened, the ion transport across the paracellular route76,77 will be faster. In this nanosystem, TEER values tend to decrease faster for the Ins@MPs than for free insulin, as expected. Some studies have reported that chitosan tends to bind with integrins from the membrane of intestinal epithelial cells, activating a signal pathway that involves protein kinases, such as focal adhesion kinase (FAK) and Src tyrosine kinase.78 In turn, these kinases induce degradation and translocation of the tight junctions proteins, such as cytoplasmic proteins, occludin and claudins (e.g., CLDN4).79,80 This activation mechanism promotes the reversible opening of the tight junctions, enhancing the permeability of the molecules through a paracellular pathway.81 In contrast, the decrease of the TEER values observed for free insulin, after 120 min, was not expected, since in the drug permeability assays, no insulin was observed in the basolateral compartment. During the insulin permeation, it was important to evaluate whether its activity remains the same. Thus, far UV region circular dichroism, a tool widely used to identify the structural secondary elements of peptides,82 was performed in order to identify the α-helix and β-sheet structures of the permeated insulin and to compare them with those of the native form of insulin (Fig. S2, ESI†). As described elsewhere, the insulin's α-helix conformation is characterized by a valley at 208 nm and a shoulder at 223 nm.27,83 These properties can also be clearly observed in the permeated free insulin and insulin released from liposomes (Fig. 2C(b)). In addition, when two insulin monomers associate themselves, antiparallel β-structures are created. This phenomenon can be observed through the ratio of θ208 nm/θ223 nm.27 The insulin dissolved in saline citric solution at pH 2 (free-insulin) presents a ratio of 0.949, whereas the insulin from Ins@MPs presents a ratio of 1.035, which is close to the value of native insulin (1.088). Therefore, regarding these observations, it is possible to conclude that the insulin's structure remained preserved after being loaded into the liposomes and permeated through the Caco-2/HT29-MTX monolayers.
In order to evaluate the Ins@MP interaction with both cell lines (Caco-2 and HT29-MTX), confocal fluorescence microscopy was used for qualitative analysis, in which a fluorescent membrane dye, 4-(4-dihexadecylaminostyryl)-N-methylpyridinium iodide (DiA), was used for imaging purposes. In this way, the experiments were carried out with 500 μg mL−1 of DiA-loaded liposomes (DiALip) and DiALip-loaded chitosan nanoparticles (DiALip-CHT) for 6 h. The nanoparticles coated with MF were not evaluated, since at this pH the polymer is completely dissolved, as shown in the release experiments. Fig. 3A shows the interaction of both nanoparticles with Caco-2 cells. It is possible to observe that DiALip were not internalized, only interacting with the cell's surface. This observation is supported by the presence of the poly(ethyleneglycol)2000 (PEG2000) in the liposomes. PEG, grafted onto the surface of the liposomes, promotes an elevated liposome blood circulation, by the decrease of macrophage recognition and opsonization.84,85 In addition, the slightly negative charge from DSPE-PEG2000 can promote a steric barrier with the cells, and thus, DiALip take more time to be internalized.86 In contrast, the liposomes coated with chitosan were easily internalized. Regarding Fig. 3A, it is possible to observe a decrease of fluorescence intensity on InsLip-CHT. This phenomenon might be due to the chitosan-based nanoparticle internalization, whereas the DiALip were on the cell membrane. This phenomenon was also observed in the quantitative flow cytometry study, indicating the chitosan capability to enhance the cellular uptake. A difference of more than 10% of the DiALip-CHT association with the cells shows the higher interactions of this system, compared with DiA-loaded liposomes (Fig. 3B). Confocal images of the HT29-MTX cells (Fig. 3C) did not show notable interactions between the cells and the nanoparticles. However, this difference was detected in the flow cytometry quantitative analysis (Fig. 3D). These different observations could be due to the confocal images showing part of the sample, whereas flow cytometry quantifies the entire sample. In this way, regarding Fig. 3D, an uptake of over 40% of the chitosan-coated liposomes was observed, whereas no uptake was detected for liposomes without chitosan. This phenomenon was also observed in previous works69,87 and it might be related to the chitosan mucoadhesion properties, driven by the electrostatic interactions between the positive charge of the chitosan and the negative charge of mucins.88
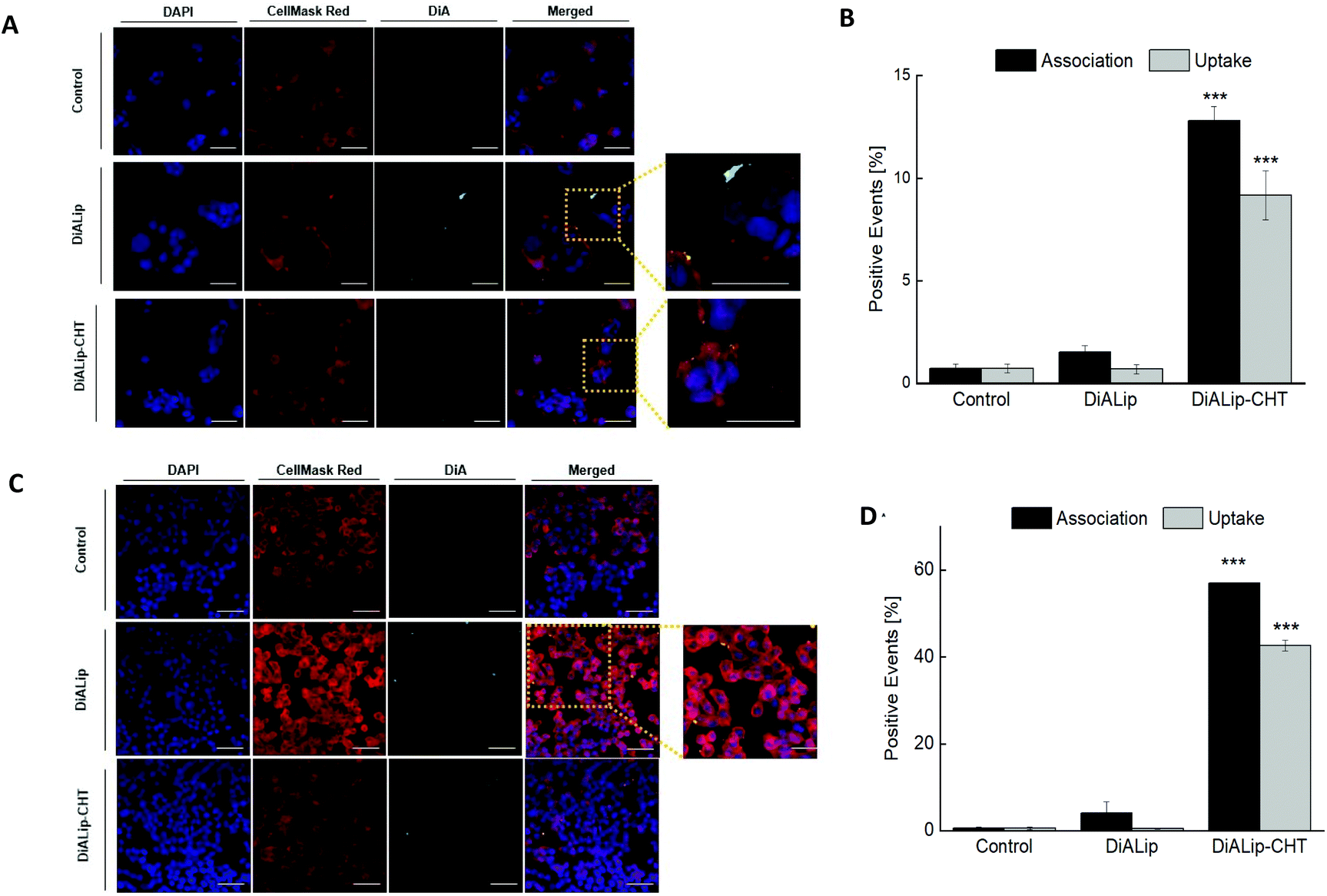 | ||
| Fig. 3 (A) and (C) 2D confocal fluorescence microscope images of the interactions of DiA-loaded liposomes (DiALip) and DiA-loaded liposomes coated by chitosan (DiALip-CHT) at a concentration of 500 μg mL−1 with Caco-2 cells and HT29-MTX cells, respectively. Blue: cell nucleus stained with DAPI (4′,6′-diamidino-2-phenyl-indole); red: cell membranes stained with CellMask Red; green: DiA (4-(4-dihexadecylaminostyryl)-N-methylpyridinium iodide)-encapsulated liposomes (scale bar: 50 μm). (B) and (D) Flow cytometry quantitative analysis of the interactions between the nanoparticles and the Caco-2 cells and HT29-MTX, respectively. The experiments were carried out at 37 °C after 6 h of incubation time. The level of significance was set at a probability of ***p < 0.001. | ||
Conclusions
In this work, a multistage oral insulin delivery system using a two-step microfluidic process was successfully designed and developed. Insulin was efficiently encapsulated (91 ± 4%), into PEGylated liposomes using a microfluidic technique, overcoming the common liposome batch production drawbacks and obtaining a narrow size distribution. This allowed the successful encapsulation of the nanoparticles in an enteric polymer, preventing their degradation under harsh gastric conditions. In vitro release studies showed insulin release starting only at pH 6.8, above the pKa of MF, demonstrating an efficient protection under gastric acidic conditions. 2D confocal microscope images and flow cytometry analysis supported that the mucoadhesive properties of the nanoparticles were enhanced by the physical coating with chitosan on the surface of the liposomes. In addition, insulin permeability and circular dichroism studies showed a strong improvement in the insulin permeation across the intestinal epithelium without compromising its activity, respectively. Moreover, low cytotoxicity of the Ins@MPs in the intestinal cells was observed. Overall, we obtained a pH-responsive mucoadhesive cell-mimicking system for protein/peptide delivery that represents a promising strategy for a continuous, scalable and high throughput method to produce oral targeted systems.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
C. Costa and A. Aguiar-Ricardo are grateful for the financial support of the Associate Laboratory for Green Chemistry (LAQV), which is financed by national funds from FCT/MCTES (UIDB/50006/2020). C. Costa also acknowledges the FCT/MCTES for the grant PD/BD/142880/2018 and for the travel grant. M. L. Corvo acknowledges the financial support from Research Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Lisbon, Portugal, which is supported in part by UID/DTP/04138/2020 from FCT/MCTES, Portugal. H. A. Santos acknowledges financial support from the HiLIFE Research Funds and the Sigrid Jusélius Foundation.Notes and references
- M. Goldberg and I. Gomez-Orellana, Nat. Rev. Drug Discovery, 2003, 2, 289–295 CrossRef CAS PubMed.
- J. Huang, Q. Shu, L. Wang, H. Wu, A. Y. Wang and H. Mao, Biomaterials, 2015, 39, 105–113 CrossRef CAS PubMed.
- T. X. Nguyen, L. Huang, M. Gauthier, G. Yang and Q. Wang, Nanomedicine, 2016, 11, 1169–1185 CrossRef CAS PubMed.
- A. Banerjee, K. Ibsen, T. Brown, R. Chen, C. Agatemor and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7296–7301 CrossRef CAS PubMed.
- L. N. Thwala, V. Préat and N. S. Csaba, Expert Opin. Drug Delivery, 2017, 14, 23–36 CrossRef CAS PubMed.
- P. Batista, P. M. Castro, A. R. Madureira, B. Sarmento and M. Pintado, Peptides, 2018, 101, 112–123 CrossRef CAS PubMed.
- E. M. Pridgen, F. Alexis and O. C. Farokhzad, Expert Opin. Drug Delivery, 2015, 12, 1459–1473 CrossRef PubMed.
- N. Shrestha, F. Araújo, M. A. Shahbazi, E. Mäkilä, M. J. Gomes, B. Herranz-Blanco, R. Lindgren, S. Granroth, E. Kukk, J. Salonen, J. Hirvonen, B. Sarmento and H. A. Santos, Adv. Funct. Mater., 2016, 26, 3405–3416 CrossRef CAS.
- Y. Xu, Y. Zheng, L. Wu, X. Zhu, Z. Zhang and Y. Huang, ACS Appl. Mater. Interfaces, 2018, 10, 9315–9324 CrossRef CAS PubMed.
- P. Fonte, F. Araújo, S. Reis and B. Sarmento, J. Diabetes Sci. Technol., 2013, 7, 520–531 CrossRef PubMed.
- W. Fan, D. Xia, Q. Zhu, X. Li, S. He, C. Zhu, S. Guo, L. Hovgaard, M. Yang and Y. Gan, Biomaterials, 2018, 151, 13–23 CrossRef CAS PubMed.
- A. Abramson, E. Caffarel-Salvador, M. Khang, D. Dellal, D. Silverstein, Y. Gao, M. R. Frederiksen, A. Vegge, F. Hubálek, J. J. Water, A. V. Friderichsen, J. Fels, R. K. Kirk, C. Cleveland, J. Collins, S. Tamang, A. Hayward, T. Landh, S. T. Buckley, N. Roxhed, U. Rahbek, R. Langer and G. Traverso, Science, 2019, 363, 611–615 CrossRef CAS PubMed.
- G. Sharma, K. Wilson, C. F. van der Waalse, N. Sattar, J. R. Petrie and M. N. V. Ravi Kumar, Eur. J. Pharm. Biopharm., 2010, 76, 159–169 CrossRef CAS PubMed.
- Y. Li, W. Yokoyama, S. Xu, S. Zhu, J. Ma and F. Zhong, J. Funct. Foods, 2017, 30, 134–141 CrossRef CAS.
- F. Araújo, N. Shrestha, M. A. Shahbazi, D. Liu, B. Herranz-Blanco, E. M. Mäkilä, J. J. Salonen, J. T. Hirvonen, P. L. Granja, B. Sarmento and H. A. Santos, ACS Nano, 2015, 9, 8291–8302 CrossRef PubMed.
- K. Sonaje, Y. J. Chen, H. L. Chen, S. P. Wey, J. H. Juang, H. N. Nguyen, C. W. Hsu, K. J. Lin and H. W. Sung, Biomaterials, 2010, 31, 3384–3394 CrossRef CAS PubMed.
- J. P. Martins, R. D. Auria, D. Liu, F. Fontana, M. P. A. Ferreira, A. Correia, M. Kemell, K. Moslova, E. Mäkilä, J. Salonen, L. Casettari, J. Hirvonen, B. Sarmento and H. A. Santos, Small, 2018, 1800462, 1–11 Search PubMed.
- T. Andreani, C. P. Kiill, A. L. R. d. Souza, J. F. Fangueiro, L. Fernandes, S. Doktorovová, D. L. Santos, M. L. Garcia, M. P. D. Gremião, E. B. Souto and A. M. Silva, Colloids Surf., B, 2014, 123, 916–923 CrossRef CAS PubMed.
- C. Y. Wong, H. Al-Salami and C. R. Dass, Int. J. Pharm., 2018, 549, 201–217 CrossRef CAS PubMed.
- M. Niu, Y. Tan, P. Guan, L. Hovgaard, Y. Lu, J. Qi, R. Lian, X. Li and W. Wu, Int. J. Pharm., 2014, 460, 119–130 CrossRef CAS PubMed.
- A. D. Bangham, M. M. Standish and J. C. Watkins, J. Mol. Biol., 1965, 13, 238–252 CrossRef CAS PubMed.
- J. Parmentier, M. M. M. Becker, U. Heintz and G. Fricker, Int. J. Pharm., 2011, 405, 210–217 CrossRef CAS PubMed.
- H. M. Patel and B. E. Ryman, FEBS Lett., 1976, 62, 10–13 CrossRef.
- G. Gregoriadis, P. D. Leathwood and B. E. Ryman, FEBS Lett., 1971, 14, 95–99 CrossRef CAS PubMed.
- S. Hu, M. Niu, F. Hu, Y. Lu, J. Qi, Z. Yin and W. Wu, Int. J. Pharm., 2013, 441, 693–700 CrossRef CAS PubMed.
- H. He, Y. Lu, J. Qi, Q. Zhu, Z. Chen and W. Wu, Acta Pharm. Sin. B, 2019, 9, 36–48 CrossRef PubMed.
- M. Niu, Y. Lu, L. Hovgaard and W. Wu, Int. J. Nanomed., 2011, 6, 1155–1166 CrossRef CAS PubMed.
- J. Yu, Y. Zhang, J. Wang, D. Wen, A. R. Kahkoska, J. B. Buse and Z. Gu, Nano Res., 2019, 12, 1539–1545 CrossRef CAS.
- K. Iwanaga, S. Ono, K. Narioka, K. Morimoto, M. Kakemi, S. Yamashita, M. Nango and N. Oku, Int. J. Pharm., 1997, 157, 73–80 CrossRef CAS.
- A. Wang, T. Yang, W. Fan, Y. Yang, Q. Zhu, S. Guo, C. Zhu, Y. Yuan, T. Zhang and Y. Gab, Adv. Healthcare Mater., 2018, 1801123, 1–11 CrossRef PubMed.
- A. Bigdeli, S. Palchetti, D. Pozzi, M. R. Hormozi-Nezhad, F. Baldelli Bombelli, G. Caracciolo and M. Mahmoudi, ACS Nano, 2016, 10, 3723–3737 CrossRef CAS PubMed.
- M. Luisa Corvo, J. C. S. Jorge, R. Van't Hof, M. E. M. Cruz, D. J. A. Crommelin and G. Storm, Biochim. Biophys. Acta, Biomembr., 2002, 1564, 227–236 CrossRef CAS.
- H. Elsana, T. O. B. Olusanya, J. Carr-wilkinson, S. Darby, A. Faheem and A. A. Elkordy, Sci. Rep., 2019, 9, 1–17 CrossRef CAS PubMed.
- C. Charcosset, A. Juban, J. P. Valour, S. Urbaniak and H. Fessi, Chem. Eng. Res. Des., 2015, 94, 508–515 CrossRef CAS.
- I. Takeuchi, N. Kishi, K. Shiokawa, H. Uchiro and K. Makino, Colloid Polym. Sci., 2018, 296, 1137–1144 CrossRef CAS.
- D. Szoka, F. Olson, F. Heath, T. Vail, W. Mayhew and E. Papahadjopoulos, Biochim. Biophys. Acta, Biomembr., 1980, 601, 559–571 CrossRef.
- R. Ran, A. P. J. Middelberg and C. X. Zhao, Colloids Surf., B, 2016, 148, 402–410 CrossRef CAS PubMed.
- D. Liu, S. Cito, Y. Zhang, C. F. Wang, T. M. Sikanen and H. A. Santos, Adv. Mater., 2015, 27, 2298–2304 CrossRef CAS PubMed.
- N. Dimov, E. Kastner, M. Hussain, Y. Perrie and N. Szita, Sci. Rep., 2017, 7, 1–13 CrossRef CAS PubMed.
- P. M. Valencia, O. C. Farokhzad, R. Karnik and R. Langer, Nat. Nanotechnol., 2012, 7, 623–629 CrossRef CAS PubMed.
- S. Joshi, M. T. Hussain, C. B. Roces, G. Anderluzzi, E. Kastner, S. Salmaso, D. J. Kirby and Y. Perrie, Int. J. Pharm., 2016, 514, 160–168 CrossRef CAS PubMed.
- J. P. Rolland, R. M. Van Dam, D. A. Schorzman, S. R. Quake and J. M. DeSimone, J. Am. Chem. Soc., 2004, 126, 2322–2323 CrossRef CAS PubMed.
- J. P. Martins, G. Torrieri and H. A. Santos, Expert Opin. Drug Delivery, 2018, 1–11 Search PubMed.
- D. Bodas and C. Khan-Malek, Sens. Actuators, B, 2007, 123, 368–373 CrossRef CAS.
- D. Liu, H. Zhang, F. Fontana, J. T. Hirvonen and H. A. Santos, Lab Chip, 2017, 17, 1856–1883 RSC.
- D. Carugo, E. Bottaro, J. Owen, E. Stride and C. Nastruzzi, Sci. Rep., 2016, 6, 1–15 CrossRef PubMed.
- V. M. Shah, D. X. Nguyen, P. Patel, B. Cote, A. Al-Fatease, Y. Pham, M. G. Huynh, Y. Woo and A. W. Alani, Nanomedicine, 2019, 18, 146–156 CrossRef CAS PubMed.
- D. Van Swaay and A. Demello, Lab Chip, 2013, 13, 752–767 RSC.
- G. T. Vladisavljevic, A. Laouini, C. Charcosset, H. Fessi, H. C. H. Bandulasena and R. G. Holdich, Colloids Surf., A, 2014, 458, 168–177 CrossRef CAS.
- W. Li, L. Zhang, X. Ge, B. Xu, W. Zhang, L. Qu, C. H. Choi, J. Xu, A. Zhang, H. Lee and D. A. Weitz, Chem. Soc. Rev., 2018, 47, 5646–5683 RSC.
- R. K. Shah, H. C. Shum, A. C. Rowat, D. Lee, J. J. Agresti, A. S. Utada, L. Y. Chu, J. W. Kim, A. Fernandez-Nieves, C. J. Martinez and D. A. Weitz, Mater. Today, 2008, 11, 18–27 CrossRef CAS.
- W. Li, L. Zhang, X. Ge, B. Xu, W. Zhang, L. Qu, C. H. Choi, J. Xu, A. Zhang, H. Lee and D. A. Weitz, Chem. Soc. Rev., 2018, 47, 5646–5683 RSC.
- W. Li, D. Liu, H. Zhang, A. Correia, E. Mäkilä, J. Salonen, J. Hirvonen and H. A. Santos, Acta Biomater., 2017, 48, 238–246 CrossRef CAS PubMed.
- C. Y. Wong, H. Al-salami and C. R. Dass, Int. J. Pharm., 2017, 537, 223–244 CrossRef PubMed.
- F. L. Lopez, T. B. Ernest, C. Tuleu and M. O. Gul, Expert Opin. Drug Delivery, 2015, 12, 1727–1740 CrossRef CAS PubMed.
- E. T. L. Lau, K. J. Steadman, M. Mak, J. A. Y. Cichero and L. M. Nissen, Aust. J. Pharm., 2015, 96, 68–71 Search PubMed.
- J. M. Zook and W. N. Vreeland, Soft Matter, 2010, 6, 1352–1360 RSC.
- E. Kastner, V. Verma, D. Lowry and Y. Perrie, Int. J. Pharm., 2015, 485, 122–130 CrossRef CAS PubMed.
- A. Jahn, W. N. Vreeland, D. L. Devoe, L. E. Locascio and M. Gaitan, Langmuir, 2007, 23, 6289–6293 CrossRef CAS PubMed.
- D. D. Lasic and F. J. Martin, J. Membr. Sci., 1990, 50, 215–222 CrossRef CAS.
- K. Muramatsu, Y. Maitani and T. Nagai, Chem. Pharm. Bull., 1996, 19, 1055–1058 CrossRef CAS PubMed.
- O. Mertins and R. Dimova, Langmuir, 2011, 27, 5506–5515 CrossRef CAS PubMed.
- J. D. Fisher, A. P. Acharya and S. R. Little, Clin. Immunol., 2015, 160, 24–35 CrossRef CAS PubMed.
- M. Fukasawa and S. Obara, Chem. Pharm. Bull., 2004, 52, 1391–1393 CrossRef CAS.
- D. T. Friesen, R. Shanker, M. Crew, D. T. Smithey, W. J. Curatolo and J. A. S. Nightingale, Mol. Pharm., 2008, 5, 1003–1019 CrossRef CAS PubMed.
- A. A. P. Mansur and H. S. Mansur, Nanoscale Res. Lett., 2015, 10, 172 CrossRef PubMed.
- S. Bertoni, Z. Liu, A. Correia, J. P. Martins, A. Rahikkala, F. Fontana, M. Kemell, D. Liu, B. Albertini, N. Passerini, W. Li and H. A. Santos, Adv. Funct. Mater., 2018, 28, 1–11 Search PubMed.
- N. Shrestha, M. A. Shahbazi, F. Araújo, E. Mäkilä, J. Raula, E. I. Kauppinen, J. Salonen, B. Sarmento, J. Hirvonen and H. A. Santos, Biomaterials, 2015, 68, 9–20 CrossRef CAS PubMed.
- J. P. Martins, D. Liu, F. Fontana, M. P. A. Ferreira, A. Correia, S. Valentino, M. Kemell, K. Moslova, E. Mäkilä, J. Salonen, J. Hirvonen, B. Sarmento and H. A. Santos, ACS Appl. Mater. Interfaces, 2018, 10, 44354–44367 CrossRef CAS.
- E. L. McConnell, A. W. Basit and S. Murdan, J. Pharm. Pharmacol., 2008, 60, 63–70 CrossRef CAS PubMed.
- A. L. Sarode, S. Obara, F. K. Tanno, H. Sandhu, R. Iyer and N. Shah, Carbohydr. Polym., 2014, 101, 146–153 CrossRef CAS PubMed.
- Y. Sambuy, I. De Angelis, G. Ranaldi, M. L. Scarino, A. Stammati and F. Zucco, Cell Biol. Toxicol., 2005, 21, 1–26 CrossRef CAS PubMed.
- E. Leteurtre, V. Gouyer, K. Rousseau, O. Moreau, A. Barbat, D. Swallow, G. Huet and T. Lesuffleur, Biol. Cell, 2004, 96, 145–151 CrossRef CAS PubMed.
- P. Opanasopit, P. Aumklad, J. Kowapradit, T. Ngawhiranpat, A. Apirakaramwong, T. Rojanarata and S. Puttipipatkhachorn, Pharm. Dev. Technol., 2007, 12, 447–455 CrossRef CAS PubMed.
- S. Y. Chae, M. K. Jang and J. W. Nah, J. Controlled Release, 2005, 102, 383–394 CrossRef CAS PubMed.
- X. Boulenc, T. Breul, J. Gautier, H. Joyeux, C. Roques, Y. Berger and G. Fabre, Int. J. Pharm., 1995, 123, 71–83 CrossRef CAS.
- B. Srinivasan, A. R. Kolli, M. B. Esch, H. E. Abaci, M. L. Shuler and J. J. Hickman, J. Lab. Autom., 2015, 20, 107–126 CrossRef CAS PubMed.
- L. W. Hsu, P. L. Lee, C. T. Chen, F. L. Mi, J. H. Juang, S. M. Hwang, Y. C. Ho and H. W. Sung, Biomaterials, 2012, 33, 6254–6263 CrossRef CAS PubMed.
- J. M. Smith, M. Dornish and E. J. Wood, Biomaterials, 2005, 26, 3269–3276 CrossRef CAS PubMed.
- Y. H. Chen, Q. Lu, E. E. Schneeberger and D. A. Goodenough, Mol. Biol. Cell, 2000, 11, 849–862 CrossRef CAS PubMed.
- M. Collado-González, Y. G. Espinosa and F. M. Goycoolea, Biomimetics, 2019, 4, 32 CrossRef PubMed.
- N. Greenfield and G. D. Fasman, Biochemistry, 1969, 8, 4108–4116 CrossRef CAS PubMed.
- D. E. S. Valerie, A. Ivancic, C. A. Krasinski, Q. Zheng, R. J. Meservier and N. D. Lazo, Biosci. Rep., 2018, 38, BSR20181555 CrossRef PubMed.
- D. Pozzi, V. Colapicchioni, G. Caracciolo, S. Piovesana, A. L. Capriotti, S. Palchetti, S. De Grossi, A. Riccioli, H. Amenitsch and A. Laganà, Nanoscale, 2014, 6, 2782–2792 RSC.
- N. Dan, Biochim. Biophys. Acta, Biomembr., 2002, 1564, 343–348 CrossRef CAS.
- C. R. Miller, B. Bondurant, S. D. McLean, K. A. McGovern and D. F. O'Brien, Biochemistry, 1998, 37, 12875–12883 CrossRef CAS PubMed.
- M. Lopes, N. Shrestha, A. Correia, M. A. Shahbazi, B. Sarmento, J. Hirvonen, F. Veiga, R. Seiça, A. Ribeiro and H. A. Santos, J. Controlled Release, 2016, 232, 29–41 CrossRef CAS PubMed.
- I. A. Sogias, A. C. Williams and V. V. Khutoryanskiy, Biomacromolecules, 2008, 9, 1837–1842 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, experimental details and additional data. See DOI: 10.1039/d0bm00743a |
| ‡ These authors have contributed for this work equally. |
| This journal is © The Royal Society of Chemistry 2020 |