
A self-healing hydrogel with pressure sensitive photoluminescence for remote force measurement and healing assessment†
Ming
Li‡
 a,
Weijun
Li‡
a,
Wei
Cai
a,
Xiaojie
Zhang
a,
Zhihang
Wang
a,
Jason
Street
b,
Wee-Jun
Ong
*cd,
Zhenhai
Xia
e and
Quan
Xu
*a
a,
Weijun
Li‡
a,
Wei
Cai
a,
Xiaojie
Zhang
a,
Zhihang
Wang
a,
Jason
Street
b,
Wee-Jun
Ong
*cd,
Zhenhai
Xia
e and
Quan
Xu
*a
aState Key Laboratory of Heavy Oil Processing, China University of Petroleum, Beijing, 102249, China. E-mail: xuquan@cup.edu.cn
bDepartment of Sustainable Bioproducts, Mississippi State University, Mississippi 39762, USA
cSchool of Energy and Chemical Engineering, Xiamen University Malaysia, Selangor Darul Ehsan 43900, Malaysia. E-mail: weejun.ong@xmu.edu.my; ongweejun@gmail.com; Web: http://sites.google.com/site/wjongresearch/
dCollege of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China
eDepartment of Materials Science and Engineering and Department of Chemistry, University of North Texas, Denton, Texas 76203, USA
First published on 17th January 2019
Abstract
Light-guiding materials capable of total internal reflection, remote mechanical force sensing and self-healing are appealing in emerging fields including robotics and optical force measuring instruments. However, achieving all these features in a single material remains challenging at present. Herein, we have fabricated a fluorescence-responsive self-healing hydrogel with a triple network structure, which exhibits a 100% recovery in tensile strength after healing in air for 30 s and a 90% recovery in tensile strength after healing in water for 60 s. Furthermore, this material can resist a rotation of 1800° without breaking at the healed site. As the fluorescence excitation intensity of the hydrogel shows a good correspondence with the forces exerted on the hydrogel, the forces and the self-healing efficiency could be determined by measuring the intensity of the excitation peak. The stress states of the hydrogel in different liquids could be remotely monitored, eliminating the effect of surface contacts. With these sensing and self-healing abilities all in one, self-healing luminescent materials could be applied to tissue engineering, photoresponsive biosensors, flexible light guiding devices, structural health monitoring, etc.
Conceptual insightsSelf-healing hydrogels are restricted by their relatively long healing times, poor healing performance in various environments and unstable optical performance. To address this paradox, we fabricated a fluorescence-responsive self-healing hydrogel, which exhibits excellent immediate self-healing properties in different media. The hydrogel could also be rotated 1800° at the damaged site after the site underwent self-healing without breaking. The hydrogel exhibited stable fluorescence response properties in different environments. Besides, the hydrogel exhibited a pressure reshaping effect. The damaged hydrogel showed good performance after reshaping under a certain pressure. Moreover, the fluorescence excitation intensity of the hydrogel revealed a unique correspondence with the external pressure that the hydrogel received and with the hydrogel's self-repairing efficiency. Therefore, the external pressure force and the self-repairing efficiency could be determined from the intensity corresponding to the hydrogel excitation peak. |
1. Introduction
Self-healing dimming materials exhibit interesting color changes when exposed to external stimuli such as light,1 temperature,2 pH,3,4 and force.5–7 These interesting properties are important for the advancement of new color-developing and fluorescent color-changing materials for applications in sensors, structural health monitoring systems, optical force measuring devices, flexible devices, tissue engineering and regenerative medicine.8–11 Hydrogels are hydrophilic polymers which swell in water but are insoluble in water. These materials have a high water content, good flexibility, and a strong ability to penetrate into small molecules. The unique network structure provides hydrogels with excellent biocompatibility and self-healing abilities.12,13 In recent years, optical self-healing hydrogels that have both self-healing properties and the ability to respond to external stimuli have attracted widespread attention.14–18 However, in practical applications, the self-healing functions of hydrogels are characterized by relatively long healing times,19–22 stimulation dependences,23–25 poor healing performance when in a humid environment or underwater,26–28 and unstable optical performance.29 It would be of great interest to prepare hydrogels with ultra-fast self-healing properties (no external stimuli, multiple environmental adaptations) and stable optical responses.Materials with self-healing properties or optical response properties have been extensively studied due to their potential applications in many fields. Currently, their self-healing is achieved through hydrogen bonding, chain entanglement, supramolecular interaction, ionic bond and other non-covalent reactions,23–25 or disulfide bonds, phenyl boronate esters, hydrazide bonds and other covalent reactions,30 whereas their optical response primarily relies on the use of organic chromophores.31 These chromophores can dynamically change their molecular structure or assembly state to achieve an optical response function when stimulated by external factors, but large-scale synthesis of these chromophores remains challenging. Compared to organic chromophores that rely on covalent bond cleavage, the optical response achieved by non-covalent metal coordination is a better alternative because of the dynamic coordination of non-covalent metals. The use of luminescent lanthanide metals (e.g., Eu and Tb)29 that are capable of dynamically coordinating with a polymer provides the polymer with reversible luminescence properties to achieve potential optical on/off effects within the material.32 However, the stimuli response of the existing non-covalent metal coordination polymers occurs in organic solvents,32–34 while the water molecules in hydrogels would hydrate with the lanthanide ions, which not only diminishes the optical response, but also weakens the structural network of the hydrogel.35–37 Due to the difficulty in the large-scale synthesis of organic chromophores and the hydration of lanthanide ions, these materials are not the best choice for the production of new self-healing dimming hydrogels.
In this work, we report a new photoluminescent self-healing hydrogel based on a multi-hydrogen bond network structure that achieves self-healing using reversible physical cross-linking capability and a photoluminescence effect through the uniform distribution of quantum dots in the hydrogel. The resultant photoluminescent hydrogel not only exhibits excellent toughness and stability, but also possesses an excellent self-healing ability. This self-healing hydrogel can work in different media (air, water, oil, salt solution), at ambient temperature (−10 to 50 °C), and in an acidic or basic solution (pH: 1–14) without any external stimulations. Moreover, the hydrogel also shows a pressure reshaping effect: the damaged hydrogel could be re-shaped into a new one at room temperature with the absence of any cracks or previous damage and no significant property degradation. In addition, the hydrogel is non-toxic and has cell compatibility. Furthermore, the stress state of the hydrogel could be detected remotely by the fluorescence effect without considering the contact situation between materials, allowing the resultant measurement to be more accurate. These properties render the photoluminescent self-healing hydrogel useful for a variety of applications including underwater engineering, integrated optics, optical force measuring materials, and biomaterials.
2. Experimental
2.1 Materials
PVA (degree of polymerization: 2699) was purchased from Anhui Wanwei Group Co. Agarose (Analytical Grade) was purchased from Life Technologies Corporation. Chitosan was purchased from Beijing Bellingway Technology Co. Ltd. Sodium tetraborate (Analytical Grade: Na2B4O7·10H2O) was purchased from Tianjin Guangfu Technology Development Co. Ltd. Glycerol was purchased from Beijing Solarbio Science Technology Co. Ltd (Analytical Grade). Sodium citrate dihydrate and ethylenediamine were purchased from Shanghai Aladdin Biochemical Technology Co. Ltd. All reagents were used directly and did not require further purification. Cells and other materials for determining the biocompatibility of the hydrogel were provided by the Institute of Chemistry, Chinese Academy of Sciences.2.2 Preparation of fluorescent quantum dots
The specific method for preparing quantum dots involves mixing 0.375 g of sodium citrate, 60 μL of ethylenediamine and 25 mL of deionized water in a container, and bringing the solution up to a temperature of 160 °C for 8 hours using a one-step hydrothermal method (this example involves the preparation of blue carbon quantum dots). The preparation of other quantum dots follows a similar methodology.2.3 Preparation of fluorescent gels
In this experiment, PVA (10 g), agarose (0.5 g), chitosan (0.5 g) and glycerin (4 mL) were dissolved in deionized water to form a 50 mL solution, and this was stirred by hand for 2 minutes. The solution was oil-bathed at 98 °C for 2 hours to form a solution with a 20 wt% PVA mass fraction. Then 50 mL of the solution was mixed with 50 mL of a borax mixed solution (0.04 mol L−1) and heated with stirring in a 90 °C oil bath for 1 h. The resulting gel was then placed in a mold previously coated with a borax solution and fluorescent quantum dot liquids (green, blue, and red) were added. Next, the mixture was pressed for 2 h to obtain a fluorescent gel material. The purpose of pressing the material was to remove air bubbles from the gel and shape the material to a desired geometry.2.4 Biocompatibility evaluation of polyvinyl alcohol hydrogels
The biocompatibility of the hydrogel was evaluated according to the standards of a cytotoxicity test in vitro, an intradermal reaction test and a delayed-type hypersensitivity assay recommended in GB/T 16886.2.5 Characterization
Scanning electron microscopy (SEM) images were taken with a JEOL JSM-6700F scanning electron microscope. Photographs and videos were taken with a SLR camera (Canon-EOS-760D). Tensile and compression experiments were performed using an Instron universal testing machine (INSTRON 5567). Fourier-transform infrared spectroscopy (FTIR) was performed on a Bruker Tensor 27 spectrophotometer. X-ray photoelectron spectroscopy (XPS) measurements were performed using a Rigaku X-ray diffractometer (D/max-2400) with a Cu Kα X-ray source. Fluorescence performance experiments were performed using UV lamps (12W-ZF-7A-254-365 nm) and purple LED chips (3W-Violet-400–405 nm). The fluorescence intensity of the hydrogel was measured with a Fluorescence Spectrometer (FS5). Cell images were taken with an inverted fluorescence microscope (OLYMPUS IX71).3. Results and discussion
We proposed a multi-network structure of a fluorescence-responsive self-healing hydrogel (hydrogen bond, Fig. S1, ESI†). The hydrogel is non-toxic and has cell compatibility (Fig. S2 and Tables S1 and S2, ESI†). The molecular structure of the hydrogel is shown in Fig. 1. The backbone network was formed during the cooling process of the sol–gel in which chitosan–agarose, chitosan–PVA, and agarose–PVA were strongly crosslinked (hydrogen bond I). These strong hydrogen bonding networks can effectively inhibit the movement of the PVA chain within a certain force range, thus allowing the hydrogel to exhibit high mechanical stability and tensile properties. The self-healing properties of the prepared hydrogel were attributed to the reversible network consisting of two dynamic covalent cross-linking reactions of PVA–borax–PVA and PVA–glycerol–PVA (hydrogen bonds II) in the hydrogel. The quantum dots were uniformly dispersed in the hydrogel to ensure stable fluorescence response (Fig. S3, ESI†) because of the static hydrogen bond network (hydrogen bond III) between PVA and the quantum dots (water-soluble/fat-soluble carbon quantum dots, MXene quantum dots).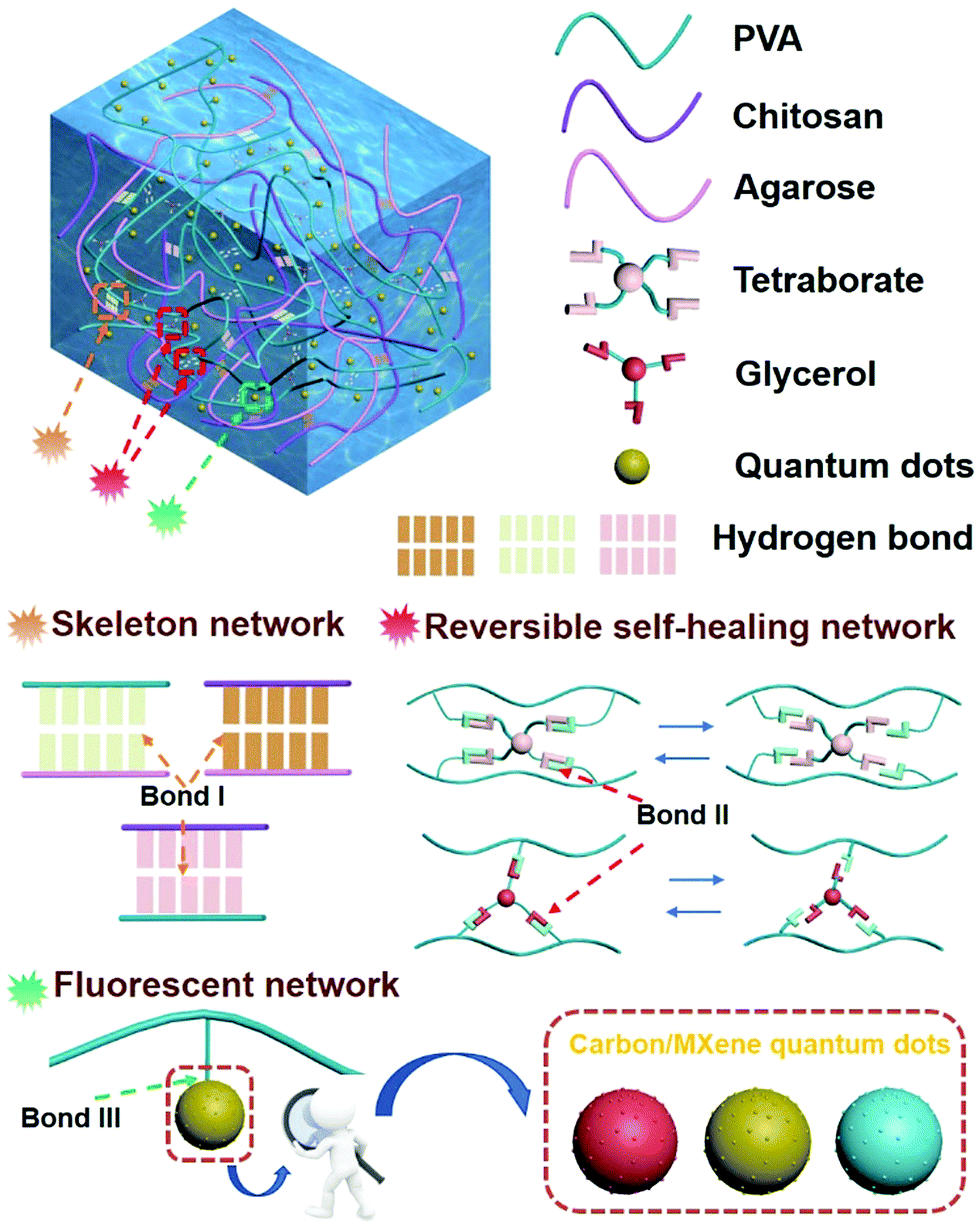 | ||
| Fig. 1 Schematic of the internal structure and bonds of the photoluminescent self-repairing hydrogel. | ||
3.1 Self-healing performance
The tensile and self-healing experiments showed that our hydrogel exhibited excellent mechanical properties (10.21 MPa, Fig. S4, ESI†) and self-healing properties (Fig. 2a and b). The self-healing properties of the hydrogel were manifested by the self-healing efficiency defined as:| R = δ1/δ0 × 100% | (1) |
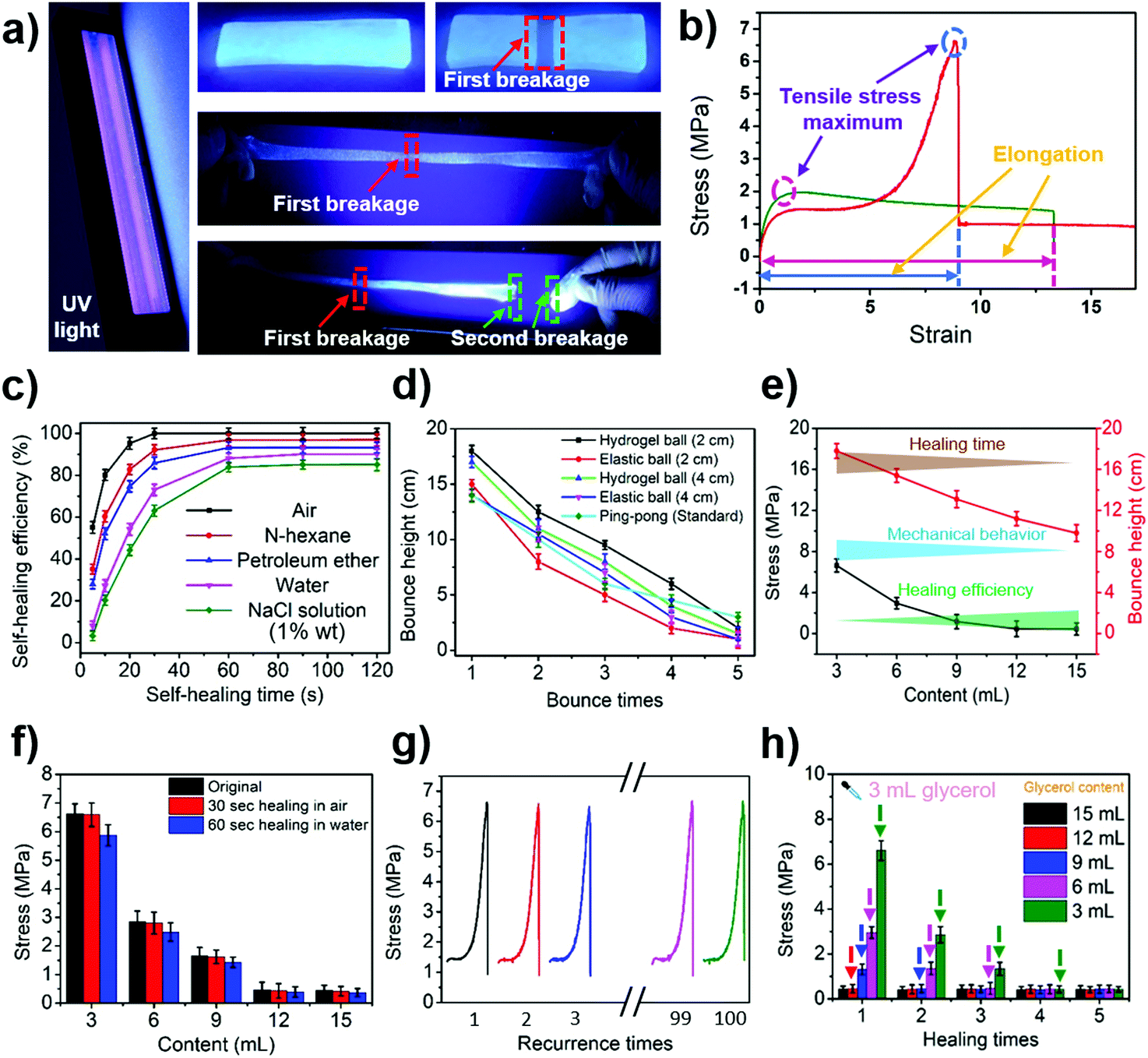 | ||
| Fig. 2 (a) Self-healing performance photos of the hydrogel under UV light. (b) Typical tensile stress–strain curves of different glycerol contents contained within the hydrogels. (c) Relationship between the self-healing efficiency and healing time of the hydrogel in different media. (d) Bounce height–time curves of different balls. (e) Relationship between the properties of the hydrogel and the glycerol content in the hydrogel. (f) Tensile stress–glycerol content curves of the hydrogel in different stages (original, self-healed) and different media. (g) Relationship between the pressure remodeling times and tensile properties of hydrogels. (h) Performance diagram of hydrogels obtained by adding glycerol at two different time periods (during preparing, during remodeling). | ||
Apart from that, the hydrogel also had excellent elasticity and its bounce performance was stronger than that of an elastic ball with the same diameter (Fig. 2d and Video S3, ESI†). The bouncing performance, the mechanical properties and the self-healing properties of the hydrogel were closely related to the volume of glycerol added to it. The higher the glycerol content in the hydrogel, the shorter the required self-healing time. The healing efficiency was also found to be higher within the same self-healing time, but the tensile strength of the hydrogel dropped (Fig. 2e and f).
The hydrogel also exhibits a pressure reshaping effect. Under the aid of an external force, the damaged hydrogel had the ability to be completely repaired (Fig. S14, ESI†). The mechanical performance of the repaired hydrogel was similar to that of the newly produced hydrogel (Fig. 2f and g). If glycerol was added to the damaged hydrogel during the pressure remodelling process, the properties of the hydrogel after reconstitution were comparable to those of the hydrogel that had the same amount of glycerol added during the synthesis process (Fig. 2h). This result demonstrates that glycerol can be used to regulate the properties of hydrogels.
3.2 Fluorescent properties and applications
The presence of fluorescent quantum dots (carbon/MXene quantum dots) allows the hydrogel to exhibit photoluminescence properties. Fig. 3a(I) shows the test schematic of a fluorescence spectrometer. The fluorescence properties of liquid quantum dots and hydrogels were tested in different environments (Fig. S15–S18, ESI†). The experimental results show that under the influence of strong acid and alkali the liquid quantum dots would be deactivated and the fluorescence performance would be significantly reduced (Fig. S19a, c, e and S20a, ESI†). In contrast, the fluorescence performance of the hydrogel always maintained the same order of magnitude (Fig. S19b, d, f and S20b, ESI†), indicating that the photoluminescence-responsive self-healing hydrogel could be maintained and continue to exhibit a long-term fluorescence response under various extreme conditions. The photoluminescence properties of the hydrogel make it useful in LED application (Fig. S21 and Video S4, ESI†). Further experiments revealed that the concentration and distribution of quantum dots in the hydrogel could be achieved by controlling the amount of quantum dots. The same type of quantum dot was concentrated in a continuous region, and only one kind of quantum dot existed in the same distribution region (Fig. S22, ESI†).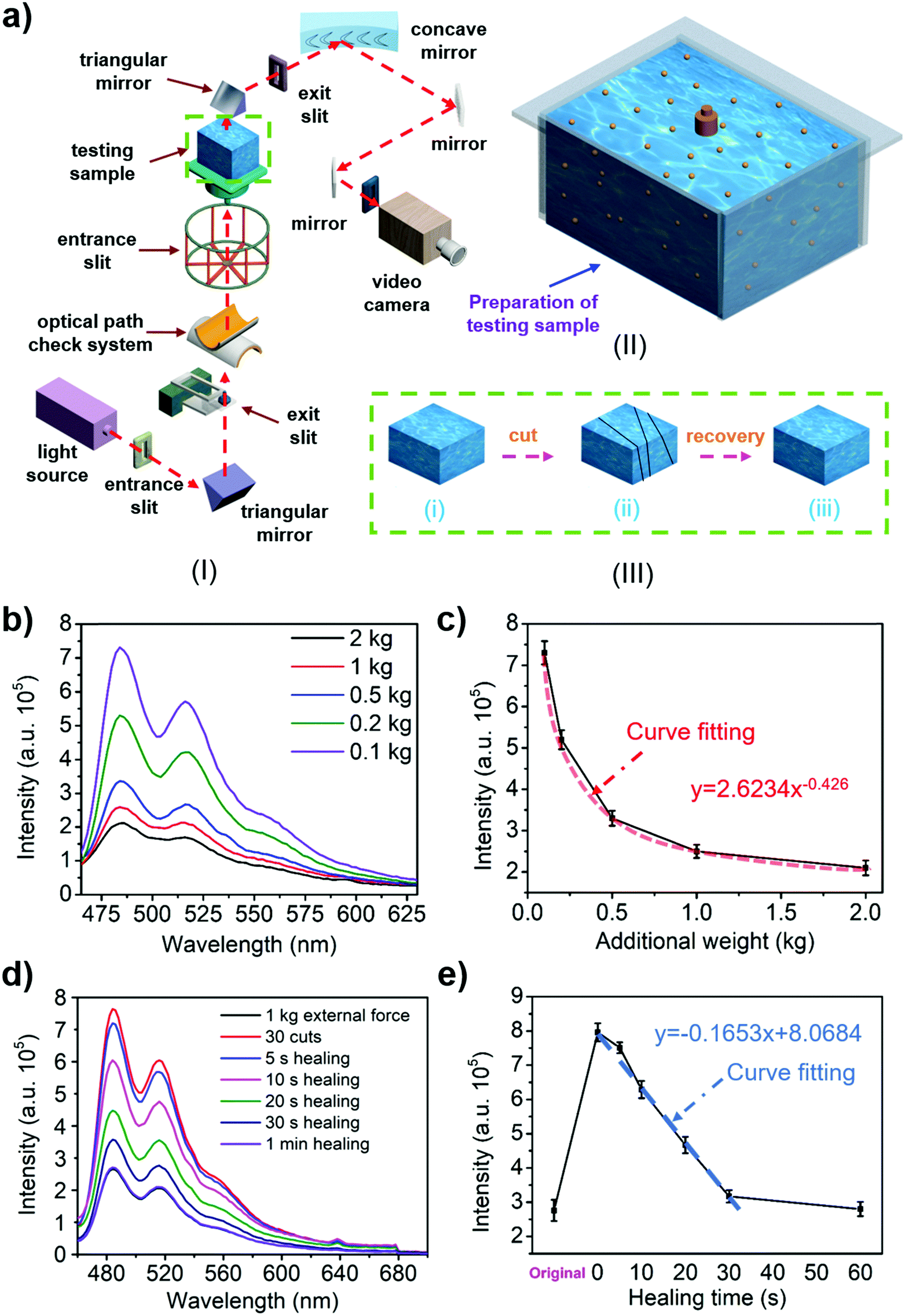 | ||
| Fig. 3 (a) Mechanism of a hydrogel fluorescence emission intensity test. (b) Relationship between the fluorescence intensity of the hydrogel (doped with carbon quantum dots) and its state (original, cut, healed). (c) A fitted curve between the fluorescence intensity of the hydrogel (doped with carbon quantum dots) and its state (original, cut, healed). (d) The relationship between the fluorescence intensity of the hydrogel (doped with carbon quantum dots) and the external force to which the hydrogel was subjected. (e) A fitted curve between the fluorescence intensity of the hydrogel (doped with carbon quantum dots) and the external force it was subjected to. | ||
Owing to its excellent self-healing properties and stable photoluminescence properties, the hydrogel has been demonstrated to achieve optical measurements for the first time. Since the spacing of quantum dots in the hydrogel is closely related to the external force, higher external force leads to smaller spacings of the quantum dots in the hydrogel (Fig. S23, ESI†). The different spacings result in different intensities of fluorescence excited in the hydrogel, such that the external force could be measured by measuring the fluorescence intensity excited during the loading. This method of force measurement is superior to the traditional one because it is not affected by contact conditions. The ability of the hydrogel to resist acidic and basic conditions and metal ions allows it to work in various liquids for force measurement. In the force measurement, the hydrogel was pressed in the mold shown in Fig. 3a(II) to avoid the shape effect. The fluorescence intensity of the hydrogels was tested under different external forces (Fig. 3b and Fig. S24a, ESI†). The data were analysed to obtain the relationship between the excitation intensity and the external force (Fig. 3c and Fig. S24b, ESI†). A standard optical comparison graph of the hydrogel's external force was obtained and the external force was determined using this relationship. The confirmatory experiment was performed in order to check the reliability of the method, and the external force measured by the optical method was similar to the actual external force (Fig. S25 and S26, ESI†).
The degree of the hydrogel's self-healing ability could also be determined by the fluorescence intensity. This remote optical measurement approach does not damage the hydrogel during the test, and in fact, it is more efficient than mechanical testing. The fluorescence excitation intensity (Fig. 3d and Fig. S24c, ESI†) of the hydrogel with different loading states were tested using a similar approach for measuring the external force exerted on the hydrogel (the original, the cut/damaged, and the healed, Fig. 3a(III)). The obtained data were fitted to establish the relationship between the excitation intensity and healing time (Fig. 3e and Fig. S24d, ESI†). Next, Fig. 2c was used to obtain the self-healing efficiency of the hydrogel. Finally, a standard optical comparison can be made for the degree of the self-healing ability, from which the self-healing degree of the hydrogel could be determined. The confirmatory experiment (Fig. S27 and S28, ESI†) was performed in order to check the effectiveness of the method, and the results showed that the self-healing efficiency measured by the optical method was comparable to the data measured by the tensile test.
3.3 Discussion
The structure of the hydrogel shown in Fig. 1 contained a variety of hydrogen bonding structures. While strong supporting hydrogen bonds exist between the chitosan–agarose–PVA chains (hydrogen bond I), reversible healing hydrogen bonds link PVA–borax–PVA to PVA–glycerol–PVA (hydrogen bond II), and dispersive hydrogen bonds appear between the PVA–quantum dots (hydrogen bond III, evenly dispersed quantum dots). Among these hydrogen bonds, the bond strength is in such order: hydrogen bond I > hydrogen bond II, and the strength of hydrogen bond III is influenced by the category of the quantum dots.10,38–44 The size of a quantum dot is on the nanoscale so it remains almost unaffected by external forces. Therefore, a macroscopic force would not directly affect the quantum points. Although hydrogen bond III is the weakest of these three bonds, the static hydrogen bond network between PVA and quantum dots is the most stable during the deformation. In fact, the main bonds subjected to external forces include hydrogen bond I and hydrogen bond II. When the external force exceeded the force that the bond could withstand, it would break, as shown in Fig. 4. Due to the weak tolerance of the linkage bond (hydrogen bond II) between PVA–borax–PVA and PVA–glycerol–PVA, it broke first (Fig. 4a: reactions 1 and 3 (Fig. 4c); from state I to state II). When most of the linkage bonds in PVA–borax–PVA and PVA–glycerol–PVA (hydrogen bond II) were broken, the connecting bond (hydrogen bond I) in chitosan–agarose–PVA began to break (Fig. 4a: reaction 5 (Fig. 4c); from state II to state III). The fixation function of the hydrogen bond I was lost, and the external force continued to act and the hydrogel therefore began to deform, and the distance between the PVA, chitosan, and agarose molecules in the hydrogel gradually decreased. At the same time, the linkage bonds in PVA–borax–PVA and PVA–glycerol–PVA (hydrogen bond II) began to recover (Fig. 4b: reactions 2 and 4 (Fig. 4c); from state III to state IV), but due to these weak forces, it was difficult to resist the external forces. Therefore, under the continuous action of an external force, these newly formed bonds were still prone to break. The distance between different molecules in the hydrogel decreased, and thus the strong supporting hydrogen bond (hydrogen bond I) in chitosan–agarose–PVA was also re-generated (Fig. 4b: reaction 6 (Fig. 4c); from state III to state IV; Fig. S29 and 30, ESI†). However, there were not enough supporting hydrogen bonds (hydrogen bond I) generated at this time to resist the external force, so the distance between molecules in the hydrogel decreased. During this process, hydrogen I and II bonds were also constantly generated and broken until the amount of the hydrogen I bonds was sufficient to resist external forces. The molecules in the hydrogel ceased moving and then the hydrogen I and II bonds were no longer broken (Fig. 4b: reactions 1–6 (Fig. 4c); from state IV to state V; Fig. S30 and 31, ESI†). A new hydrogel was generated through the “pressure reshaping effect” (Video S5, ESI†). Although the quantum dots were never separated from the PVA molecular chain throughout the process, the movement of the PVA molecular chain simultaneously drove the movement of the quantum dots, which caused the spacing of the quantum dots in the hydrogel to change. Such changes caused modifications to occur in the fluorescence response (Fig. S32, ESI†) to the forces.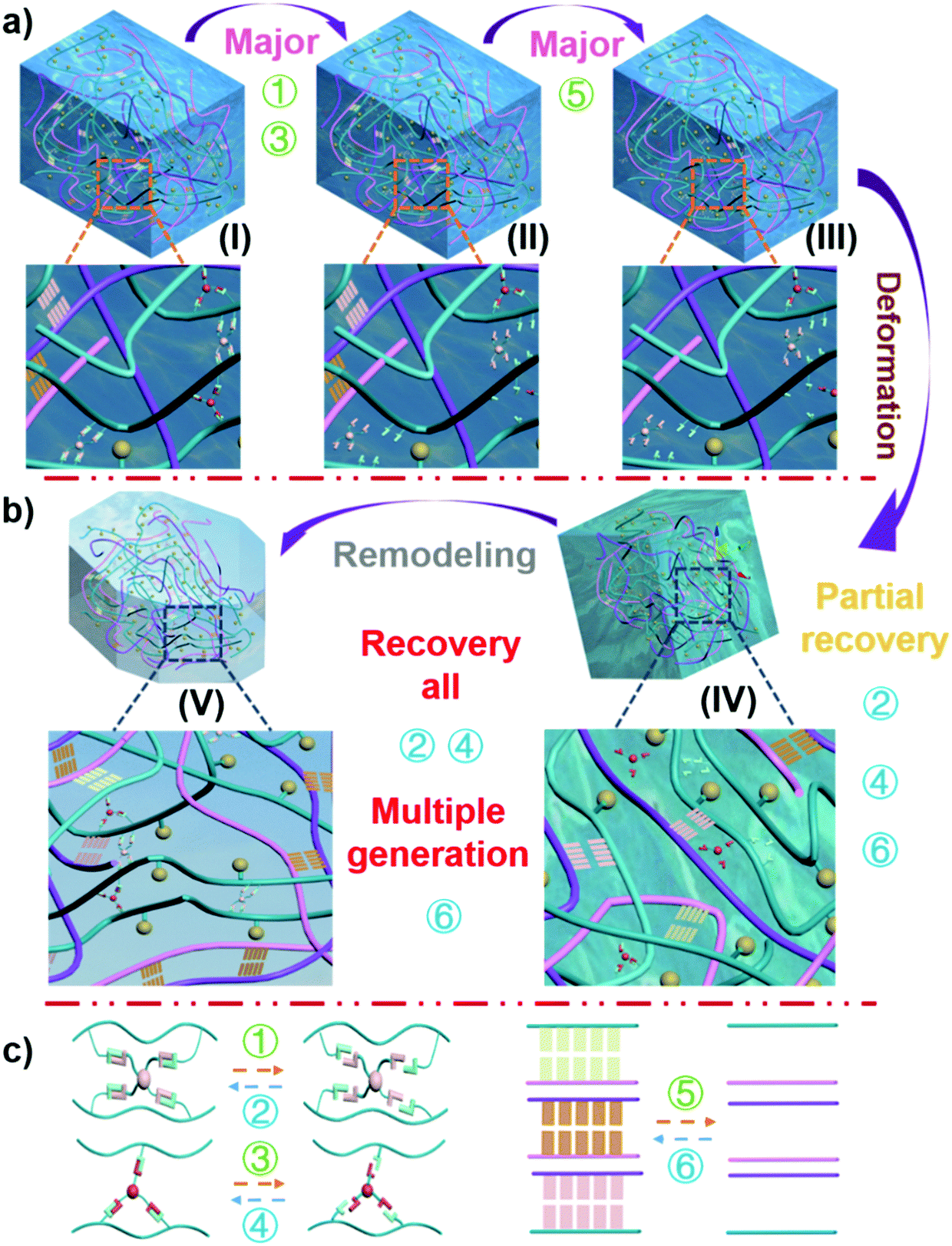 | ||
| Fig. 4 Schematic diagram of the number of bonds and state changes in the hydrogel when subjected to external forces. (a) Bond breaking period in the hydrogel. (b) Deformation and reconstruction periods of the hydrogel. (c) Bond breaking and formation reactions in the hydrogel. | ||
The arrangement of the quantum dots in the hydrogel had a one-to-one correspondence with the external force exerted on them, and the arrangement of the quantum dots had a one-to-one correspondence with the fluorescence excitation intensity of the hydrogel. Therefore, the fluorescence excitation intensity of the hydrogel had a one-to-one correspondence with the external force. Thus, the external force exerted on the hydrogel could be measured by analysing the fluorescence excitation intensity of the hydrogel. When the external force increases, the content of the hydrogel in the mold (Fig. 3a(II)) increases, and the quantum dot density also increases. The quantum dots in the front region would block the quantum dots in the rear region; thus the quantum dots in the rear region would not receive the excitation light. Meanwhile, the quantum dots in the rear region would also block the fluorescence excited by the front quantum dots (making them unable to be perceived by the detector). Based on these double blocking effects, the measured fluorescence intensity becomes weaker under a larger compressive force because of the smaller spacing of quantum dots in the hydrogel. When it comes to the self-healing condition of the hydrogel, as the self-healing time is increased, the self-healing area of the hydrogel is gradually increased, so the double blocking effects caused by the quantum dots in the hydrogel are more intensive. This also lowers the fluorescence excitation intensity.
4. Conclusions
In summary, we have prepared a new type of photo-fluorescent hydrogel that exhibits excellent self-healing properties in various environments (air, water, petroleum ether, and salt solution). In a gaseous medium, the hydrogel exhibited a 100% tensile strength after 30 seconds of healing compared to an original, unbroken hydrogel sample. After the damaged area self-repaired, the same sample area could be rotated 1800° without breaking. In a liquid medium, the hydrogel had 90% of its original tensile strength after healing for 60 seconds compared to an unbroken hydrogel sample. The fluorescence performance was shown not to be affected by external conditions (temperature, humidity, and pH). The hydrogel had a pressure remodelling effect. There was no substantial difference in the tensile strength performance and self-healing performance between the reshaped hydrogel obtained after pressing for 10 minutes under an external force of 2 kg and the newly made hydrogel. The unique relationship between the fluorescence excitation intensity of the hydrogel and the external pressure exerted on it allowed for external pressure measurements to be taken by determining the fluorescence intensity variation. The self-healing efficiency could be measured using an optical measurement method without damaging the material. As such, the as-developed hydrogels demonstrate a wide variety of potential applications because of their excellent self-healing properties, pressure remodelling ability and fluorescence properties. These hydrogels can be used as light-responsive biosensors, optical force measuring devices, plastic tissue, smart self-healing devices, and biomedical materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51875577), the Beijing Nova Program (No. Z171100001117058), the Tribology Science Fund of State Key Laboratory of Tribology (SKLTKF16A06), and the Science Foundation of China University of Petroleum Beijing (No. 2462018BJC004). We are grateful to Prof. Zhenan Bao (Department of Chemical Engineering, Stanford University), Prof. Shouceng Tian, Prof. Mao Sheng, and Xiaoxiao Zhen (China University of Petroleum, Beijing) for their selfless dedication, discussion and assistance in this work. W.-J. Ong acknowledges financial assistance and start-up grants/support (Grant number: XMUMRF/2019-C3/IENG/0013) from Xiamen University Malaysia. W.-J. Ong would also like to thank Petronas, ExxonMobil and Shell Malaysia for granting him the ‘2018 Merdeka Award Grant’.References
- A. H. Gelebart, D. J. Mulder, G. Vantomme, A. Schenning and D. J. Broer, Angew. Chem., Int. Ed., 2017, 56, 13436–13439 CrossRef CAS PubMed.
- J. H. Kim, Y. Jung, D. Lee and W. D. Jang, Adv. Mater., 2016, 28, 3499–3503 CrossRef CAS PubMed.
- S. Tokunaga, Y. Itoh, Y. Yaguchi, H. Tanaka, F. Araoka, H. Takezoe and T. Aida, Adv. Mater., 2016, 28, 4077–4083 CrossRef CAS PubMed.
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS PubMed.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- X. Han, Y. Liu and Y. Yin, Nano Lett., 2014, 14, 2466–2470 CrossRef CAS PubMed.
- H. Zhang, F. Gao, X. Cao, Y. Li, Y. Xu, W. Weng and R. Boulatov, Angew. Chem., Int. Ed., 2016, 55, 3040–3044 CrossRef CAS PubMed.
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed.
- Y. Matsunaga and J. S. Yang, Angew. Chem., Int. Ed., 2015, 54, 7985–7989 CrossRef CAS PubMed.
- W. P. Chen, D. Z. Hao, W. J. Hao, X. L. Guo and L. Jiang, ACS Appl. Mater. Interfaces, 2018, 10, 1258–1265 CrossRef CAS PubMed.
- Z. Wei, J. H. Yang, J. Zhou, F. Xu, M. Zrínyi, P. H. Dussault, Y. Osada and Y. M. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- Z. Hu and G. Chen, Adv. Mater., 2014, 26, 5950–5956 CrossRef CAS PubMed.
- K. Luo, Y. Yang and Z. Shao, Adv. Funct. Mater., 2016, 26, 872–880 CrossRef CAS.
- M. J. Glassman, J. Chan and B. D. Olsen, Adv. Funct. Mater., 2013, 23, 1182–1193 CrossRef CAS PubMed.
- S. Chen, X. Li, Y. Li and J. Sun, ACS Nano, 2015, 9, 4070–4076 CrossRef CAS PubMed.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed.
- X. Dai, Y. Zhang, L. Gao, T. Bai, W. Wang, Y. Cui and W. Liu, Adv. Mater., 2015, 27, 3566–3571 CrossRef CAS PubMed.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- T. Kakuta, Y. Takashima, M. Nakahata, M. Otsubo, H. Yamaguchi and A. Harada, Adv. Mater., 2013, 25, 2849–2853 CrossRef CAS PubMed.
- B. Rybtchinsk, ACS Nano, 2011, 5, 18 Search PubMed.
- E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251–14260 CrossRef CAS PubMed.
- Y. Guo, X. Zhou, Q. Tang, H. Bao, G. Wang and P. Saha, J. Mater. Chem. A, 2016, 4, 8769–8776 RSC.
- Z. Li, Z. Hou, H. Fan and H. Li, Adv. Funct. Mater., 2017, 27, 1604379 CrossRef.
- Q. Chen, L. Zhu, H. Chen, H. Yan, L. Huang, J. Yang and J. Zheng, Adv. Funct. Mater., 2015, 25, 1598–1607 CrossRef CAS.
- B. C. Tee, C. Wang, R. Allen and Z. Bao, Nat. Nanotechnol., 2012, 7, 825–832 CrossRef CAS PubMed.
- I. Jeon, J. Cui, W. R. Illeperuma, J. Aizenberg and J. J. Vlassak, Adv. Mater., 2016, 28, 4678–4683 CrossRef CAS PubMed.
- S. Fu, H. Zhou, H. Wang, H. Niu, W. Yang, H. Shao and T. Lin, Mater. Horiz., 2019, 6, 122–129 RSC.
- G. Weng, S. Thanneeru and J. He, Adv. Mater., 2018, 30, 1706526 CrossRef PubMed.
- Z. Wei, J. H. Yang, Z. Q. Liu, F. Xu, J. X. Zhou, M. Zrínyi, Y. Osada and Y. M. Chen, Adv. Funct. Mater., 2015, 25, 1352–1359 CrossRef CAS.
- Y. Zhao, H. Gao, Y. Fan, T. Zhou, Z. Su, Y. Liu and Y. Wang, Adv. Mater., 2009, 21, 3165–3169 CrossRef CAS.
- P. Chen, Q. Li, S. Grindy and N. Holten-Andersen, J. Am. Chem. Soc., 2015, 137, 11590–11593 CrossRef CAS PubMed.
- D. W. Balkenende, S. Coulibaly, S. Balog, Y. C. Simon, G. L. Fiore and C. Weder, J. Am. Chem. Soc., 2014, 136, 10493–10498 CrossRef CAS PubMed.
- M. Martinez-Calvo, O. Kotova, M. E. Mobius, A. P. Bell, T. McCabe, J. J. Boland and T. Gunnlaugsson, J. Am. Chem. Soc., 2015, 137, 1983–1992 CrossRef CAS PubMed.
- Z. Sun, F. Lv, L. Cao, L. Liu, Y. Zhang and Z. Lu, Angew. Chem., Int. Ed., 2015, 54, 7944–7948 CrossRef CAS PubMed.
- S. C. Grindy, R. Learsch, D. Mozhdehi, J. Cheng, D. G. Barrett, Z. Guan, P. B. Messersmith and N. Holten-Andersen, Nat. Mater., 2015, 14, 1210–1216 CrossRef CAS PubMed.
- L. Li, B. Yan, J. Yang, L. Chen and H. Zeng, Adv. Mater., 2015, 27, 1294–1299 CrossRef CAS PubMed.
- S. W. Sinton, Macromolecules, 1987, 20, 2430–2441 CrossRef CAS.
- C. Qiang, Z. Lin, Z. Chao, W. Qiuming and Z. Jie, Adv. Mater., 2013, 25, 4171–4176 CrossRef PubMed.
- S. Spoljaric, A. Salminen, N. D. Luong and J. Seppälä, Eur. Polym. J., 2014, 56, 105–117 CrossRef CAS.
- L. He, D. Szopinski, Y. Wu, G. A. Luinstra and P. Theato, ACS Macro Lett., 2015, 4, 673–678 CrossRef CAS.
- B. Lu, F. Lin, X. Jiang, J. Cheng, Q. Lu, J. Song, C. Chen and B. Huang, ACS Sustainable Chem. Eng., 2016, 5, 948–956 CrossRef.
- P. Zhu, M. Hu, Y. Deng and C. Wang, Adv. Eng. Mater., 2016, 18, 1799–1807 CrossRef CAS.
- T. Liu, X. Peng, Y. N. Chen, Q. W. Bai, C. Shang, L. Zhang and H. Wang, Macromol. Rapid Commun., 2018, 39, 1800050 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The description of various testing methodologies, characterization studies, experimental mechanisms, and explanations of this material (Tables S1–S3, Fig. S1–S32 and Videos S1–S5). See DOI: 10.1039/c8mh01441h |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |