
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCorrelative light and electron microscopy for complex cellular structures on PDMS substrates with coded micro-patterns†
Rong
Sun
 ab,
Xi
Chen
*c,
Chun-Ying
Yin
ab,
Lei
Qi
b,
Pak-Ming
Lau
b,
Hua
Han
c and
Guo-Qiang
Bi
*abd
ab,
Xi
Chen
*c,
Chun-Ying
Yin
ab,
Lei
Qi
b,
Pak-Ming
Lau
b,
Hua
Han
c and
Guo-Qiang
Bi
*abd
aCenter for Integrative Imaging, National Laboratory for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei, China
bCAS Key Laboratory of Brain Function and Disease, and School of Life Sciences, University of Science and Technology of China, No. 443 Huangshan Road, 230027, Hefei, Anhui, China. E-mail: gqbi@ustc.edu.cn
cInstitute of Automation, Chinese Academy of Sciences, No. 95 Zhongguancun East Road, 100190, Beijing, China. E-mail: xi. chen@ia.ac.cn
dCAS Center for Excellence in Brain Science and Intelligence Technology, University of Science and Technology of China, Hefei, China
First published on 12th November 2018
Abstract
Fluorescence light microscopy (FLM) is commonly used for localizing specific cellular and subcellular targets. Electron microscopy (EM), on the other hand, can reveal ultrastructural details of cellular architectures beyond the limit of optical resolution. Correlative light and electron microscopy (CLEM) that combines the two techniques has proven valuable in various cell biological applications that require both specificity and resolution. Here, we report an efficient and easy-to-use CLEM system, and its applications in studying neuronal synapses. The system utilizes patterned symbols to encode coordinates on micro-fabricated polydimethylsiloxane (PDMS) substrates, on which dissociated primary hippocampal neurons grow and form synaptic connections. After imaging and localizing specifically labeled synapses with FLM, samples are embedded in resin blocks and sectioned for EM analysis. The patterned symbols on PDMS substrates provide coordinate information, allowing efficient co-registration between FLM and EM images with high precision. A custom-developed software package achieves automated EM image collection, FLM/EM alignment, and EM navigation. With this CLEM system, we have obtained high quality electron tomograms of fluorescently labeled synapses along dendrites of hippocampal neurons and analyzed docking statistics of synaptic vesicles (SVs) in different subtypes of excitatory synapses. This technique provides an efficient approach to combine functional studies with ultrastructural analysis of heterogeneous neuronal synapses, as well as other subcellular structures in general.
1. Introduction
Electron microscopy (EM), especially electron tomography (ET), resolves 3D details of cellular architectures at nanometer resolution.1–4 Although immuno-EM has been used to label certain proteins in biological samples with high resolution, it still suffers from relatively low labeling density and poor ultrastructure preservation.5,6 On the other hand, the use of light microscopy (LM) to image fluorescently labeled molecules has revealed significant information regarding the organization and function of subcellular structures.7–9 However, direct observation of ultrastructures is still limited by the resolution of light microscopy. To combine the advantages of LM and EM techniques, correlative light and electron microscopy (CLEM) has been developed.10–12 As CLEM is a combination of two imaging modalities, a series of correlative methods have been developed based on varieties of LM and EM techniques, respectively,13–16 and also based on different probes to correlate LM and EM images.17–19 By such correlative methods, specific cellular features or components localized by fluorescence light microscopy (FLM) can then be examined by EM to achieve high resolution details in the same biological sample.For complex cellular structures, however, such as processes and synapses of neuronal cells which form extensive networks in culture, it remains a challenge to efficiently and accurately target fluorescence in certain areas in EM with existing CLEM methods. Although commercial photoetched glass gridded coverslips for FLM/EM correlation have been applied in mammalian cell lines or yeast cells,20,21 this kind of method would have low efficiency for long stretching neuropils and tiny synapses in cultured neurons inside each square of the coverslip. Custom micro-patterning substrates were reported to be adopted for correlative confocal microscopy and scanning electron microscopy (SEM) in the cultured neuronal system.22 However, the patterns themselves do not encode the coordinates, while the laser-cut alphabets on the substrates do. In addition, SEM could only provide surface information of samples. Another limitation of current CLEM methods concerns available software. Although several software solutions to CLEM workflows have been developed,19,23,24 a software package that includes all aspects of the CLEM procedure in EM is still requisite.
To overcome these limitations, we developed a system of correlative light and electron microscopy. Our CLEM system is based on custom-designed coded micro-patterning PDMS substrates, on which dissociated primary hippocampal neurons grow and form synaptic connections. A CLEM software package has also been developed to achieve automated EM image collection, FLM/EM alignment, and EM navigation. With this CLEM system, including both hardware and software, we have obtained high quality electron tomograms of excitatory synapses along dendrites of hippocampal neurons and analyzed docking statistics of synaptic vesicles (SVs) in different subtypes of excitatory synapses.
2. Materials and methods
All animal experiments were approved and conducted according to protocols approved by the Animal Experiments Committee at the University of Science and Technology of China. All animal procedures were performed following the guidelines of the Animal Experiment Committee at the University of Science and Technology of China. Low density cultures of dissociated embryonic rat hippocampal neurons were prepared according to a previously described protocol26 with few modifications.2.1. PDMS substrate fabrication and coded micro-patterning design
The patterned PDMS substrate was fabricated by conventional soft lithography according to a previously described protocol25 with some modifications (Fig. 1a). SU-8 3005 photoresist (MicroChem) was spin-coated on a cleaned Si wafer. The coated wafer was baked on a hot plate at a temperature of 95 °C for 2 min. The SU-8 3005 photoresist was exposed to UV light under a photomask containing designed patterns. After UV exposure, the coated wafer was again baked on a hot plate at a temperature of 95 °C for 2 min. The exposed SU-8 photoresist was developed by a SU-8 developer, and the SU-8 mold on the Si wafer was completed. The mixture of the PDMS pre-polymer and curing agent (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w), Dow Corning Corp.) was poured on the SU-8 mold and cured on a hot plate at 80 °C for 40 min. After curing, the patterned PDMS was removed from the SU-8 mold.
:1 (w/w), Dow Corning Corp.) was poured on the SU-8 mold and cured on a hot plate at 80 °C for 40 min. After curing, the patterned PDMS was removed from the SU-8 mold.
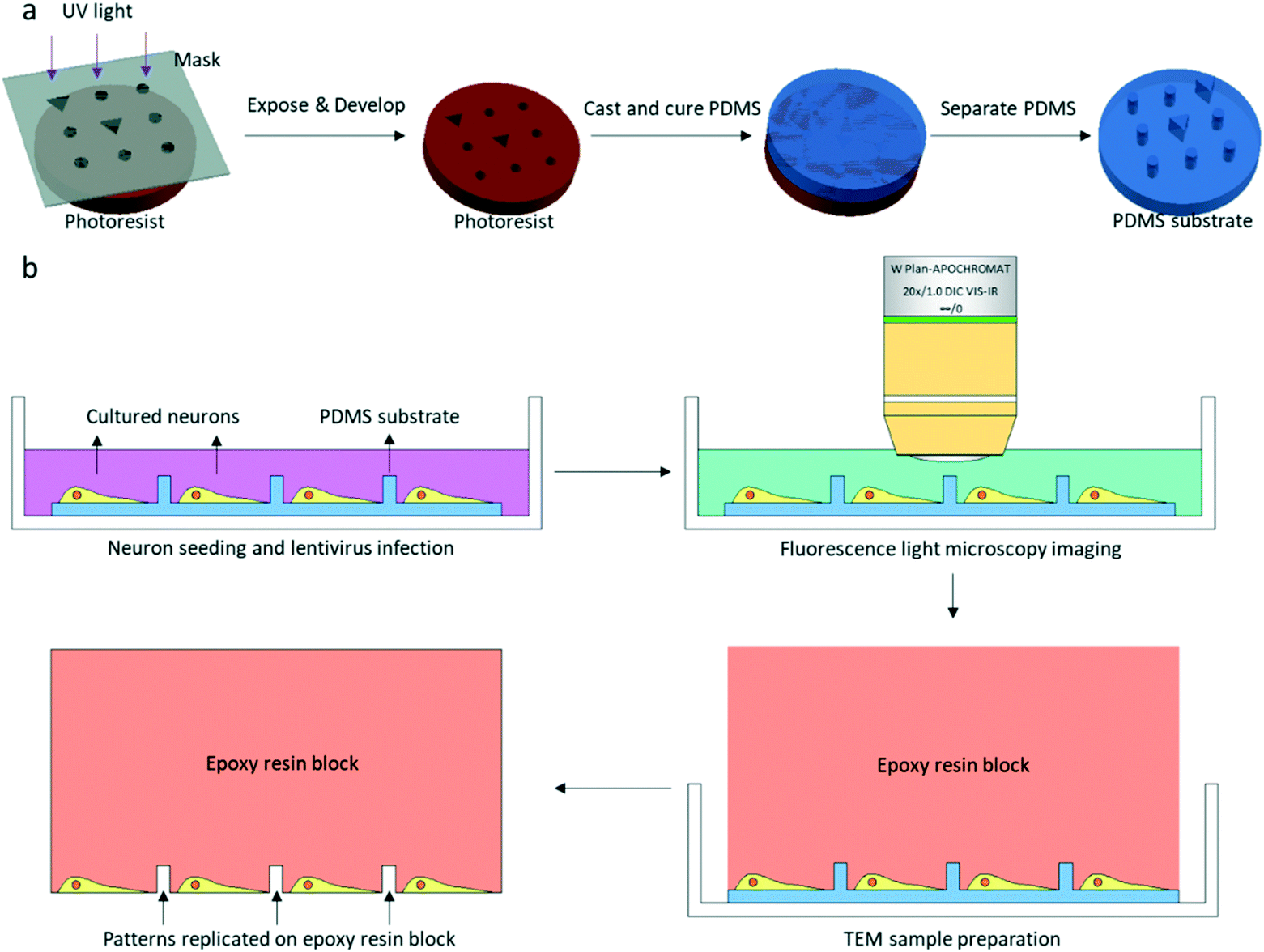 | ||
| Fig. 1 Schematic of PDMS substrate fabrication, fluorescence light microscopy imaging, and transmission electron microscopy (TEM) sample preparation. (a) The conventional soft lithography method is used for PDMS substrate fabrication, and the patterns are custom-designed for FLM and EM correlation. (b) FLM imaging and TEM sample preparation. After FLM imaging, the TEM sample is prepared in the dish. Patterns designed on PDMS are replicated on the epoxy resin block. | ||
The entire photomask consisted of 24 repeated elements (Fig. S1†). Each element represented one piece of PDMS substrate used for neuron culture, which is shown in Fig. 2a. The PDMS substrate comprises a two-tier hierarchical coordinate system. The first-tier hierarchical coordinates were letters and digits along the left and top side of the substrate. These letters and digits encoded the coordinates of the squares (Fig. 2a and b). In this way, the patterned PDMS substrate constituted 10 × 10 identical squares from A1 to J10. The secondary-tier hierarchical coordinates were patterned geometrical symbols, including circles and triangles, within each square. A square was divided into 36 symbol sets, each of which consisted of 3 × 3 symbols (Fig. 2c and d). The set coordinate was defined according to the combination of symbols inside the symbol set: a triangle occupied the center site of the 3 × 3 set of geometrical symbols. And the remaining eight sites were one reversed triangle and seven circles, or two reversed triangles and six circles. Thus, there were exactly (81) + (82) different sets from set 1 to set 36. These pattern-coded coordinates would allow rapid addressing in both the LM image and EM image (Fig. 2e–h), and further led to efficient LM/EM correlation.
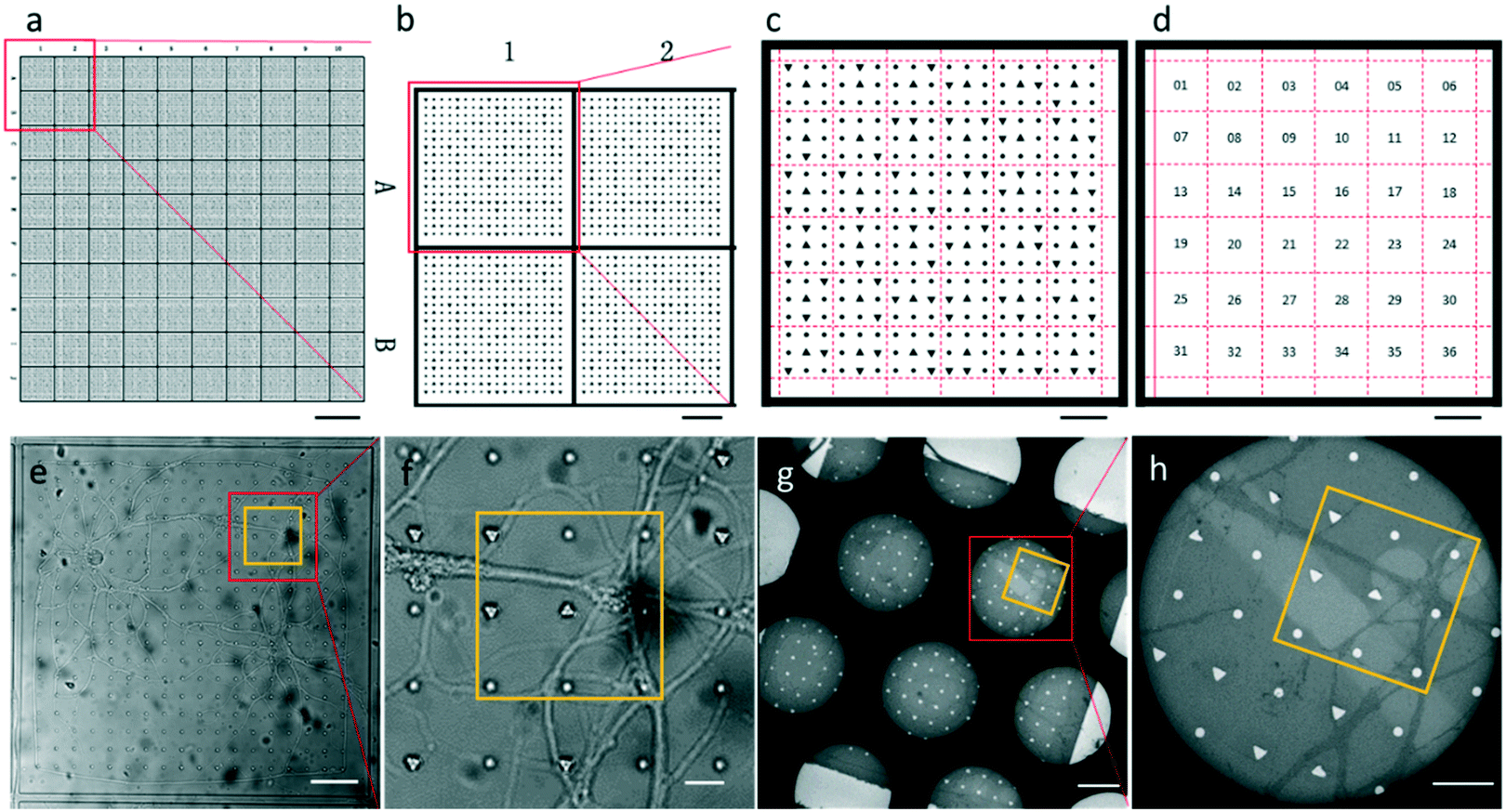 | ||
| Fig. 2 Patterned PDMS design and EM section with replicated patterns. (a–d) Design for the micro-fabricated PDMS substrate with a two-tier hierarchical coordinate system. (a and b) The square coordinates are defined by letters and digits along the sides of the PDMS substrate. (c and d) Each square is divided into 36 sets whose coordinates are encoded by 3 × 3 geometrical symbols. (c) Shows geometrical symbols inside square A1, which is boxed in (b). (e–h) The PDMS substrate and EM section of a square. Set 11 is yellow-boxed. Dark circles in (g) and (h) are holes of the EM grid where the section was deposited. Scale bars: (a) 500 μm, (b) 100 μm, (c–e and g) 50 μm, (f) 10 μm, and (h) 20 μm. | ||
2.2. Primary culture of hippocampal neurons and lentivirus infection
Low density cultures of dissociated embryonic rat hippocampal neurons were prepared according to a previously described protocol26 with few modifications. Patterned PDMS substrates for cell culture were glow-discharged with H2 and O2 for 1 min by a Gatan plasma cleaning machine (Gatan, Inc.), and sterilized with UV light for 30 min. These PDMS substrates were coated with 1 mg ml−1 poly-L-lysine (Sigma) in sodium borate buffer for approximately 24 h, followed by washing with ddH2O for approximately another 24 h. Hippocampi were removed from embryonic day-18 (E18) rats and were treated with trypsin for 15 min at 37 °C, followed by washing and gentle trituration. The dissociated cells were plated on poly-L-lysine coated patterned PDMS substrates attached on the bottom of 35 mm Petri dishes with a density of 40000–60000 cells per ml.
The PSD95-EGFP construct was produced based on GW-PSD95-EGFP (a generous gift from Dr. Weidong Yao) and plentivirus-CaMKIIp-mKate2. Plasmids were packaged into lentivirus following a protocol from the laboratory of Dr. Karl Deisseroth.27 For fluorescence light microscopy imaging, cultures were infected by lentivirus encoding PSD95-EGFP for 5–7 d in vitro (DIV). Twelve hours after the infection, half of the culture medium was replaced by fresh medium as previously described.28
2.3. Fluorescence light microscopy imaging
Cultures were used for FLM imaging at 16-18DIV. LM Imaging was performed with a 20× water objective lens in a Zeiss 710 microscope (Fig. 1b). After replacing the culture medium with extracellular solution (ECS, containing 150 mM NaCl, 3 mM KCl, 3 mM CaCl2·2H2O, 10 mM HEPES and 5 mM glucose, pH 7.3), the dish was placed in the microscope. One bright field image and z-stack fluorescence images of the neurons were collected for one square of the PDMS substrate. The fluorescence image used for CLEM would be a z-projection image of the z-stack images. The cultured neurons were immediately fixed with 4% paraformaldehyde (PFA, SPI supplies) and 2.5% glutaraldehyde (GA, Alfa Aesar) in 0.1 M cacodylate buffer/HCl (Ted Pella) after fluorescence light microscopy imaging.2.4. EM sample preparation
Samples were fixed in a refrigerator overnight at 4 °C. Fixative solution was discarded and neurons were stained with 1% OsO4 (Ted Pella) and 1.5% potassium ferrocyanide (Sigma-Aldrich) in 0.1 M cacodylate/HCl buffer for 40 min after three washes with 0.1 M cacodylate/HCl buffer. The samples were post-fixed with 1% OsO4 in 0.1 M cacodylate/HCl buffer for 40 min followed by three washes with 0.1 M cacodylate/HCl buffer. The samples were then stained with 2% uranyl acetate (SPI supplies) in water for 1 h. After staining, samples were dehydrated in a graded series of ethanol and infiltrated by Spi-Pon 812 resin (SPI supplies). A Spi-Pon 812 resin-filled tube was placed on top of the PDMS substrate before the resin was polymerized in an oven at 60 °C for 48 h. The PDMS was peeled off from the resin block after resin polymerization. Neurons were left in the resin, and patterns of the PDMS were also transferred to the resin block (Fig. 1b and 2g and h). The resin block surface was trimmed using a razor blade so that only one square imaged by FLM was at the apex of the resin block. 200 nm sections were acquired in an ultramicrotome (Leica EM UC7). All of the serial sections containing patterns and neuropils were collected on EM grids. The sections were contrasted with uranyl acetate and lead citrate (Alfa Aesar) prior to EM imaging.2.5. Correlative EM imaging by the custom-developed software package
A software package was developed to acquire EM images and perform LM/EM correlation automatically (Fig. S2†). By this CLEM software, automatic EM image collection, EM image montaging, FLM/EM alignment, and EM navigation could be achieved. The CLEM software, which allowed us to work with correlative EM imaging in the graphical user interface (Fig. S3†), consisted of an image capture program and a mosaic program. The image capture program was running on the computer which controlled the EM. It could record the current position of the EM stage, move the EM stage to the specified position and save the EM image. With these basic operations on the EM, the function of collecting the tile images, which covered the region of interest automatically, was also incorporated in the program. The mosaic program was running on another computer and communicated with the image capture program using the TCP/IP protocol. The tile EM images were transferred from the image capture program to the mosaic program to be montaged. And the LM image was aligned with the montaged EM image in the mosaic program. Then the positions of the corresponding points in the montaged EM image and the EM stage were recorded to establish the mapping function between the pixel coordinates of the montaged EM image and the physical coordinates of the EM stage. With the calculated mapping function, the user could use the mosaic program to move the EM stage by specifying the point in the spliced EM image. We provide the CLEM software which can be downloaded with detailed instructions for installation, configuration and usage. The following is the correlative EM imaging procedure achieved by our CLEM software.Once the EM grid with sections was transferred into the transmission electron microscope (TEM), a series of images with overlap were acquired at relatively low magnification to cover the region of interest (ROI), which was defined by a rectangle using the start point and the end point. After the low magnification scanning, a navigation map was generated by montaging the acquired images using the CLEM software. The montaged EM image and the LM image were correlated by selecting the corresponding geometrical symbols that appeared in both images in the CLEM software. The current physical coordinates of the EM stage and its corresponding location in the EM image were recorded as a correspondence by the CLEM software during scanning. At least four correspondences were obtained at different positions during scanning. The transformation between the EM stage and the navigation map was calculated. Then, the physical coordinates of the EM stage and the pixel positions of the montaged navigation map were also correlated. With the correspondence between the LM image, the montaged EM image and the EM stage, the EM stage could be moved to the desired position of the LM image using the CLEM software. Fluorescent puncta were selected in the optical image in the software to move the EM stage to the actual position. Tilt series were taken after all of the fluorescent puncta of interest were selected.
2.6. Electron tomography imaging and reconstruction
Electron tomography data were obtained using a Tecnai F20 transmission electron microscope (Thermo Fisher) equipped with a 4 K × 4 K CCD camera (Eagle, Thermo Fisher). Tilt series were automatically collected with Thermo Fisher Xplore 3D software from −60° to +60° at 2° intervals. The defocus value was set at −15 μm. The final pixel size was 1.12 nm. Tilt series were aligned and reconstructed with IMOD.29 The tilt series were aligned by a cross correlation algorithm without fiducial markers because of the high contrast of the heavy metal stained sample. Reconstruction was performed using the simultaneous iterative reconstruction (SIRT) technique with five iterations.3. Results and discussion
3.1. Fluorescence imaging of cultured hippocampal neurons grown on patterned PDMS substrates and correlation with EM imaging
We attached our micro-patterning coded PDMS substrate to the bottom of the dish, planted the disassociated hippocampal neurons on the PDMS substrates, and transfected the neurons with lentivirus to express PSD95-EGFP for live cell fluorescence imaging. One example of a bright field and fluorescence image shows the typical morphology of the cultured hippocampal neurons on PDMS substrates (Fig. 3a–c). Two cell bodies were localized in one square with extended axons and dendrites. Some neuropils made a loop in the square to appear as if they were trapped by the PDMS bars between squares. Meanwhile, some neuropils could grow over the square bars (arrows in Fig. 3a), indicating that neurons in different squares might form connections. These neurons labeled by PSD95-EGFP showed green fluorescent puncta at the processes, indicative of excitatory synapses (arrows in Fig. 3d). Moreover, the bright field image was taken to reveal the PDMS patterns by which we could perform the correlation procedure with EM images.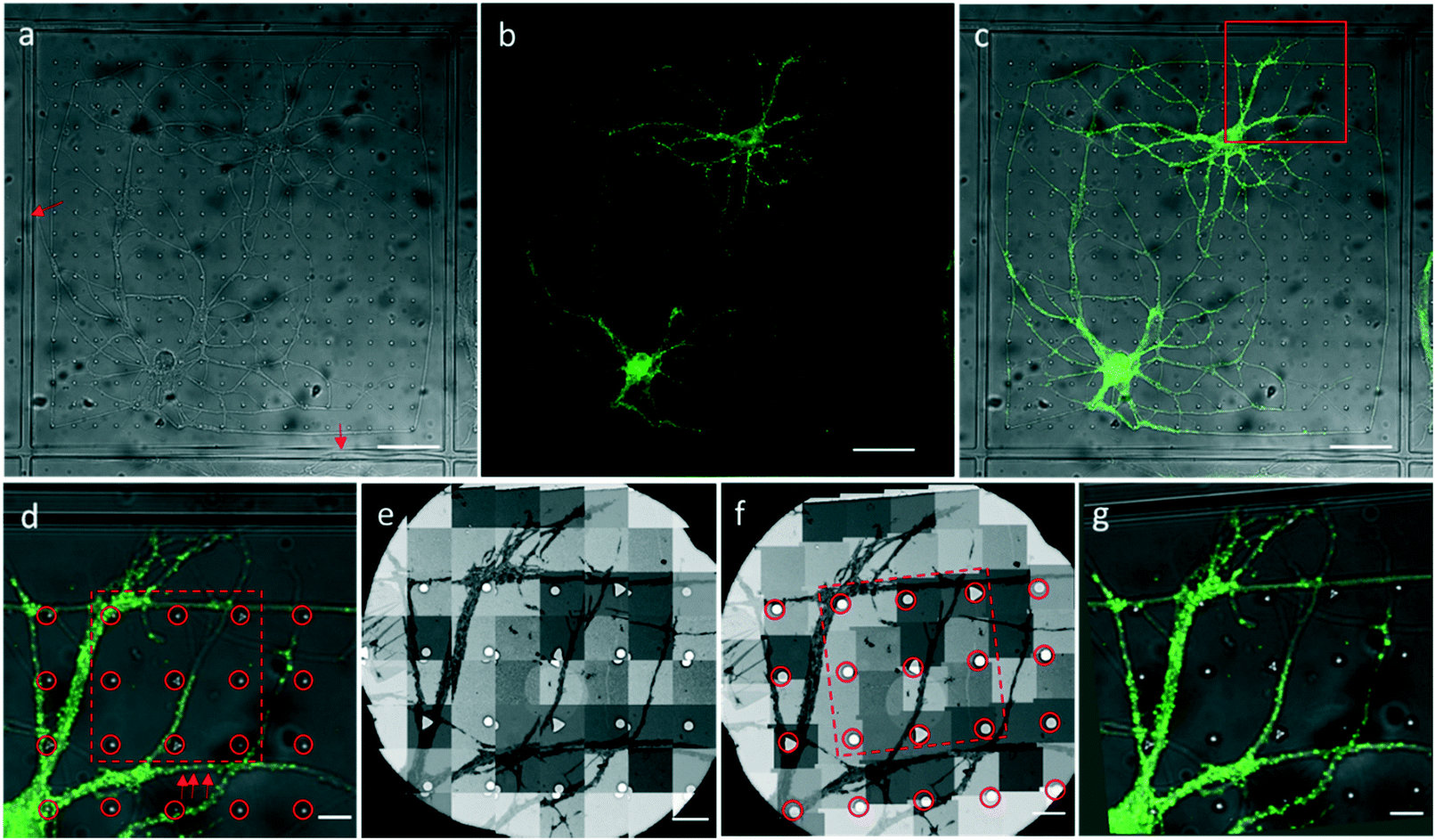 | ||
| Fig. 3 An example of optical imaging of cultured hippocampal neurons on the PDMS substrate and the CLEM procedure. (a–c) Bright field (a), fluorescence (b), and overlaid (c) images of cultured neurons expressing PSD95-EGFP on the PDMS substrate. Arrows indicate neuropils growing over the square bars. (d) Zoomed-in view of the boxed area in (c). Arrows indicate PSD95-EGFP puncta along the dendrite. (e) Sequential EM images taken by the CLEM software. Overlap between images is preset for montaging. (f) Montaged image of the sequential EM images. (g) Warped LM image to correlate the EM image (f) by the circled patterns in the EM (f) and LM (d) images. Scale bars: (a–c) 50 μm, (d–g) 10 μm. | ||
After FLM imaging, we prepared samples following the conventional TEM sample preparation protocols as described in Methods. After peeling off the PDMS from the resin block, the patterns were transferred onto the resin block along with the neurons. We cut samples into 200 nm sections, and loaded them into the electron microscope. Then, 3500× EM images with overlap (Fig. 3e) were acquired to cover a hole of the 150 mesh EM grid automatically. We generated the navigation map (Fig. 3f) by montaging the acquired images using the CLEM software. The montaged navigation map offered a larger field of view with acceptable resolution compared with just one low magnification image, and provided adequate localization information to establish the correlation between the EM image and the LM image. Therefore, both neuropils and designed patterns could be clearly observed in the montaged navigation map. And the boxed area (set 30) in Fig. 3f would allow quick registration of the same set of symbols in the LM image (Fig. 3d). We correlated the montaged EM image and the LM image by selecting the corresponding symbols (circles in Fig. 3d and f). By aligning selected symbols, the light image was warped to the EM image (Fig. 3g) using non-linear transformation thin plate splines.30 The accuracy of the correlation could be improved with more corresponding points involved.
3.2. Electron tomography of identified excitatory synapses through CLEM
Through the CLEM procedure, we acquired the electron tomograms of excitatory synapses identified by PSD95-EGFP labeling. When we selected the PSD95-EGFP punctum (box 1 in Fig. 4e) in the optical image in the software, the EM stage moved to the actual position of the synapse in the EM section. In order to show the localization of the synapse clearly, it is also box-marked in the contrast-adjusted image (Fig. 4c) of the navigation map and overlaid image (Fig. 4f) of the FLM/EM image.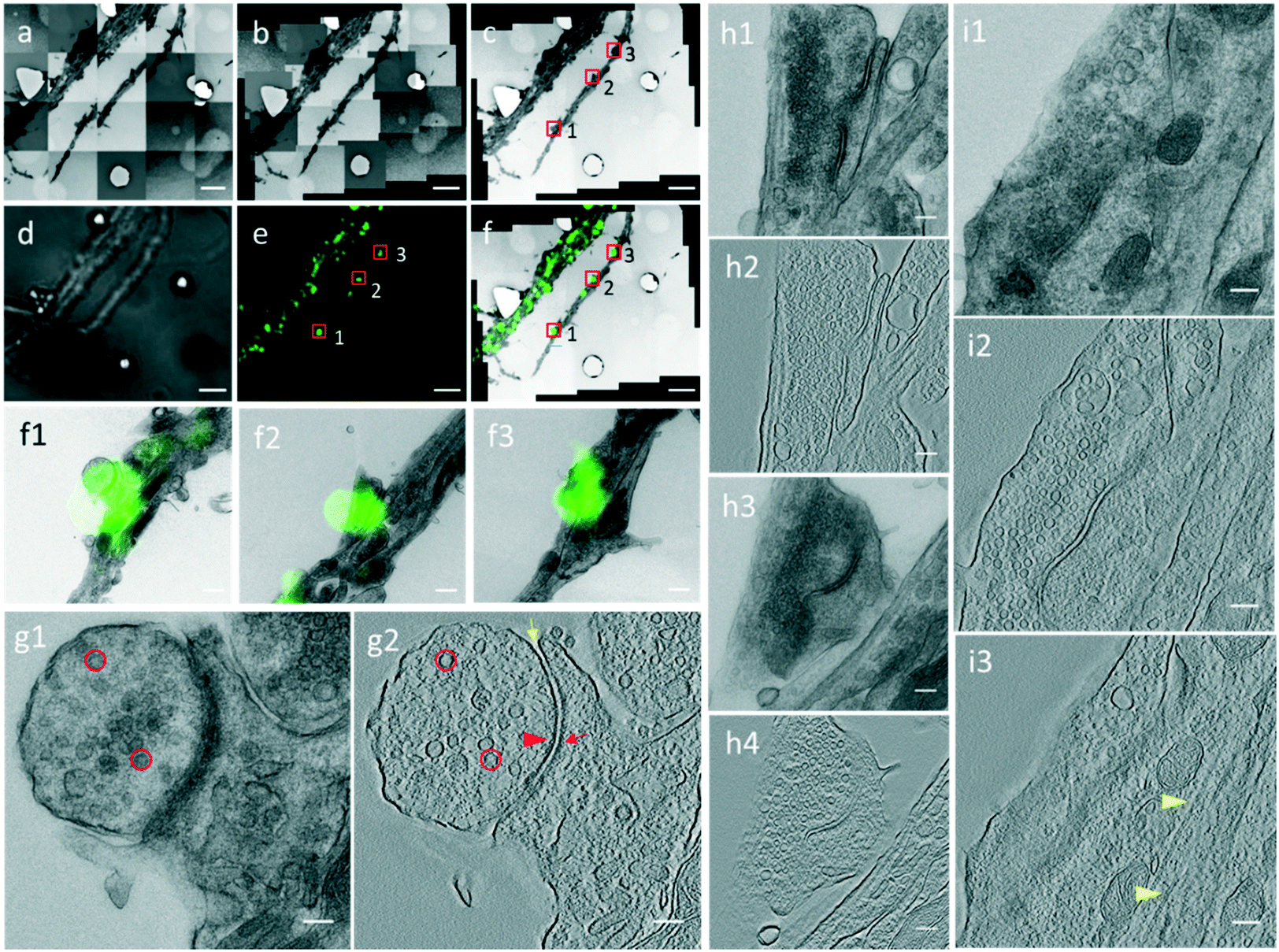 | ||
| Fig. 4 Electron tomogram of identified excitatory spine and shaft synapses. (a–c) Raw (a), montaged (b), and contrast-adjusted (c) images of sequential EM images. (d and e) Bright field (d) and fluorescence (e) images of the same view field with the EM image. (f) EM and LM images are correlated and merged by aligning designed PDMS patterns. (f1–f3) Zoomed-in views of boxed synapses in (f) from area 1–3. (g1 and g2) A single projection image and a tomographic slice of the same synapse boxed in area 1 in (c), (e), and (f). (h1–h4) Single projection images and tomographic slices on two serial EM sections showing the same spine synapse boxed in area 2 in (c), (e), and (f). (i1–i3) A single projection image and two tomographic slices showing the same shaft synapse boxed in area 3 in (c), (e), and (f). Red circle, SV; red arrowhead, AZ; red arrow, PSD; yellow arrow, synaptic cleft; yellow arrowhead, microtubule. Scale bars: (a–f) 5 μm, (f1–f3) 100 μm, (g1–i3) 100 nm. | ||
After moving the EM stage to the selected synapse, we took a single projection image and a tilt-series. The single projection image at high magnification shows the typical ultrastructure of an excitatory synapse (Fig. 4g1). There was a population of SVs in the presynaptic compartment. The synaptic cleft, which was covered by an electron-dense area, was difficult to observe in the single projection image in this 200 nm section. In the 3D tomogram reconstructed based on the tilt series collected for the synapse (Fig. 4g2), individual SVs were distinguishable. Electron-dense areas in both presynaptic and postsynaptic compartments were recognized as the active zone (AZ) and postsynaptic density (PSD), respectively. A relatively uniform synaptic cleft appeared between the pre- and postsynaptic membrane. These results demonstrate that the high resolution electron tomograms of the excitatory synapses enable us to conduct quantitative analysis of the synaptic ultrastructure.
By this CLEM routine, we generated a data set containing 42 electron tomographic reconstructions of excitatory synapses. Among these synapses, in most cases, whether it was a spine or shaft synapse could be determined in a single projection image of one EM section. For example, the synapse in Fig. 4g1 was a typical spine synapse and the synapse in Fig. S4† was a typical shaft synapse as the synapse formed directly on the dendritic shaft, with microtubules readily visible in the postsynaptic compartment.
However, in some cases, a spine or shaft synapse was difficult to distinguish in the single projection image because the postsynaptic compartment was too dense to observe microtubules or the postsynaptic compartment was cut too small in a single EM ultrathin section. By our CLEM system, two excitatory synapses were detected in the same dendrite in the PSD95-EGFP labeled cultured neurons (boxes 2 and 3 in Fig. 4e), and whether they were spine or shaft synapses was determined by information in multiple serial EM sections or by information in multiple tomographic slices of the 3D tomograms. For the synapse boxed in area 2 in Fig. 4e, the single projection image (Fig. 4h1) or the tomogram (Fig. 4h2) in one EM section only reveals a very small postsynaptic compartment, from which it is unable to determine whether or not it is a spine. We then performed the CLEM procedure on the neighboring EM section and localized the same synapse indicated by the same fluorescent punctum (Fig. S5†). Next, a high resolution single projection image and an electron tomogram of the synapse were acquired, which clearly contained a dendritic spine without microtubules as the postsynaptic compartment (Fig. 4h3 and h4). Thus, according to the synaptic structure in the two serial sections, we verified the presence of a spine synapse. Regarding the synapse boxed in area 3 in Fig. 4e, although microtubules were not visible in the single projection image (Fig. 4i1), the 3D reconstructed tomogram presented a typical presynaptic ultrastructure, including SVs and a synaptic cleft in one tomographic slice (Fig. 4i2), and microtubules along the postsynaptic dendrite in another tomographic slice (Fig. 4i3), showing that it was a shaft synapse. According to the above described process, the 42 excitatory synapses were subdivided into 27 spine synapses and 15 shaft synapses.
3.3. Quantitative analysis of docked SVs in identified excitatory spine and shaft synapses
High resolution electron tomograms allowed us to detect SV docking (Fig. 5), a preceding step of SV fusion upon Ca2+ stimulation.31 SVs that were in direct physical contact with the presynaptic membrane in the AZ were considered to be docked (Fig. 5e). In contrast, some SVs appeared linked to the presynaptic membrane by short filaments, and these SVs were termed tethered SVs (Fig. 5f). In our analysis, these tethered SVs were not considered docked. Fused SVs (Fig. 5g) were excluded from the analysis, as well.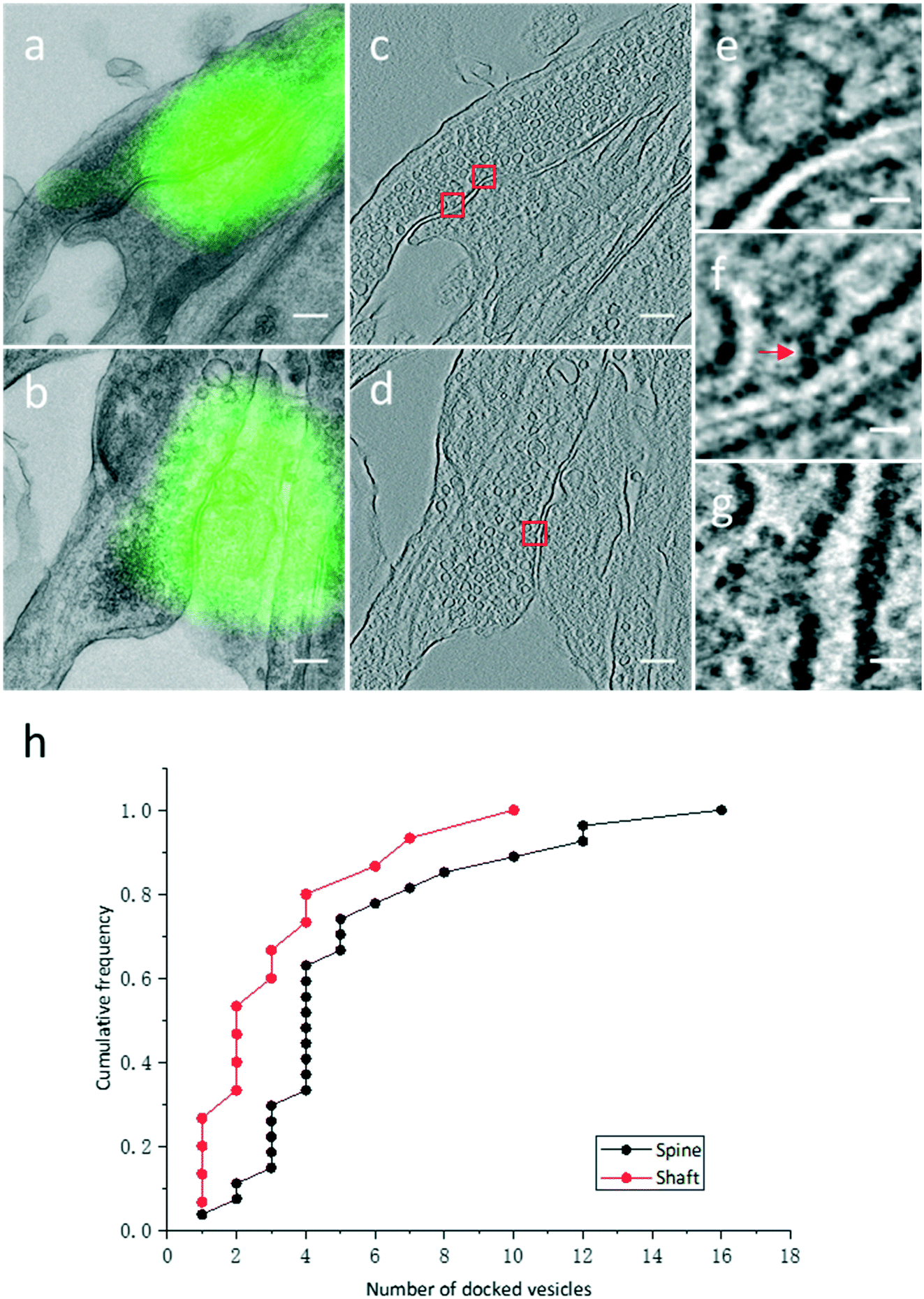 | ||
| Fig. 5 Quantitative analysis of docked vesicles in excitatory spine and shaft synapses. (a and b) Single projection images of two excitatory synapses co-localized with PSD95-EGFP puncta. (c) A tomographic slice showing the same synapse in (a). (d) A tomographic slice showing the same synapse in (b). (e–g) Vesicles fixed at multiple stages of exocytosis boxed in (c) and (d). (e) Vesicle directly in contact with the AZ. (f) Vesicle tethered to the AZ membrane by filaments (arrow). (g) Vesicle fused to the AZ membrane. (h) Cumulative frequency of the number of docked vesicles in excitatory spine and shaft synapses. Scale bars: (a–d) 200 nm, (e–g) 20 nm. | ||
With structural differences between spine and shaft synapses in their postsynaptic compartments, the number of docked SVs might also differ in these two subtypes of synapses. Thus, we analyzed the number of docked vesicles in 27 excitatory spine synapses and 15 excitatory shaft synapses obtained by our CLEM system (Fig. 5h). We noted that every synapse analyzed here was just a part of the intact synapse. More docked SVs were found in excitatory spine synapses (5.3 ± 0.7 (Mean ± SEM), n = 27 in excitatory spine synapses and 3.3 ± 0.7 (Mean ± SEM), n = 15 in excitatory shaft synapses; p < 0.05, two-sample t-test). This ultrastructural difference implies different sizes of the readily releasable pool between excitatory spine and shaft synapses.
3.4. Discussion
CLEM methods are applied as a powerful tool to connect ultrastructure information and functional study in cell biology. Combining multiple forms of LM and EM methods, a series of hybrid correlative approaches show great potential in neuroscience study.12,32 However, some limitations to resolve the ultrastructure of crowded synapses still remain. For example, the CLEM method, by preserving fluorescence in the resin-embedded sample, which often requires reduced or no osmium tetroxide during sample preparation, leads to poor ultrastructure in EM.33,34 The CLEM method using anatomical landmarks, such as vessels or cell bodies,35 which are randomly distributed, is incapable of providing efficient and accurate correlative probes for the neuronal culture system.In this article, we describe an automated CLEM system based on the micro-patterning coded PDMS and a custom-developed software package. With this hardware and software together, we utilize this CLEM system for the observation of excitatory synapses in cultured hippocampal neurons. PDMS is a suitable substrate for culturing mammalian cells, and possesses the characteristic of being able to be made into different shapes and sizes by the conventional soft lithography method.36–38 Thus, this kind of PDMS substrate offers a series of advantages for CLEM in cultured neuronal networks. First, cultured neurons grow very well on PDMS substrates after proper treatment. Second, compared to the laser-etched gridded glass coverslip, PDMS is inexpensive, can be rapidly produced, and is very easy to separate from the EM resin block without other operations. Third, our micro-patterning PDMS substrate is coded by a two-tier hierarchical coordinate system, which provides high resolution information for the LM/EM correlation.
The CLEM software constitutes another major advantage of our system. The reported software solutions covered only parts of the CLEM procedure, while our software package offers a high integration level, including the functions of automatic scanning, navigation and LM/EM correlation, which greatly improves the efficiency of CLEM experiments. The image capture program was developed using AutoIt,39 a BASIC-like scripting language which can simulate keystrokes and mouse movement. This means that the acquiring software is not restricted by the API from electron microscope manufacturers and can be adopted by other microscopes easily, because it just fills in input boxes and clicks buttons in the control software of the electron microscope. We also provide the source code of the image capture program with the CLEM software so that the other laboratories could adjust the code to accord with their EM control GUI easily.
Using the CLEM system, we have successfully obtained a batch of identified excitatory synaptic tomograms. Although the majority of excitatory synapses are spine synapses, excitatory terminals make synaptic contacts on dendritic shafts.6,40,41 The hypothesis that shaft synapses represent immature excitatory synapses and eventually become spine synapses was proposed because of the downregulation of excitatory shaft synapses and the increase of excitatory spine synapses during development.40,42,43 We analyze the number of docked SVs and find more docked SVs in excitatory spine synapses than those in excitatory shaft synapses. As the number of docked vesicles is changing during synaptic maturation,44 our result may imply that the two subtypes of excitatory synapses are in different developmental stages. By our CLEM system, we use PSD95-EGFP labeled FLM to exclude inhibitory synapses and high resolution 3D tomograms to detect docked vesicles specifically, confirming the power of our CLEM method.
In conclusion, our CLEM system, which is highly integrative, efficient and easy-to-use, provides an overall solution for ultrastructural studies guided by fluorescence imaging in a cultured neuronal system and other culture cell systems.
Author contributions
R. S., X. C., P.-M. L., H. H. and G.-Q. B. designed the research; R. S., C.-Y. Y. and L. Q. performed the experiments; X. C. developed the CLEM software; R. S. and X. C. analyzed the data; R. S., X. C., H. H. and G.-Q. B. wrote the paper.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported in part by grants from the National Key R&D Program of China (No. 2016YFA0400900), CAS (No. XDB02050000), and the National Natural Science Foundation of China (No. 31630030). We acknowledge the use of instruments at the Center for Integrative Imaging of the Hefei National Laboratory for Physical Sciences at the Microscale. We thank Chang-Lu Tao and Yun-Tao Liu for technical advice on imaging and data processing. We thank Fang Xu for assistance in setting up the soft lithography platform.References
- W. Baumeister, Curr. Opin. Struct. Biol., 2002, 12, 679–684 CrossRef CAS PubMed.
- A. Hoenger and J. R. McIntosh, Curr. Opin. Cell Biol., 2009, 21, 89–96 CrossRef CAS PubMed.
- S. Watanabe, B. R. Rost, M. Camacho-Perez, M. W. Davis, B. Sohl-Kielczynski, C. Rosenmund and E. M. Jorgensen, Nature, 2013, 504, 242–247 CrossRef CAS PubMed.
- C. Imig, S. W. Min, S. Krinner, M. Arancillo, C. Rosenmund, T. C. Sudhof, J. Rhee, N. Brose and B. H. Cooper, Neuron, 2014, 84, 416–431 CrossRef CAS PubMed.
- M. Megias, Z. Emri, T. F. Freund and A. I. Gulyas, Neuroscience, 2001, 102, 527–540 CrossRef CAS PubMed.
- K. M. Harris and R. J. Weinberg, Cold Spring Harbor Perspect. Biol., 2012, 4, 30 Search PubMed.
- B. N. G. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217–224 CrossRef CAS PubMed.
- J. W. Lichtman and J. A. Conchello, Nat. Methods, 2005, 2, 910–919 CrossRef CAS PubMed.
- D. J. Stephens and V. J. Allan, Science, 2003, 300, 82–86 CrossRef CAS PubMed.
- J. Caplan, M. Niethammer, R. M. Taylor, 2nd and K. J. Czymmek, Curr. Opin. Struct. Biol., 2011, 21, 686–693 CrossRef CAS PubMed.
- D. S. Lidke and K. A. Lidke, J. Cell Sci., 2012, 125, 2571–2580 CrossRef CAS PubMed.
- I. Begemann and M. Galic, Front. Synaptic Neurosci., 2016, 8, 28 Search PubMed.
- W. Kukulski, M. Schorb, M. Kaksonen and J. A. G. Briggs, Cell, 2012, 150, 508–520 CrossRef CAS PubMed.
- E. Johnson, E. Seiradake, E. Y. Jones, I. Davis, K. Grunewald and R. Kaufmann, Sci. Rep., 2015, 5, 9583 CrossRef CAS PubMed.
- V. Lucic, A. H. Kossel, T. Yang, T. Bonhoeffer, W. Baumeister and A. Sartori, J. Struct. Biol., 2007, 160, 146–156 CrossRef PubMed.
- Y.-W. Chang, S. Chen, E. I. Tocheva, A. Treuner-Lange, S. Loebach, L. Sogaard-Andersen and G. J. Jensen, Nat. Methods, 2014, 11, 737–739 CrossRef CAS PubMed.
- M. Grabenbauer, W. J. C. Geerts, J. Fernadez-Rodriguez, A. Hoenger, A. J. Koster and T. Nilsson, Nat. Methods, 2005, 2, 857–862 CrossRef CAS PubMed.
- D. Bishop, I. Nikic, M. Brinkoetter, S. Knecht, S. Potz, M. Kerschensteiner and T. Misgeld, Nat. Methods, 2011, 8, 568–570 CrossRef CAS PubMed.
- M. Schorb and J. A. G. Briggs, Ultramicroscopy, 2014, 143, 24–32 CrossRef CAS PubMed.
- L. E. Reddick and N. M. Alto, J. Visualized Exp., 2012, e3650 Search PubMed.
- H. Asakawa, Y. Hiraoka and T. Haraguchi, Micron, 2014, 61, 53–61 CrossRef CAS PubMed.
- L. Benedetti, E. Sogne, S. Rodighiero, D. Marchesi, P. Milani and M. Francolini, Sci. Rep., 2014, 4, 7033 CrossRef CAS PubMed.
- P. Paul-Gilloteaux, X. Heiligenstein, M. Belle, M. C. Domart, B. Larijani, L. Collinson, G. Raposo and J. Salamero, Nat. Methods, 2017, 14, 102–103 CrossRef CAS PubMed.
- W. Kukulski, M. Schorb, S. Welsch, A. Picco, M. Kaksonen and J. A. G. Briggs, J. Cell Biol., 2011, 192, 111–119 CrossRef CAS PubMed.
- Y. C. Li, R. Sun, B. Zhang, Y. C. Wang and H. Y. Li, PLoS One, 2015, 10, 15 Search PubMed.
- G. Q. Bi and M. M. Poo, J. Neurosci., 1998, 18, 10464–10472 CrossRef CAS PubMed.
- F. Zhang, V. Gradinaru, A. R. Adamantidis, R. Durand, R. D. Airan, L. de Lecea and K. Deisseroth, Nat. Protoc., 2010, 5, 439–456 CrossRef CAS PubMed.
- C.-L. Tao, Y.-T. Liu, R. Sun, B. Zhang, L. Qi, S. Shivakoti, C.-L. Tian, P. Zhang, P.-M. Lau, Z. H. Zhou and G.-Q. Bi, J. Neurosci., 2018, 38, 1493–1510 CrossRef CAS PubMed.
- J. R. Kremer, D. N. Mastronarde and J. R. McIntosh, J. Struct. Biol., 1996, 116, 71–76 CrossRef CAS PubMed.
- F. L. Bookstein, IEEE Trans. Pattern Anal. Mach. Intell., 1989, 11, 567–585 CrossRef.
- T. C. Südhof, Neuron, 2013, 80, 675–690 CrossRef PubMed.
- S. Wang, S. Li, G. Ji, X. Huang and F. Sun, Biophys. Rep., 2017, 3, 8–16 CrossRef CAS PubMed.
- O. Avinoam, M. Schorb, C. J. Beese, J. A. G. Briggs and M. Kaksonen, Science, 2015, 348, 1369–1372 CrossRef CAS PubMed.
- S. Watanabe, J. Richards, G. Hollopeter, R. J. Hobson, W. M. Davis and E. M. Jorgensen, J. Visualized Exp., 2013, e3995 Search PubMed.
- W. C. Lee, V. Bonin, M. Reed, B. J. Graham, G. Hood, K. Glattfelder and R. C. Reid, Nature, 2016, 532, 370–374 CrossRef CAS PubMed.
- J. N. Lee, X. Jiang, D. Ryan and G. M. Whitesides, Langmuir, 2004, 20, 11684–11691 CrossRef CAS PubMed.
- D. B. Weibel, W. R. DiLuzio and G. M. Whitesides, Nat. Rev. Microbiol., 2007, 5, 209–218 CrossRef CAS PubMed.
- A. Vishwanathan, G. Q. Bi and H. C. Zeringue, Lab Chip, 2011, 11, 1081–1088 RSC.
- A. Flesner, AutoIt v3: Your Quick Guide, O'Reilly Media, 2007 Search PubMed.
- J. C. Fiala, M. Feinberg, V. Popov and K. M. Harris, J. Neurosci., 1998, 18, 8900–8911 CrossRef CAS PubMed.
- J. C. Anderson and K. A. C. Martin, J. Comp. Neurol., 2006, 495, 709–721 CrossRef PubMed.
- C. Boyer, T. Schikorski and C. F. Stevens, J. Neurosci., 1998, 18, 5294–5300 CrossRef CAS PubMed.
- I. M. Ethell and E. B. Pasquale, Prog. Neurobiol., 2005, 75, 161–205 CrossRef CAS PubMed.
- M. G. Mozhayeva, Y. Sara, X. R. Liu and E. T. Kavalali, J. Neurosci., 2002, 22, 654–665 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8lc00703a |
| This journal is © The Royal Society of Chemistry 2018 |