
Trisubstituted olefin synthesis via Ni-catalyzed hydroalkylation of internal alkynes with non-activated alkyl halides†
Xiao-Yu
Lu
 *ab,
Mei-Lan
Hong
a,
Hai-Pin
Zhou
a,
Yue
Wang
a,
Jin-Yu
Wang
a and
Xiu-Tao
Ge
a
*ab,
Mei-Lan
Hong
a,
Hai-Pin
Zhou
a,
Yue
Wang
a,
Jin-Yu
Wang
a and
Xiu-Tao
Ge
a
aSchool of Materials and Chemical Engineering, ChuZhou University, Chuzhou, 239000, China. E-mail: xiaoyulu@mail.ustc.edu.cn
bSchool of Chemistry and Chemical Engineering, AnHui University, Hefei, 230601, China
First published on 28th March 2018
Abstract
The stereoselective synthesis of tri-substituted alkenes is challenging. Herein, we report a Ni-catalyzed regio- and stereo-selective hydroalkylation of internal alkynes with non-activated alkyl halides. This method does not use any sensitive organometallic reagents and shows good functional group compatibility, which enables the efficient synthesis of many tri-substituted olefins from readily available coupling partners. It also provides a straightforward method for the modification of bioactive organic molecules.
Alkenes are one of the most widely occurring and important class of organic compounds, which are widely used in the chemical, materials, and pharmaceutical industries.1 Over the past century, numerous methods, such as elimination reactions, the reduction of alkynes, Wittig-type reaction, and the Julia–Kocienski, have been developed for the synthesis of olefins. In addition, many significant catalytic methods using transition metals have been reported, for example, the Heck reaction, the semi-hydrogenation of alkynes, olefin metathesis, and the cross-coupling reactions of alkenyl metal reagents or alkenyl halides.2 Recently, intermolecular hydrocarbonation reactions between alkynes and electrophilic reagents have been considered as one of the most attractive methods for the synthesis of alkenes because these can synthesize a diverse array of substituted alkenes by controlling the regio- and stereoselectivity. However, most of the hydrocarbonation reactions of alkynes use π-electrophiles.3–8 Hydrocarbonation cross-coupling reactions of alkynes with alkyl electrophiles are less. In 2015, the Lalic group reported the copper-catalyzed hydroalkylation of terminal alkynes with alkyl triflates (Scheme 1a).9 The Hu group has reported the iron-catalyzed reductive coupling of terminal aryl alkynes with alkyl halides (Scheme 1b).10 In 2016, Fu realized the Ni-catalyzed Markovnikov hydroalkylation of alkynes with alkyl halides (Scheme 1c).11 The Nishikata group has reported that Cu-catalyzed tandem reactions enable trans- and cis-hydro-tertiary-alkylations (Scheme 1d).12 However, these elegant studies can only utilize terminal alkynes in the hydroalkylation reaction to afford di-substituted olefins.13
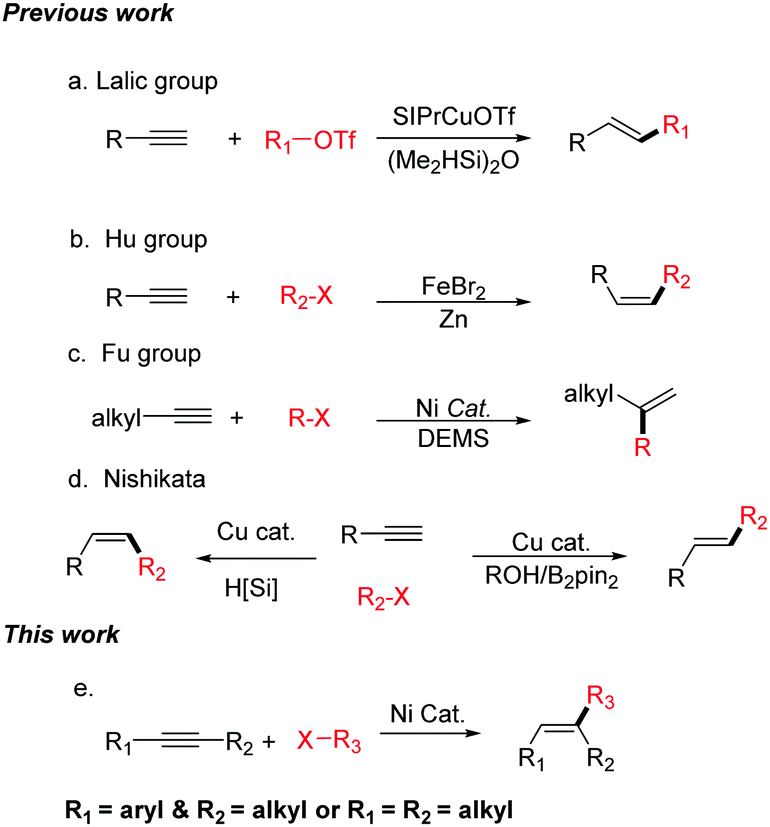 | ||
| Scheme 1 The hydroalkylation of alkynes with alkyl electrophiles. | ||
Tri-substituted alkenes are in high demand, and the efficient regio- and stereoselective synthesis of tri-substituted alkenes bearing three different carbon-linked groups presents a particular challenge in modern organic synthesis. Over the past few decades, transition metal-catalyzed cross-coupling reactions of tri-substituted alkenyl halides or tri-substituted alkenyl metal reagents are regarded as a versatile and straightforward method for the synthesis of tri-substituted alkenes.14 These coupling partners need to have the corresponding stereo-configurations. However, the stereoselective synthesis of these coupling partners is difficult.15 Herein, we report the first example of a nickel-catalyzed regio- and stereo-selective hydroalkylation of non-functionalized internal alkynes with non-activated alkyl halides (Scheme 1e). Not only aryl–alkyl substituted alkynes, but also alkyl–alkyl substituted alkynes can be successfully transformed into the desired products. This method does not use any sensitive organometallic reagents and both of the starting materials are readily available, thus enabling the efficient synthesis of many tri-substituted alkenes. Due to the mild reaction conditions, this new approach shows good functional group compatibility. In addition, it provides a method to modify complex organic molecules.
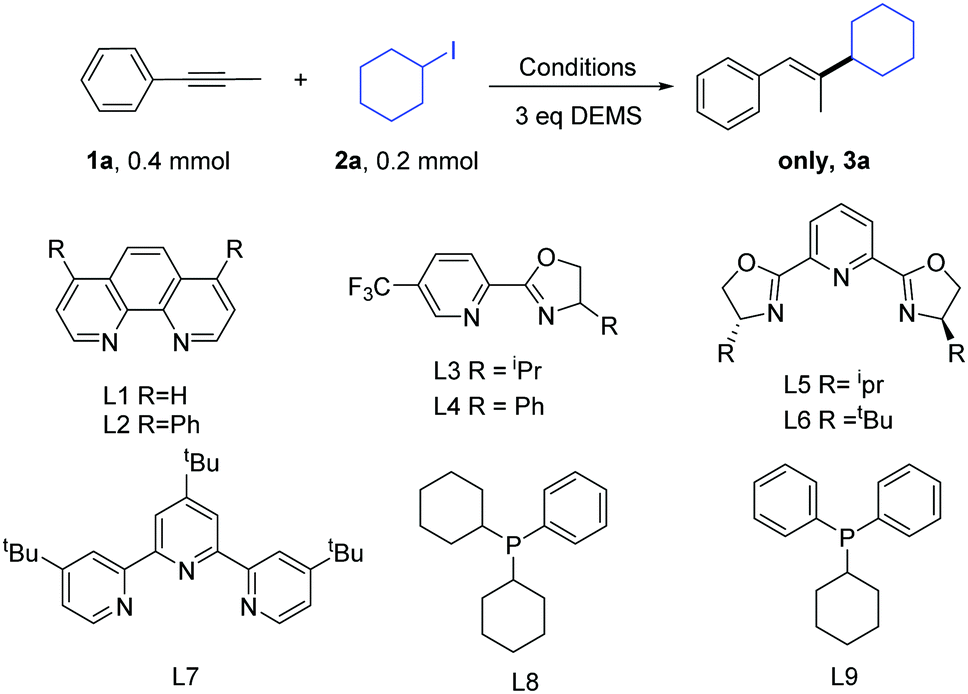
We began our study by choosing the commercially available 1-phenyl-1-propyne (1a) and iodocyclohexane (2a) as the model substrates (Table 1). On the basis of the previous study on the Ni-catalyzed hydroalkylation of alkynes with alkyl halides, we first examined the previously reported catalytic conditions used for the reaction.11 Gratifyingly, we obtained the product in a moderate yield (entry 1). The results showed that the previously reported conditions were not suitable. Next, we examined other bidentate nitrogen ligands such as the phenanthroline family of ligands (L1–L2) and pyrox family of ligands (L3–L4). Disappointingly, these ligands did not increase the yield. Then, we used tri-nitrogen ligands instead of the bidentate nitrogen ligands (L5–L7). However, the yields obtained for the desired product remained very poor. We also tested some phosphine ligands (L8–L9). Disappointingly, the reactions did not afford any desired product. Consequently, we screened a series of bases (entries 11–14). Gratifyingly, when K2CO3 was used as a base, we obtained the optimal reaction conditions (85% GC yield and 81% isolated yield, entry 14, product ratio >30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Finally, the control experiments indicated that the reaction almost shut down without the use of a nickel catalyst (entry 15).
:1). Finally, the control experiments indicated that the reaction almost shut down without the use of a nickel catalyst (entry 15).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. (10 mol%) | Ligand (12 mol%) | Base (2.5 eq.) | Solvent (0.6 mL) | Yield (%) 3aa |
| a The reaction was carried out at 30 °C for 10 h under an Ar atmosphere. 3 equiv. of diethoxymethylsilane was used as a hydride donor. The yields were determined by GC analysis using biphenyl as an internal standard (the average of two GC runs). b Performed without NiBr2·diglyme. c The yield of isolated product. DMAC = N,N-dimethylacetamide. | |||||
| 1 | NiBr2·diglyme | dtbbpy | Cs2CO3 | DMAc | 32 |
| 2 | NiBr2·diglyme | L1 | Cs2CO3 | DMAc | 5 |
| 3 | NiBr2·diglyme | L2 | Cs2CO3 | DMAc | Trace |
| 4 | NiBr2·diglyme | L3 | Cs2CO3 | DMAc | 4 |
| 5 | NiBr2·diglyme | L4 | Cs2CO3 | DMAc | 8 |
| 6 | NiBr2·diglyme | L5 | Cs2CO3 | DMAc | 7 |
| 7 | NiBr2·diglyme | L6 | Cs2CO3 | DMAc | 5 |
| 8 | NiBr2·diglyme | L7 | Cs2CO3 | DMAc | 4 |
| 9 | NiBr2·diglyme | L8 | Cs2CO3 | DMAc | Trace |
| 10 | NiBr2·diglyme | L9 | Cs2CO3 | DMAc | Trace |
| 11 | NiBr2·diglyme | dtbbpy | NaOAc | DMAc | 15 |
| 12 | NiBr2·diglyme | dtbbpy | CsF | DMAc | 38 |
| 13 | NiBr2·diglyme | dtbbpy | LiOMe | DMAc | 50 |
| 14 | NiBr 2 ·diglyme | dtbbpy | K 2 CO 3 | DMAc | 85(81) |
| 15b | — | dtbbpy | K2CO3 | DMAc | Trace |
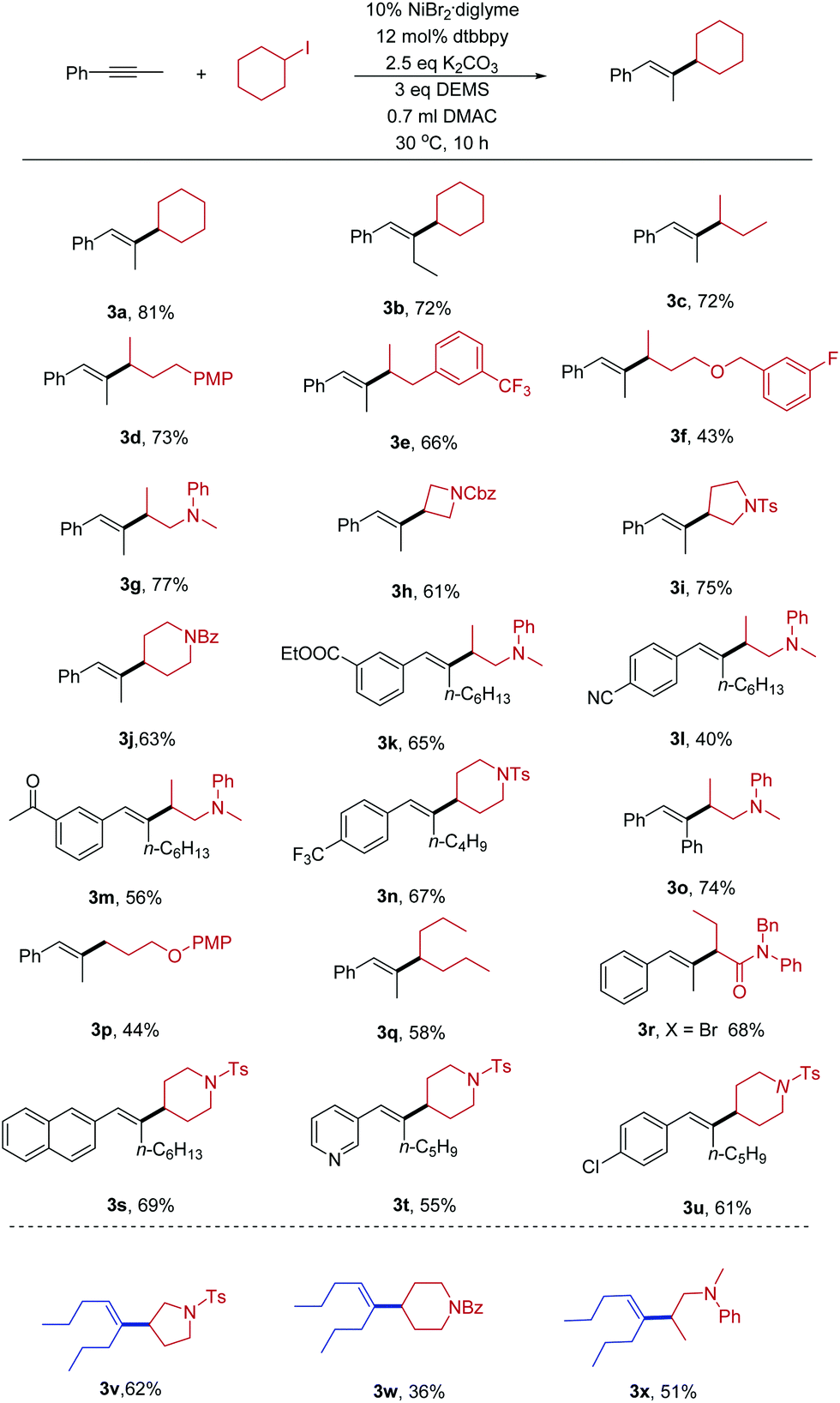
With the optimized conditions in hand, we explored the scope of the hydroalkylation reaction of internal alkynes. As shown in Table 2, our protocol exhibited excellent regio- and stereoselectivity (product ratio >25:1, as determined by GC and 1H-NMR spectroscopy). The coupling partners with different functional groups can be successfully converted into the desired products in modest to excellent yields. Both cyclic and acyclic alkyl halides can be transformed. Due to the mild reaction conditions, the hydroalkylation of internal alkynes is compatible with lots of synthetically relevant functional groups such as trifluoromethyl (3e), amine (3g), fluoride (3f), amide (3h, 3j), and sulfonamide (3i). Some base-sensitive functional groups such as ester (3k) and nitrile (3l) groups can be well tolerated. Even more active groups, such as ketone (3m), were compatible in the reaction. The success of the reaction inspired us to apply them to the cross-coupling of primary alkyl halides. For example 3p, we successfully obtained product when using a primary alkyl halide as the substrate. Activated secondary α-bromo amide (3r) was also a good substrate except alkyl iodides. Furthermore, heterocycles, such as pyrrolidine (3i), piperidine (3j), naphthaline (3s), and pyridine (3t) are tolerated in either of the two coupling substrates. Aryl–Cl bonds (3u) did not hinder the reaction.
| a The reactions were conducted on a 0.2 mmol scale at 30 °C. The yields of the isolated products after 10 h. Bz = benzoyl, DEMS = diethoxymethylsilane and Ts = 4-toluenesulfonyl. |
|---|
|
Next, we examined whether alkyl–alkyl substituted alkynes with lower activity could participate in the reaction. Fortunately, the present conditions were applicable for these substrates. Alkyl halides bearing sulfonamide (3v), amide (3w), and amine (3x) groups react under these conditions to afford the desired products in moderate yields.
We next demonstrated the efficiency of this regio- and stereoselective hydroalkylation of internal alkynes in the late-stage modification of complex active molecules (Scheme 2). Modification of fructose derivative (1aa), which is of great interest in life sciences, with 1ba results in the formation of 1ca in a moderate yield (Scheme 2a). Treatment of pregnenolone derivative (1ab), tolerating ketone and alkenes, with 2ab afforded the product 3aa in a moderate yield (Scheme 2b).
 | (1) |
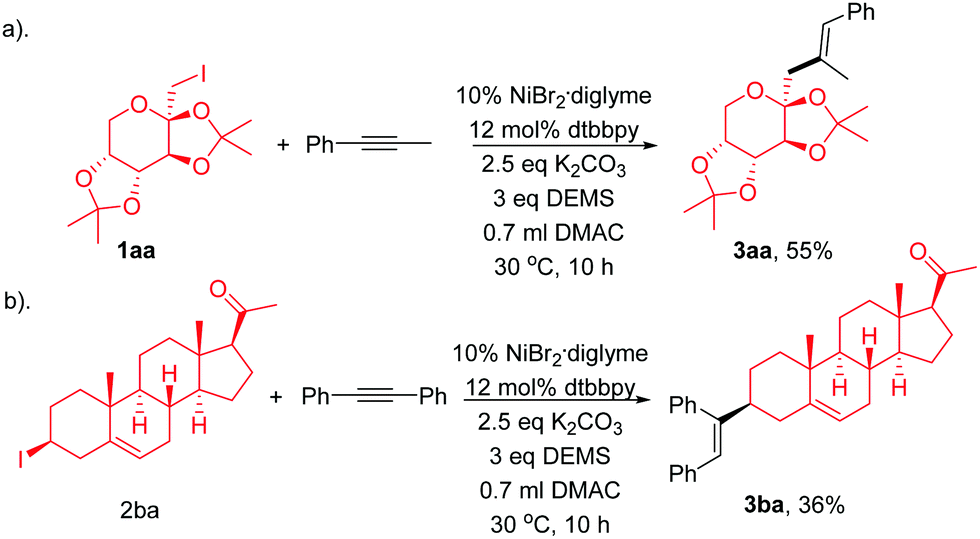 | ||
| Scheme 2 The modification of complex molecules. | ||
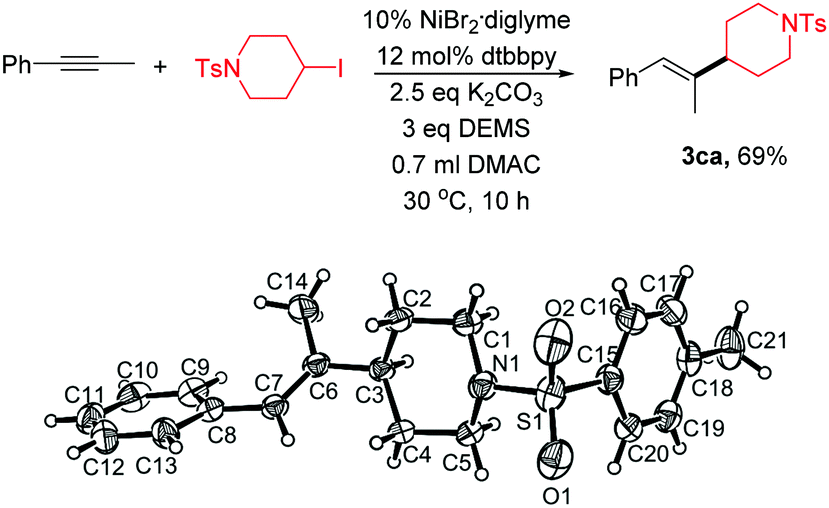
Single-crystal XRD analysis of 3ca confirmed the regio- and stereoselectivity of the hydroalkylation of internal alkynes (eqn (1)). The reaction provides an efficient method for the synthesis of many of the single configuration tri-substituted alkenes.
To explore the reaction mechanism, several experiments were conducted. Initially, when we added 1.0 equiv. 2,2,6,6-tetramethylpiperidinyloxy (TEMPO), the reaction was completely inhibited. Next, we performed an experiment between 2da and phenyl-1-propyne (eqn (2)). A mixture of linear coupling product (3da) and ring-cyclized product (4da) was obtained. The abovementioned results were consistent with a radical-type mechanism of alkyl halides.10,16 However, the detailed reaction process was not clear at this point. Our preliminary view is that the reaction goes through the LnNiH intermediate, and then, the intermediate reacts with the internal alkyne via a cis-addition. The detailed mechanism is under study.
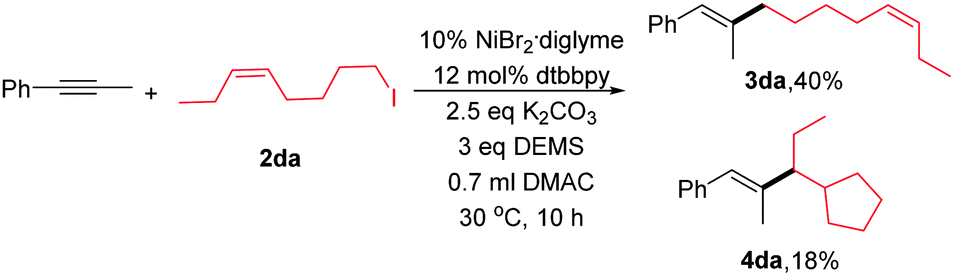 | (2) |
In summary, we have developed nickel-catalyzed regio- and stereoselective hydroalkylation of non-functionalized internal alkynes with non-activated alkyl halides for the first time. This method does not use sensitive organometallic reagents, and both of the starting materials are readily available, thus enabling the efficient synthesis of many single configuration tri-substituted alkenes. Due to the mild reaction conditions, this new approach shows good functional group compatibility. Not only aryl–alkyl substituted alkynes, but also alkyl–alkyl substituted alkynes can be successfully converted into the desired products.
We are very grateful for the support of our PhD supervisor. This work was supported by 2017qd11.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) C. Oger, L. Balas, T. Durand and J. M. Galano, Chem. Rev., 2013, 113, 1313–1350 CrossRef CAS PubMed; (b) R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783–1826 CrossRef CAS PubMed; (c) D. J. Pasto, in Comprehensive Organic Synthesis, ed. B. M. T. Fleming, Pergamon, Oxford, 1991, pp. 471–488 Search PubMed; (d) K. N. Campbell and L. T. Eby, J. Am. Chem. Soc., 1941, 63, 216–219 CrossRef CAS.
- (a) C. M. McMahon and E. J. Alexanian, Angew. Chem., Int. Ed., 2014, 53, 5974–5977 CrossRef CAS PubMed; (b) C. Wu and J. S. Zhou, J. Am. Chem. Soc., 2014, 136, 650–652 CrossRef CAS PubMed; (c) D. Mc Cartney and P. J. Guiry, Chem. Soc. Rev., 2011, 40, 5122–5150 RSC; (d) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (e) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS PubMed; (f) A. M. Lozano-Vila, S. Monsaert, A. Bajek and F. Verpoort, Chem. Rev., 2010, 110, 4865–4909 CrossRef CAS PubMed; (g) R. J. Haines and G. J. Leigh, Chem. Soc. Rev., 1975, 4, 155–188 RSC.
- Y. Nakao, H. Idei, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2009, 131, 5070–5071 CrossRef CAS PubMed.
- (a) L. Zhang, J. Cheng, B. Carry and Z. Hou, J. Am. Chem. Soc., 2012, 134, 14314–14317 CrossRef CAS PubMed; (b) S. Saito, S. Nakagawa, T. Koizumi, K. Hirayama and Y. Yamamoto, J. Org. Chem., 1999, 64, 3975–3978 CrossRef CAS; (c) M. Aoki, M. Kaneko, S. Izumi, K. Ukai and N. Iwasawa, Chem. Commun., 2004, 2568–2569 RSC; (d) K. Shimizu, M. Takimoto, Y. Sato and M. Mori, Org. Lett., 2005, 7, 195–197 CrossRef CAS PubMed; (e) T. Fujihara, Y. Tani, K. Semba, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2012, 51, 11487–11490 CrossRef CAS PubMed; (f) T. Fujihara, T. Xu, K. Semba, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2011, 50, 523–527 CrossRef CAS PubMed; (g) S. Li, W. Yuan and S. Ma, Angew. Chem., Int. Ed., 2011, 50, 2578–2582 CrossRef CAS PubMed; (h) X. Wang, M. Nakajima and R. Martin, J. Am. Chem. Soc., 2015, 137, 8924–8927 CrossRef CAS PubMed.
- (a) K. M. Partridge, S. J. Bader, Z. A. Buchan, C. E. Taylor and J. Montgomery, Angew. Chem., Int. Ed., 2013, 52, 13647–13650 CrossRef CAS PubMed; (b) E. Oblinger and J. Montgomery, J. Am. Chem. Soc., 1997, 119, 9065–9066 CrossRef CAS; (c) K. M. Miller, W.-S. Huang and T. F. Jamison, J. Am. Chem. Soc., 2003, 125, 3442–3443 CrossRef CAS PubMed; (d) G. M. Mahandru, G. Liu and J. Montgomery, J. Am. Chem. Soc., 2004, 126, 3698–3699 CrossRef CAS PubMed; (e) A. Herath, B. B. Thompson and J. Montgomery, J. Am. Chem. Soc., 2007, 129, 8712–8713 CrossRef CAS PubMed; (f) R. D. Baxter and J. Montgomery, J. Am. Chem. Soc., 2008, 130, 9662–9663 CrossRef CAS PubMed; (g) R. L. Patman, M. R. Chaulagain, V. M. Williams and M. J. Krische, J. Am. Chem. Soc., 2009, 131, 2066–2067 CrossRef CAS PubMed; (h) H. A. Malik, G. J. Sormunen and J. Montgomery, J. Am. Chem. Soc., 2010, 132, 6304–6305 CrossRef CAS PubMed; (i) K. Nakai, Y. Yoshida, T. Kurahashi and S. Matsubara, J. Am. Chem. Soc., 2014, 136, 7797–7800 CrossRef CAS PubMed; (j) E. P. Jackson and J. Montgomery, J. Am. Chem. Soc., 2015, 137, 958–963 CrossRef CAS PubMed; (k) H. Wang, S. Negretti, A. R. Knauff and J. Montgomery, Org. Lett., 2015, 17, 1493–1496 CrossRef CAS PubMed.
- (a) M.-Y. Ngai, A. Barchuk and M. J. Krische, J. Am. Chem. Soc., 2007, 129, 280–281 CrossRef CAS PubMed; (b) E. L. McInturff, K. D. Nguyen and M. J. Krische, Angew. Chem., Int. Ed., 2014, 53, 3232–3235 CrossRef CAS PubMed.
- (a) C.-C. Wang, P.-S. Lin and C.-H. Cheng, J. Am. Chem. Soc., 2002, 124, 9696–9697 CrossRef CAS PubMed; (b) W. Li, A. Herath and J. Montgomery, J. Am. Chem. Soc., 2009, 131, 17024–17029 CrossRef CAS PubMed; (c) C.-H. Wei, S. Mannathan and C.-H. Cheng, J. Am. Chem. Soc., 2011, 133, 6942–6944 CrossRef CAS PubMed; (d) A. D. Jenkins, A. Herath, M. Song and J. Montgomery, J. Am. Chem. Soc., 2011, 133, 14460–14466 CrossRef CAS PubMed; (e) D. P. Todd, B. B. Thompson, A. J. Nett and J. Montgomery, J. Am. Chem. Soc., 2015, 137, 12788–12791 CrossRef CAS PubMed.
- C.-Y. Zhou, S.-F. Zhu, L.-X. Wang and Q.-L. Zhou, J. Am. Chem. Soc., 2010, 132, 10955–10957 CrossRef CAS PubMed.
- (a) M. R. Uehling, A. M. Suess and G. Lalic, J. Am. Chem. Soc., 2015, 137, 1424–1427 CrossRef CAS PubMed; (b) A. M. Suess, M. R. Uehling, W. Kaminsky and G. Lalic, J. Am. Chem. Soc., 2015, 137, 7747–7753 CrossRef CAS PubMed.
- C. W. Cheung, F. E. Zhurkin and X. Hu, J. Am. Chem. Soc., 2015, 137, 4932–4935 CrossRef CAS PubMed.
- X. Y. Lu, J. H. Liu, X. Lu, Z. Q. Zhang, T. J. Gong, B. Xiao and Y. Fu, Chem. Commun., 2016, 52, 5324–5327 RSC.
- K. Nakamura and T. Nishikata, ACS Catal., 2017, 7, 1049–1052 CrossRef CAS.
- Lalic group realized copper-catalyzed hydro-alkylation of functionalized internal alkynes with allyl electrophilic reagents: M. Mailig, A. Hazra, M. K. Armstrong and G. Lalic, J. Am. Chem. Soc., 2017, 139, 6969–6977 CrossRef CAS PubMed.
- (a) D. J. Faulkner, Synthesis, 1971, 175 CrossRef CAS; (b) A. B. Flynn and W. W. Ogilvie, Chem. Rev., 2007, 107, 4698 CrossRef CAS PubMed; (c) E.-I. Negishi, Z. Huang, G. Wang, S. Mohan, C. Wang and H. Hattori, Acc. Chem. Res., 2008, 41, 1474 CrossRef CAS PubMed; (d) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (e) S. R. Chemler, D. Trauner and S. J. Danishefsky, Angew. Chem., Int. Ed., 2001, 40, 4544 CrossRef CAS; (f) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed; (g) G. Cahiez and A. Moyeux, Chem. Rev., 2010, 110, 1435 CrossRef CAS PubMed; (h) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed; (i) J. Terao, F. Bando and N. Kambe, Chem. Commun., 2009, 7336–7338 RSC; (j) K. Komeyama, Y. Okamoto and K. Takaki, Angew. Chem., 2014, 53, 11325–11328 CrossRef CAS PubMed.
- (a) The synthesis of substituted alkenyl halides and alkenyl metal reagents usually involves multistep reactions and the use of sensitive reagents; (b) C. W. Cheung and X. Hu, Chem. – Eur. J., 2015, 21, 18439–18444 CrossRef CAS PubMed.
- X. Lu, B. Xiao, Z. Zhang, T. Gong, W. Su, J. Yi, Y. Fu and L. Liu, Nat. Commun., 2016, 7, 11129 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1576631. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc01577e |
| This journal is © The Royal Society of Chemistry 2018 |