
Near-infrared luminescence of Nd3+ and Yb3+ complexes using a polyfluorinated pyrene-based β-diketonate ligand†
T. M. Georgeab,
S. Varugheseb and
M. L. P. Reddy*ab
aAcSIR-Academy of Scientific & Innovative Research, CSIR-NIIST Campus, Thiruvananthapuram, India
bMaterials Science and Technology Division, National Institute for Interdisciplinary Science and Technology (NIIST), Council of Scientific and Industrial Research (CSIR), Thiruvananthapuram-695 019, India. E-mail: mlpreddy55@gmail.com
First published on 18th July 2016
Abstract
A new polyfluorinated β-diketonate ligand containing a pyrene chromophore, namely, 4,4,5,5,6,6,6-heptafluoro-3-hydroxy-1-(pyren-1-yl)hex-2-en-1-one (Hhfpyr) has been designed and employed for the development of a series of near-infrared (NIR) emitting lanthanide complexes (Nd3+ and Yb3+) in the absence and presence of an ancillary ligand, 4,7-diphenyl-1,10-phenanthroline (bath). The isolated NIR-emitting lanthanide complexes [Nd(hfpyr)3(H2O) 1, Nd(hfpyr)3(bath) 2, Yb(hfpyr)3(H2O) 3 and Yb(hfpyr)3(bath) 4] have been characterized by various spectroscopic techniques and evaluated their photoluminescence properties. The photophysical properties disclosed that the developed pyrene-based β-diketonate ligand is well suited for the sensitization of Nd3+ as well as Yb3+ emissions, thanks to the favourable position of the triplet state (T1) of the ligand (ΔE = T1–4F3/2 = 4700 cm−1 for Nd3+ and ΔE = T1–2F5/2 = 6200 cm−1 for Nd3+), as evidenced from the phosphorescence spectra of the corresponding Gd3+ complexes. Most importantly, the displacement of solvent molecules from the coordination sphere of the NIR emitting lanthanide binary complexes (1 and 3) with an ancillary ligand markedly enhances the quantum yields (Φoverall = 0.45 for 1 to 1.07% for 2 and from 1.69 for 3 to 3.08% for 4) and excited state lifetime values (τ = 2.80 for 1 to 6.16 μs for 2 and from 6.88 for 3 to 13.45 μs for 4). Notably, Yb3+ ternary compound 4 with promising NIR luminescence properties was embedded into PMMA matrices, giving rise to a series of PMMA-supported hybrid materials (PMMA@4), where the thermal stability and the film-forming properties were significantly enhanced.
Introduction
Near-infrared (NIR) luminescence, especially from lanthanide complexes such as Nd3+ and Yb3+ has emerged as an area of paramount interest due to its pioneering technological applications in fields ranging from bioimaging to optical communications.1 However, since f–f transitions are parity forbidden, unligated Ln3+ ions have strikingly low molar absorption coefficients and hence direct excitation of lanthanide ions always leads to modest luminescence intensities.2 As a consequence, many efforts have been made in order to enhance the absorption coefficients of Ln3+ ions and thereby achieved efficient photoluminescence.3 Fortunately, this objective can be easily realized by prudent selection of new antenna chromophores with suitable conjugated motifs.4 In this context, β-diketonates are particularly important because such ligands can efficiently absorb ultraviolet light and transfer the absorbed energy to the central Ln3+ ions in an appropriately effective manner.4c,5 Indeed, there exists an absolute challenge to design and develop a novel β-diketonate ligand with relatively low triplet energy level, which matches well with the first excited state of NIR luminescent Ln3+ ions1a,6 (4F3/2 for Nd3+ = 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 257 cm−1 or 2F5/2 for Yb3+ = 10400 cm−1) as compared with visible luminescent Ln3+ ions having relatively high first excited state energy levels (5D0 for Eu3+ = 17286 cm−1 or 5D4 for Tb3+ = 20545 cm−1). Recently, some feasible strategies have been proposed by many researchers to improve the NIR luminescence of Ln3+ ions.7 For example, the fluorination of the β-diketonate ligand for minimizing the non-radiative decay pathways or molecular engineering of the β-diketonate ligand with appended suitable extended π-conjugated chromophore moieties to achieve efficient sensitization of the NIR luminescence of Ln3+ ions.8 Also, the replacement of coordinated solvent molecules around the central Ln3+ ion with an appropriate ancillary ligand, avoids the quenching effects due to the presence of high-frequency oscillators.9 Bünzli and co-workers has reported a new antenna molecule containing four benzoyltrifluoroacetone moieties anchored to single carbon atom and connected through four flexible methoxy groups namely as a sensitizer for NIR emitting lanthanides.10 New bis-β-diketonate ligand by coupling two mono-diketonate ligands (2-theonyltrifluoroacetone) has also been proposed for the sensitization of Yb3+ ions with higher quantum yields as compared with the mononuclear analogue.11
257 cm−1 or 2F5/2 for Yb3+ = 10400 cm−1) as compared with visible luminescent Ln3+ ions having relatively high first excited state energy levels (5D0 for Eu3+ = 17286 cm−1 or 5D4 for Tb3+ = 20545 cm−1). Recently, some feasible strategies have been proposed by many researchers to improve the NIR luminescence of Ln3+ ions.7 For example, the fluorination of the β-diketonate ligand for minimizing the non-radiative decay pathways or molecular engineering of the β-diketonate ligand with appended suitable extended π-conjugated chromophore moieties to achieve efficient sensitization of the NIR luminescence of Ln3+ ions.8 Also, the replacement of coordinated solvent molecules around the central Ln3+ ion with an appropriate ancillary ligand, avoids the quenching effects due to the presence of high-frequency oscillators.9 Bünzli and co-workers has reported a new antenna molecule containing four benzoyltrifluoroacetone moieties anchored to single carbon atom and connected through four flexible methoxy groups namely as a sensitizer for NIR emitting lanthanides.10 New bis-β-diketonate ligand by coupling two mono-diketonate ligands (2-theonyltrifluoroacetone) has also been proposed for the sensitization of Yb3+ ions with higher quantum yields as compared with the mononuclear analogue.11
Pyrene, a well-known organic hydrocarbon was extensively employed as a fluorophore of choice in the field of photochemistry and photophysics.12 In addition, some of the pyrene-derivatives have been used in Organic Light Emitting Diodes intending to improve the hole transporting ability because of its electron-rich property.13 There are also a few reported examples that use pyrene as a sensitizer for lanthanide emission. The ‘antenna effect’ in europium complexes involving a pyrene-based triacid ligand was first disclosed by Fages et al.14 Later, near-IR emission was noted in ytterbium and neodymium complexes containing a pyrene chromophore linked to a macrocycle via different tether lengths.15 In the later studies, Pope reported Yb3+, Nd3+ and Er3+ complexes with two pyrene chromophores tethered by a diethylene triaminepentaacetic acid chelate.16 These investigations have inspired us to develop a new antenna ligand for the sensitization of Nd3+ and Yb3+ ions by anchoring a pyrene chromophore to the β-diketonate ligand (Fig. 1).
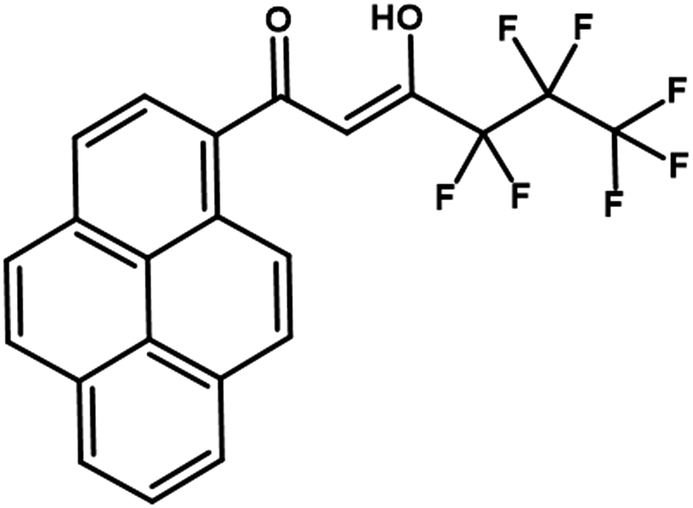 | ||
| Fig. 1 Structure of the ligand Hhfpyr. | ||
It is well documented that the NIR-emitting Ln3+ ions are especially inclined to vibrational deactivation.9 Organic chromophores containing high-energy oscillators, such as C–H and O–H bonds are able to quench the Ln3+ excited states non-radiatively, thus exhibiting weak luminescence intensities and shorter excited-state lifetimes. The replacement of C–H bonds with C–F bonds is an important strategy for the design and development of novel Ln3+ complexes with efficient photophysical properties.7h,17 As per the earlier literature reports, the replacement of C–H bonds in a β-diketonate ligand with low energy C–F oscillators is able to lower the vibrational energy of the ligand and thereby enhances the emission intensity of the Ln3+ ion.18 In addition, due to heavy atom effect, which facilitate the intersystem crossing and as a result the lanthanide-centered luminescent properties are improved.19 Therefore in the present work, a new β-diketonate molecule, namely, 4,4,5,5,6,6,6-heptafluoro-3-hydroxy-1-(pyren-1-yl)hex-2-en-1-one (Hhfpyr) has been designed by simultaneously incorporating pyrene moiety as well as heptafluorinated alkyl chain. The developed β-diketonate ligand has been utilized for the construction of a series of lanthanide complexes (Ln3+ = Nd, Yb and Gd) in the absence and presence of an ancillary ligand, 4,7-diphenyl-1,10-phenanthroline. The synthesized lanthanide complexes were characterized by various spectroscopic techniques and evaluated their photophysical properties.
Nevertheless, NIR emitting Ln3+–β-diketonate complexes typically exhibit low thermal-stability, limited photostability and poor mechanical properties. These inherent limitations hinder the practical application of NIR emitters in many of the optoelectronic technologies. It is well-known that the blending of luminescent near-IR emitting Ln3+ compounds in polymeric materials renders a series of advantages for the development of molecular materials, for instance, thermal, chemical and mechanical stability, biocompatibility and the photoluminescence properties.6a,7e,20 To the best of our knowledge, there are only few examples in the literature dealing with the incorporation of NIR emitting ternary-β-diketonate complexes into the PMMA materials.6c,21 Hence in the present study the newly developed luminescent NIR emitting Yb3+ complex has been incorporated into a host polymer matrix, such as poly(methyl methacrylate) films and investigated their photoluminescence behaviour.
Experimental
Materials and characterization
Ytterbium(III) nitrate hexahydrate (99.99%), neodymium(III) nitrate hexahydrate (99.99%), gadolinium(III) nitrate hexahydrate (99.99%), lanthanum(III) nitrate hexahydrate (99.99%), 1-acetylpyrene (97%), ethyl perfluorobutyrate (97%), sodium hydride (60% dispersion in mineral oil), poly(methyl methacrylate) (98%) and bathophenanthroline (97%) were purchased from Sigma-Aldrich and used without further purification. All the other chemicals employed were of analytical reagent grade.Single-crystal XRD data for complex 2 were collected with a Rigaku Saturn 724+ diffractometer using graphite-monochromated Mo Kα radiation, and the data were processed using Rigaku Crystal Clear software. The molecular structure of the complex was solved and refined by the SHELXTL suite of programs.22
Elemental analyses were carried out on Elementar – vario MICRO cube elemental analyzer. A Perkin-Elmer Spectrum two FT-IR spectrometer was used to record the infra-red spectral data and a Bruker Avance II 500 MHz NMR spectrometer was used to record the 1H NMR (500 MHz) and 13C NMR (125.7 MHz) spectra with tetramethylsilane as the internal standard. The electrospray ionization (ESI) mass spectra were measured with a Thermo Scientific Exactive Benchtop LC/MS Orbitrap Mass Spectrometer. The thermogravimetric analyses were performed on a TG/DTA-6200 (SII Nano Technology Inc., Japan). The optical spectra of the synthesized ligand and its corresponding metal complexes were recorded with a Shimadzu UV-3600 UV-vis spectrophotometer.
Photophysical measurements were carried out in the solid state at room temperature. Emission spectra were obtained with an Edinburgh FLS 980 spectrofluorometer equipped with a 450 W xenon arc lamp. Emission spectra were corrected for source intensity (lamp and grating) and emission spectral response (detector and grating) by standard correction curves. The absolute fluorescence quantum yields were measured on an Edinburgh FLS 980 steady state spectrometer using an integrating sphere. Luminescent excited state lifetimes in the range from 0.5 ns to 50 μs were measured by an Edinburgh FLS 980 spectrofluorometer equipped with a digital oscilloscope (Tektronix) for data acquisition in time-correlated single-photon counting experiments with a pulsed microsecond xenon flashlamp. The estimated experimental errors are 2 nm on the photoluminescence bands maxima, 5% on the luminescence quantum yield. The lifetime measurements carried out at low temperature using a Spex 1040D phosphorimeter.
Synthesis of the ligand 4,4,5,5,6,6,6-heptafluoro-3-hydroxy-1-(pyren-2-yl)hex-2-en-1-one (Hhfpyr)
The new β-diketonate ligand (Hhfpyr) was prepared according to a modified Claisen condensation method as detailed in Scheme 1. 1-Acetylpyrene (1.0 mmol) and ethyl perfluorobutyrate (1.0 mmol) were dissolved in dry tetrahydrofuran (25 mL) and the resultant mixture was stirred for 15 min in an ice bath at 0 °C. To this reaction mixture, sodium hydride (2.0 mmol) was added dropwise in nitrogen atmosphere and stirred for 20 min, followed by further stirring for 12 h at 70 °C. To the above reaction mixture, 40 mL of 2 M HCl was added and extracted twice into dichloromethane (2 × 30 mL). Then the organic layer was collected and dried over Na2SO4, and the solvent was removed by evaporation. The product obtained was then purified by column chromatography on silica gel with a solvent mixture consisting of hexane and ethyl acetate (10:1) as an eluent. Yield: 80%. Elemental analysis (%): calculated for C22H11F7O2 (440.06): C 60.01, H 2.52; found: C 60.23, H 2.63. 1H NMR (CDCl3, 500 MHz) δ (ppm): 15.53 (broad, enol–OH), 8.78 (d, 1H, J = 9 Hz), 8.23 (m, 6H), 8.05 (m, 2H), 6.62 (s, 1H). 13C NMR (125.7 MHz, CDCl3) δ (ppm): 190.58, 176.78, 134.71, 131.14, 130.51, 130.21, 129.90, 128.09, 127.05, 126.78, 126.70, 126.64, 126.55, 125.01, 124.38, 125.01, 124.38, 124.26, 124.11, 99.48, 77.16–76.05 (CDCl3). FT-IR (KBr) νmax (cm−1): 3427 (O–H), 1596, 1508, 1346, 1226, 1069, 962, 898, 763, 680, 539. m/z = 463.05 (M + Na)+.
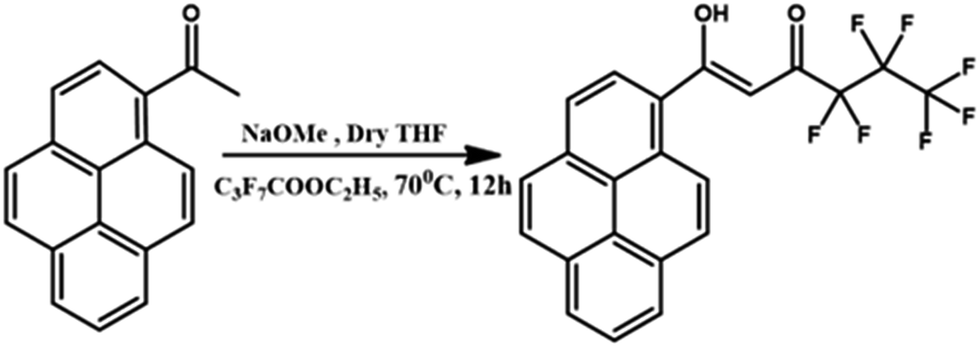 | ||
| Scheme 1 Synthetic procedure for the ligand Hhfpyr. | ||
Synthesis of complexes Ln(hfpyr)3(H2O) [Ln = Nd (1), Yb (3) and Gd (5)]
To a methanolic solution of Hhfpyr (12 mmol), 12 mmol of NaOH in water was added and stirred for 5 min. Ln(NO3)3·6(H2O) in 3 mL of water (4 mmol) was added drop-wise to the above reaction mixture and stirred for 24 h at 298 K (Scheme 2). The resultant crude precipitate was filtered, washed with water and dried. The obtained metal complex was recrystallized from chloroform solution.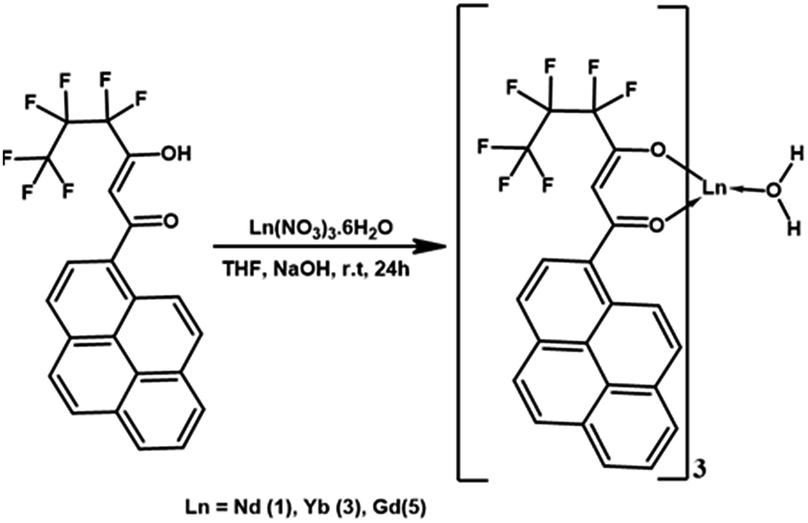 | ||
| Scheme 2 Synthesis of the Ln3+ (Ln = Nd, Yb and Gd) binary complexes. | ||
Synthesis of Ln3+ ternary complexes Ln(hfpyr)3(bath) [Ln = Nd (2), Yb (4)]
The ternary Ln3+ compounds were synthesized by mixing equimolar solutions of the corresponding binary complexes and an ancillary ligand; bathophenanthroline (bath) in CHCl3 solution and the resultant mixture was stirred for 12 h at 70 °C. The metal complexes were then isolated after the removal of solvent by evaporation process. Finally, the ternary lanthanide complexes were obtained by recrystallization from chloroform solution (as described in Scheme 3).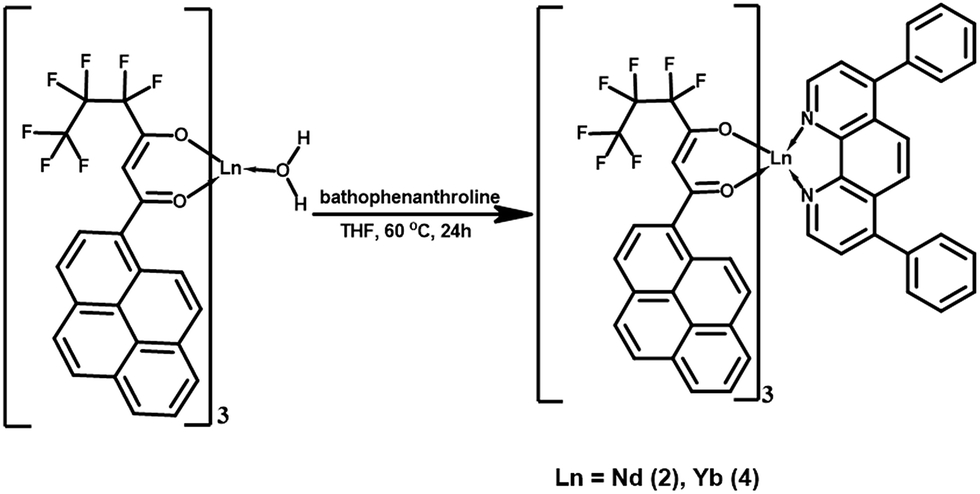 | ||
| Scheme 3 Synthesis of the Ln3+ (Ln = Nd, Yb) ternary complexes. | ||
Synthesis of Yb3+ complex doped PMMA polymer films
The PMMA polymer was doped with the Yb3+ complex 4 in the proportions 1, 3, 5, 7 and 9% (w/w). The PMMA powder was dissolved in chloroform, followed by addition of the required amount of complex 4 in chloroform solution, and the respective mixture was heated at 40 °C for 35 min. The polymer films were then obtained after evaporation of excess solvent at 60 °C.Results and discussion
Synthesis and characterization of the Hhfpyr ligand and Ln3+ complexes 1–5
The β-diketonate ligand 4,4,5,5,6,6,6-heptafluoro-3-hydroxy-1-(pyren-1-yl)hex-2-en-1-one (Hhfpyr) was synthesized in 80% yield by a modified Claisen condensation reaction of 1-acetylpyrene with the ethylperfluorobutyrate ester in the presence of sodium hydride in THF medium. The corresponding one pot synthesis of the ligand is described in Scheme 1. The developed ligand has been characterized by the 1H NMR, 13C NMR, FT-IR and electron spray ionisation mass spectroscopic (ESI-MS) methods (Fig. S1–S3 and S9 in the ESI†) as well as by elemental analysis. The developed β-diketonate ligand mainly exists as enol form in CDCl3 solution, as evident from the 1H NMR spectrum of the compound. In the 1H NMR spectrum of Hhfpyr, a broad peak at δ 15.64 ppm corresponding to enolic –OH has been noted. Further, the absence of methyne protons at δ 3.70 ppm confirms the existence of the ligand in enolic form. The synthesis routines for Ln3+ binary and ternary complexes are detailed in Schemes 2 and 3, respectively. The elemental analyses and ESI-MS studies (Fig. S4–S8 in the ESI†) of Ln3+ complexes (1–5) revealed that the central lanthanide ion is coordinating to three β-diketonate ligands. On the other hand, in the case of Ln3+ ternary complexes (2 and 4), one molecule of the bidentate nitrogen donor, 4,7-diphenyl-1,10-phenanthroline (bath), is also present in the coordination sphere. The FT-IR spectra of the binary Ln3+ complexes (1, 3 and 5) display a broad absorption in the 3000–3500 cm−1 region, thereby illustrating the presence of water molecule in the coordination sphere of the metal ions (Fig. S10, S12 and S14 in the ESI†). The absence of this broad band in the case of ternary Nd3+ and Yb3+ complexes (2 and 4) inferred that the water molecule is successfully displaced by the bidentate bathophenanthroline ligand (Fig. S11 and S13 in the ESI†). The carbonyl stretching frequency (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of free ligand Hhfpyr (1596 cm−1) was shifted to higher wave numbers in complexes 1–5 (1609 cm−1 for 1; 1610 cm−1 for 2; 1607 cm−1 for 3; 1608 cm−1 for 4; 1609 cm−1 for 5) demonstrating the involvement of carbonyl oxygen in the complex formation with the Ln3+ ion. The bands assigned to bathophenanthroline ring stretching modes CN and CC can be observed in the 1540–1500 cm−1 range and in the 1030–1000 cm−1 range, respectively. These bands are shifted in comparison with that of free ancillary ligand, suggesting that bathophenanthroline is coordinating to Ln3+ ion.
O) of free ligand Hhfpyr (1596 cm−1) was shifted to higher wave numbers in complexes 1–5 (1609 cm−1 for 1; 1610 cm−1 for 2; 1607 cm−1 for 3; 1608 cm−1 for 4; 1609 cm−1 for 5) demonstrating the involvement of carbonyl oxygen in the complex formation with the Ln3+ ion. The bands assigned to bathophenanthroline ring stretching modes CN and CC can be observed in the 1540–1500 cm−1 range and in the 1030–1000 cm−1 range, respectively. These bands are shifted in comparison with that of free ancillary ligand, suggesting that bathophenanthroline is coordinating to Ln3+ ion.
To further understand the coordination behaviour of the ligands with the lanthanide ions, in the current study, anti-paramagnetic lanthanum complexes have been synthesized and characterized by various spectroscopic techniques (experimental procedure and characterizations are given in the ESI†). The 1H NMR spectrum (Fig. S15 in the ESI†) of the lanthanum binary β-diketonate complex, La(hfpyr)3(H2O) is consistent with the presence of three Hhfpyr units coordinated to the lanthanide ion. The signal for methine proton (–CH) of Hhfpyr appears at 6.51 ppm (δ) and the aromatic protons of the pyrene moiety resonates in the range 8.49 to 7.02 ppm (δ). The upfield shift of the β-diketonate resonances, in the complex, substantiates coordination of ligands with the lanthanide ion. The proton signals of the coordinated water molecule with the metal ion can be noted at 2.59 ppm (δ). In the ternary lanthanum complex, La(hfpyr)3(bath), the methine proton appears at 6.48 ppm (δ). The signals due to aromatic protons of pyrene and bathophenanthroline moiety appear in the range 9.57 to 6.78 ppm (δ) (Fig. S16 in the ESI†). The proton signals appeared in the ternary complex indicates the presence of three Hhfpyr units and one bathophenanthroline moiety in the coordinated complex. Further, no signals for coordinated water molecule noted in the La(hfpyr)3(bath), which indicates the displacement of a coordinated water molecule with the ancillary ligand in the corresponding ternary complex.
The thermal behaviour of Nd3+ and Yb3+ β-diketonate complexes (1–5) was evaluated by means of thermogravimetric analysis (TGA) under nitrogen atmosphere and the results are given in Fig. S21–S24 in the ESI.† It is clear from the TGA data that the complexes 1, 3 and 5 undergo mass loss approximately 1.19% (calcd: 1.20%) in the first step upto 160 °C, corresponding to the elimination of coordinated water molecule. On the other hand, in the case of ternary Ln3+ complexes (2 and 4), no weight loss was noted in the range of 120–160 °C, which indicates that these complexes exist as anhydrous in nature. The above results are in accordance with the FT-IR spectral data. The weight loss noted in the thermal analyses of these complexes is found to be much lower than the calculated value for the non-volatile lanthanide oxide, indicating the partial sublimation of these compounds under atmospheric pressure which is well documented in many of the lanthanide fluorinated complexes.5b,23
X-ray crystal structure of 3
Slow diffusion of hexane into a solution of the Yb3+ binary β-diketonate complex in methanol resulted in the growth of single crystals of 3. However, our efforts to grow single-crystals of the other Ln3+ complexes were not fruitful. The molecular structure of the Yb3+-pyrene anchored β-diketonate complex (3) obtained by X-ray single-crystal diffraction technique is shown in Fig. 2. The pertinent data collection parameters and a list of significant bond distances and angles are presented in Tables 1 and 2, respectively. Yb3+ binary β-diketonate complex was found to crystallize in the triclinic crystal system with a P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The structure reveals that the Yb3+ center adopts a distorted-trigonal prismatic coordination geometry, comprising of three β-diketonate ligands and one solvent water molecule. These results are in good agreement with the crystal structure of tris(acetylacetonato)aquoytterbium reported elsewhere.24 In general, most of the lanthanide–β-diketonate complexes are eight-coordinated and the coordination sphere features three bidentate β-diketonate ligands and two solvent molecules.5a Unusually in the present study, the binary Yb-β-diketonate complex is seven-coordinated and the coordination sphere consisting of three bidentate β-diketonate ligands and one water molecule. This can be attributed to the presence of three bulky conjugated pyrene appended β-diketonate ligands in the coordination sphere of the metal ion, which may sterically hinders the presence of water molecule. The average metal–oxygen distance (2.245 Å) of the β-diketonate ligands is found to be shorter than of the coordinated water molecule (2.298 Å). This observation could be attributed to the presence of a formal negative charge on the β-diketonate oxygen atoms which could enhance the binding to the Yb3+ cation due to electrostatic effects.5a,25 Further, it is interesting to note that one of the metal–oxygen distance of the β-diketonate ligand is surprisingly larger (Yb1–O(7) = 2.298 Å) as compared to the remaining metal–oxygen distances of the β-diketonates [Yb1–O(2) = 2.269 Å; Yb1–O(3) = 2.235 Å; Yb1–O(4) = 2.221 Å; Yb1–O(5) = 2.213 Å; Yb1–O(6) = 2.233 Å]. This may be due to the strong intermolecular hydrogen bonding formation between the coordinated water molecule of Yb1 and one of the β-diketonate oxygen atom coordinated to the neighbouring metal centre as shown in Fig. 3 [the O–H⋯O (H⋯O distance = 2.10 Å) with an angle of 133.0° falls within the typical range hydrogen bonding interactions].25 In addition, there also exists a strong intermolecular hydrogen bonding interaction between the coordinated water molecule and the fluorine atom of the β-diketonate ligand coordinated to the adjacent Yb1 centre [O–H⋯F (H⋯F = 2.18 Å) with an angle of 143.0°]. These intermolecular interactions combine to form an interesting dimeric unit between the two coordinated metal centres.
space group. The structure reveals that the Yb3+ center adopts a distorted-trigonal prismatic coordination geometry, comprising of three β-diketonate ligands and one solvent water molecule. These results are in good agreement with the crystal structure of tris(acetylacetonato)aquoytterbium reported elsewhere.24 In general, most of the lanthanide–β-diketonate complexes are eight-coordinated and the coordination sphere features three bidentate β-diketonate ligands and two solvent molecules.5a Unusually in the present study, the binary Yb-β-diketonate complex is seven-coordinated and the coordination sphere consisting of three bidentate β-diketonate ligands and one water molecule. This can be attributed to the presence of three bulky conjugated pyrene appended β-diketonate ligands in the coordination sphere of the metal ion, which may sterically hinders the presence of water molecule. The average metal–oxygen distance (2.245 Å) of the β-diketonate ligands is found to be shorter than of the coordinated water molecule (2.298 Å). This observation could be attributed to the presence of a formal negative charge on the β-diketonate oxygen atoms which could enhance the binding to the Yb3+ cation due to electrostatic effects.5a,25 Further, it is interesting to note that one of the metal–oxygen distance of the β-diketonate ligand is surprisingly larger (Yb1–O(7) = 2.298 Å) as compared to the remaining metal–oxygen distances of the β-diketonates [Yb1–O(2) = 2.269 Å; Yb1–O(3) = 2.235 Å; Yb1–O(4) = 2.221 Å; Yb1–O(5) = 2.213 Å; Yb1–O(6) = 2.233 Å]. This may be due to the strong intermolecular hydrogen bonding formation between the coordinated water molecule of Yb1 and one of the β-diketonate oxygen atom coordinated to the neighbouring metal centre as shown in Fig. 3 [the O–H⋯O (H⋯O distance = 2.10 Å) with an angle of 133.0° falls within the typical range hydrogen bonding interactions].25 In addition, there also exists a strong intermolecular hydrogen bonding interaction between the coordinated water molecule and the fluorine atom of the β-diketonate ligand coordinated to the adjacent Yb1 centre [O–H⋯F (H⋯F = 2.18 Å) with an angle of 143.0°]. These intermolecular interactions combine to form an interesting dimeric unit between the two coordinated metal centres.
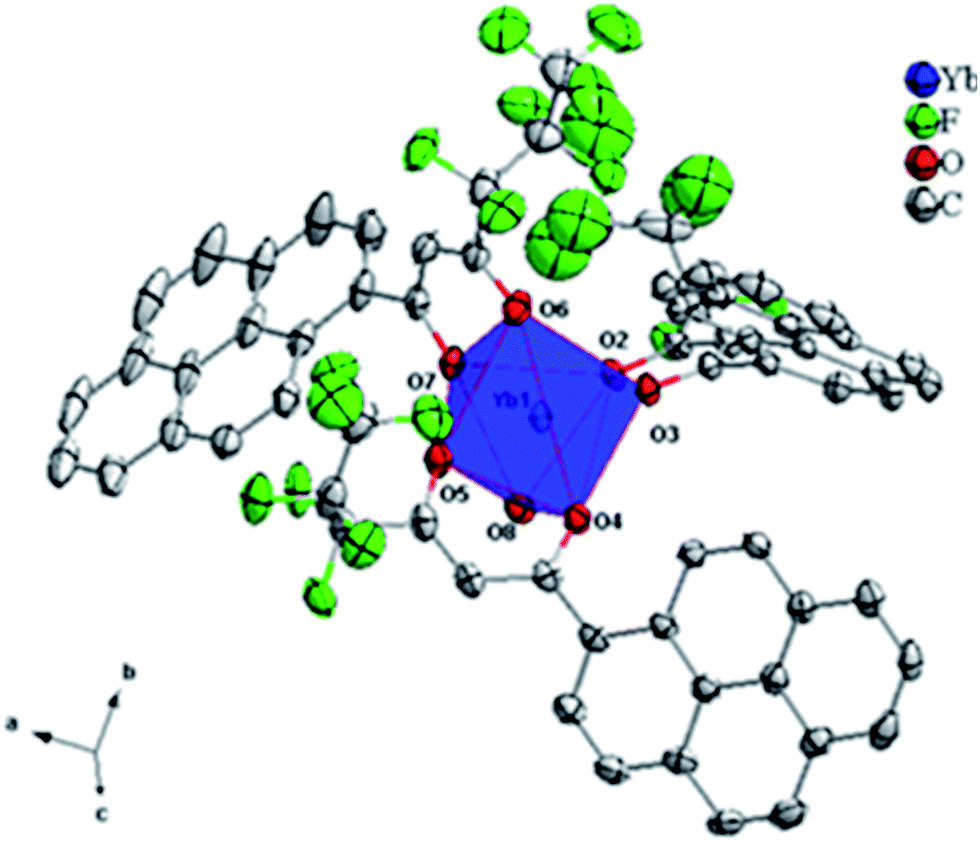 | ||
| Fig. 2 ORTEP diagram of [Yb(hfpyr)3(H2O)] 3 with the thermal ellipsoids drawn at 30% probability level and the hydrogen atoms removed for clarity. | ||
| Formula | C66H32F21O7Yb |
| Formula weight | 1508.96 |
| Crystal system | Triclinic |
| Space group | P |
| Crystallite size (mm3) | 0.20 × 0.20 × 0.15 |
| Temperature (K) | 123 K |
| a (Å) | 14.204(3) |
| b (Å) | 15.184(3) |
| c (Å) | 17.459(4) |
| α (deg) | 113.56(1) |
| β (deg) | 108.085(15) |
| γ (deg) | 94.308(12) |
| V (Å3) | 3194.2(12) |
| Z | 2 |
| Dcalcd (g cm−1) | 1.569 |
| μ (Mo Kα) (mm−1) | 1.577 |
| F(000) | 1486.0 |
| R1 [I > 2σ(I)] | 0.0606 |
| wR2 [I > 2σ(I)] | 0.1638 |
| R1 (all data) | 0.0677 |
| wR2 (all data) | 0.1696 |
| GOF | 1.178 |
| CCDC | 1473942 |
| Bond lengths (Å) | Bond angles (°) | ||
|---|---|---|---|
| Yb1–O2 | 2.2685 | O5–Yb1–O4 | 75.928 |
| Yb1–O3 | 2.2352 | O3–Yb1–O2 | 75.222 |
| Yb1–O4 | 2.2209 | O3–Yb1–O7 | 139.825 |
| Yb1–O5 | 2.2130 | O2–Yb1–O8 | 80.475 |
| Yb1–O6 | 2.2335 | O6–Yb1–O8 | 153.723 |
| Yb1–O7 | 2.2980 | O5–Yb1–O6 | 88.143 |
| Yb1–O8 | 2.2980 | O4–Yb1–O3 | 73.531 |
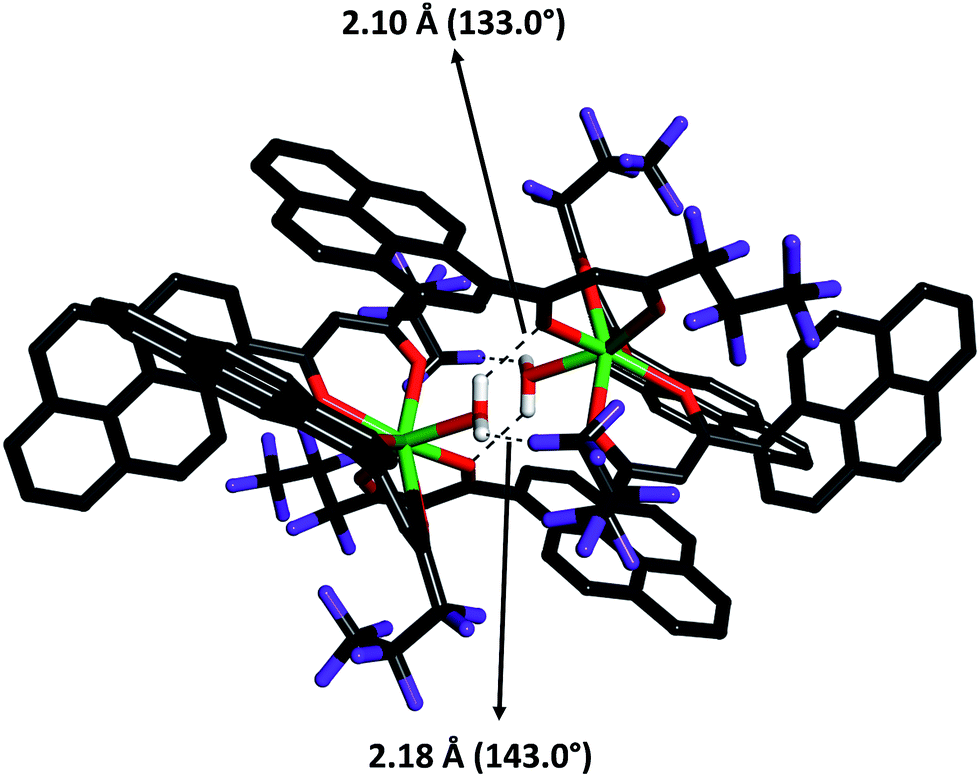 | ||
| Fig. 3 Intermolecular hydrogen bond present in 3 between water oxygen atoms and β-diketonate oxygen and fluorine atoms of Hhfpyr (shown with broken lines). | ||
Optical spectra of the Ln3+ complexes
The room-temperature optical spectra of the ligands and the corresponding Nd3+ and Yb3+ complexes (1–4) recorded in THF solution (c = 1 × 10−5 M) are shown in Fig. 4 and 5 respectively. The spectral shapes of the complexes are similar to that of the free Hhfpyr, indicating that the coordination of Ln3+ ion does not have significant influence on the energy of the singlet state of the β-diketonate. The ligand displays a composite broad band in the wavelength region 325–475 nm (λmax = 370 nm), which can be assigned to the singlet–singlet n–π* enolic transition of the β-diketonate. In addition, a high energy absorption band noted in the region 275–325 nm can be attributed to the π–π* transition of the aromatic moiety of the β-diketonate. The molar absorption coefficient (ε) of the developed β-diketonate ligand was found to be 15800 L mol−1 cm−1 at λmax = 370 nm, which highlights that the β-diketonate ligand has an ability to absorb light. The magnitudes of the molar absorption coefficient values of Nd3+ complexes 1 (ε = 49800 L mol−1 cm−1 at λmax = 370 nm) and 2 (ε = 50200 L mol−1 cm−1 at λmax = 370 nm) were found to be approximately three-fold higher than that of the β-diketonate ligand. This is in consistent with the presence of three β-diketonate ligands in the respective complexes as observed from the elemental analysis data. Similar trends have been noticed in the case of Yb3+ complexes 3 (ε = 49500 L mol−1 cm−1 at λmax = 370 nm) and 4 (ε = 49300 L mol−1 cm−1).
 | ||
| Fig. 4 UV-vis absorption spectra of the ligands Hhfpyr, bath and complexes 1 and 2 in THF (c = 1 × 10−5 M) solution at 298 K. | ||
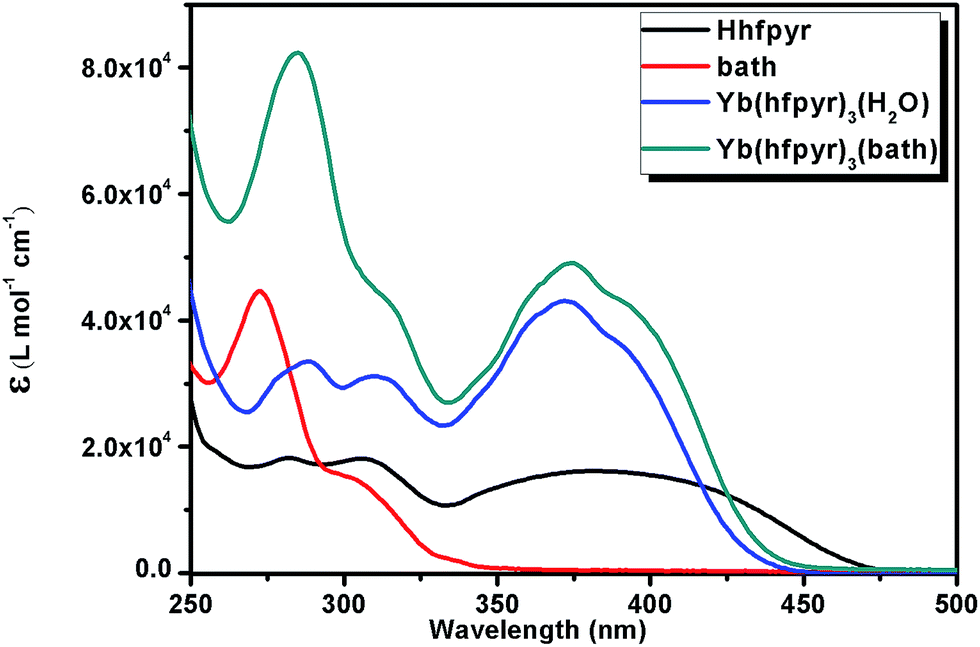 | ||
| Fig. 5 UV-vis absorption spectra of the ligands Hhfpyr, bath and complexes 3 and 4 in THF (c = 1 × 10−5 M) solution at 298 K. | ||
Photophysical properties
NIR luminescent Nd3+ and Yb3+ complexes possess considerable promise for practical applications, as their photophysical properties have several distinct advantages over organic fluorophores and semiconductor nanoparticles.1c For near-infrared Ln3+, their lowest excited states, and their ground states are primarily close in their energy, and hence, the emission often occurs in the infrared region and their intensities are weaker by several orders of magnitude, compared to that of visible light emitting Ln3+ ions.8c Moreover, their deactivation process often occurs easily through a non-radiative transition. Therefore, a fundamental challenge is to develop an appropriate antenna molecule for the sensitization of the near-infrared lanthanide ions. Thus, in the current study, a new β-diketonate molecule has been developed by anchoring pyrene as a chromophore group.To understand the energy transfer processes in the newly isolated NIR emitting Nd3+ and Yb3+ β-diketonate complexes, it is necessary to determine the singlet (S1) and triplet (T1) energy levels of the synthesized new β-diketonate ligand. The singlet energy level of the ligand was determined by reference to the UV-vis upper absorption edge of the Gd(hfpyr)3(H2O) 5 (Fig. 6), and the value was found to be 22935 cm−1 (436 nm).26 The triplet energy level of the β-diketonate ligand was determined by referring to the lower wavelength emission edge from the low-temperature phosphorescence spectrum of (Fig. 6) the Gd(hfpyr)3(H2O) 5.27 The efficient ligand-to-metal energy transfer requires a good intersystem-crossing efficiency, which is maximized when the energy difference between singlet and triplet states, ΔE (S1–T1), is closed to 5000 cm−1 as coined by Reinhoudt's empirical rule.28 In the present system, it amounts to 6728 cm−1, and therefore the newly developed β-diketonate ligand exhibits a good intersystem-crossing efficiency. It is well recognized that the Gd3+ complexes are a popular choice for elucidating the triplet energy level of a newly developed antenna molecule due to the following reason: (i) the first excited state energy levels of Gd3+ are situated at high energies (5IJ = 36900 cm−1), and hence there is no Gd3+ emission in the visible region and all the emissions noted is due to the ligand part of the complex. Therefore, the lower emission edge of the 77 K phosphorescence spectrum of the Gd3+ complex designates the triplet energy level of the ligand.27 Thus the triplet energy level of the developed β-diketonate ligand (T1 = 16207 cm−1 617 nm) lie well above the energy of the main emitting level of 4F3/2 for Nd3+ (11257 cm−1) or 2F5/2 for Yb3+ (10400 cm−1), implying that the developed β-diketonate ligand can act as an efficient antenna molecule for the sensitization of both trivalent Nd3+ or Yb3+ ions. The room-temperature lifetime experiment of Gd(hfpyr)3(H2O) shows that the decay curve can be fitted to a bi-exponential decay with τ1 = 1.58 ns and τ2 = 6.66 ns (Fig. S25 in the ESI†). This indicates that the main energy transfer in the present complexes may be through the triplet state of the ligand.29 The long lifetime value (typically 522 μs) measured for Gd(hfpyr)3(H2O) at 77 K is consistent with the emission from characteristic triplet state (Fig. S26 in the ESI†).30
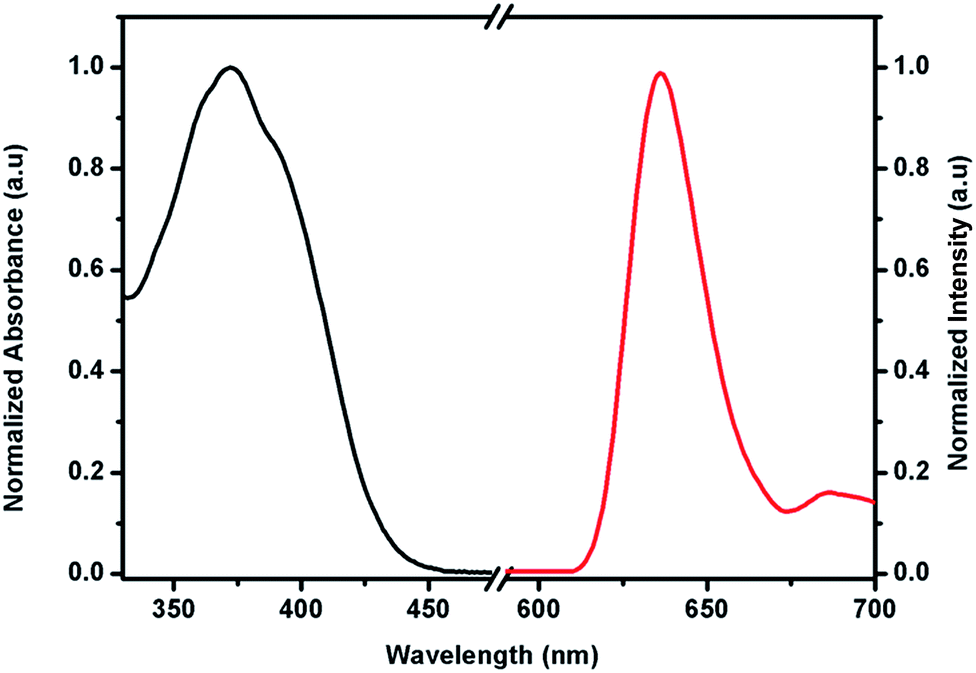 | ||
| Fig. 6 UV-vis absorption spectrum at 298 K (black) and 77 K phosphorescence spectra (red) of the Gd(hfpyr)3(H2O) complex. | ||
The excitation and emission profiles for the Nd3+ complexes (1 and 2) in the solid state at room temperature are depicted in Fig. 7. The excitation spectra of these complexes monitored around the intense 4F3/2 → 4I11/2 transition (1068 nm) of the Nd3+ ion consist of a broad band in the region 250–500 nm (λex = 400 nm) and several weak intra-configurational f–f transitions (Fig. 7). The broad band is due to the excitation of the organic chromophores (Hhfpyr and bath) and the weak intraconfigurational f–f transitions originating from the ground state of the Nd3+ ion. The f–f transitions could be assigned to 4I9/2 → 2K15/2, 4G9/2, 2(D,P)3/2, 4G11/2 (509 nm), 4I9/2 → 4G9/2, 4G7/2, 2K13/2 (528 nm), and 4I9/2 → 4G5/2, 2G7/2 (584 nm).8g However, these f–f transitions are weaker than the absorption of the organic ligands, which proves that the luminescence sensitization via excitation of the pyrene-based polyfluorinated-β-diketonate ligand is efficient. Moreover, the excitation spectra of these complexes show a good overlap with ligand-centred ππ* absorption band of the complex which reflects that energy transfer takes place from ligands to Nd3+ ion (antenna effect).
 | ||
| Fig. 7 Room temperature excitation and emission spectra of complexes 1 and 2 in the solid-state. | ||
Under the ligand excitation (λex = 400 nm), the emission spectra of the Nd3+ complexes (1 and 2) exhibit characteristic sharp bands of the Nd3+ ion in the range 850–1400 nm spectral range (Fig. 7). The emission spectra essentially display three emission peaks that are assigned to 4F3/2 → 4I9/2 (891 nm), 4F3/2 → 4I11/2 (1068 nm) and 4F3/2 → 4I13/2 (1331 nm).31 It is interesting to note that some crystal field fine structure can be observed from the emission profiles of these complexes, which illustrates that the Nd3+ ion occupies well-defined crystallographic sites in the complexes. Among these transitions, the intensity of the 4F3/2 → 4I11/2 transition is strongest, which has potential application in laser systems.1a On the other hand, the longer emission wavelength line of Nd3+ (4F3/2 → 4I13/2) at 1331 nm may find applications in the development of new optical amplification materials for telecommunications.1a Further, a moderate residual ligand emission has been observed in Nd3+ binary complex (Fig. S27 in ESI†). On the other hand, negligible residual ligand emission can be noted from ternary Nd3+ compound. The results demonstrated that the displacement of a water molecule in the coordination sphere of the Nd3+ in Nd(hfpyr)3(H2O) by an ancillary ligand, 4,7-diphenyl-1,10-phenanthroline remarkably enhances (4-fold) the emission intensity of the transition 4F3/2 → 4I11/2.
The solid-state room temperature (298 K) excitation spectra of Yb3+ complexes (3 and 4) obtained by monitoring the characteristic emission of the Yb3+ ion at 979 nm are given in Fig. 8. The excitation profiles are dominated by a broad band ranging from 250–550 nm for both the binary and ternary complexes. This broad band can be accredited to the absorption of the organic chromophores (Hhfpyr and bath) employed for the synthesis of the Yb3+ ion complexes. The emission spectra of the Yb3+ complexes derived from pyrene-based β-diketonate ligand (3 and 4), upon ligand-mediated excitation at 400 nm, clearly shows the characteristic emission bands for Yb3+ ion at 979 nm, which are assigned to 2F5/2 → 2F7/2 transition.31d,e Further, it can be noted that the primary emission band of Yb3+ ion has been split into an envelope of bands arising at the lower energy side (1006 nm and 1033 nm). These spectral features can be attributed to the splitting of the emitting levels as a consequence of ligand field effects.32 The emission intensity at 979 nm of Yb3+ ternary complex (4) has been significantly enhanced (about three fold) as compared to Yb3+ binary complex (2). Accordingly, negligible residual ligand emission has been noted in Yb3+ ternary complex as compared to binary counterpart (Fig. S28 in ESI†).
 | ||
| Fig. 8 Room temperature excitation and emission spectra of complexes 3 and 4 in the solid state. | ||
Thus, the Yb3+ ion emission in the NIR region is important because in these regions biological tissues and fluids are relatively transparent, and the development of new Yb3+ ion complexes may find as bioprobes in fluoroimmunoassay and in vivo applications.1a,33
The excited state 4F3/2 (Nd3+) and 2F5/2 (Yb3+) lifetime values (τobs) of the Ln3+ complexes 1–4 were determined at ambient temperature (298 K), by monitoring within the intense lines of the 4F3/2 → 4I11/2 and 2F5/2 → 2F7/2 transitions, respectively (Fig. 9 and 10), and the pertinent values are given in Table 3. The observed luminescent decay profiles correspond to mono-exponential functions, highlighting the presence of single emissive Ln3+ center. The shorter lifetime values noted in the case of binary lanthanide complexes (1 and 3) may be due to the dominant non-radiative decay channels associated with the vibronic coupling due to the presence of solvent molecules in the coordination sphere of these respective complexes.17b On the other hand, a two-fold enhancement in the excited state lifetime values have been observed in the case of ternary Ln3+ complexes as compared to corresponding binary complexes.
 | ||
| Fig. 9 Experimental luminescence decay profiles for complexes 1 and 2 in solid state monitored at approximately 1069 nm and excited 400 nm. | ||
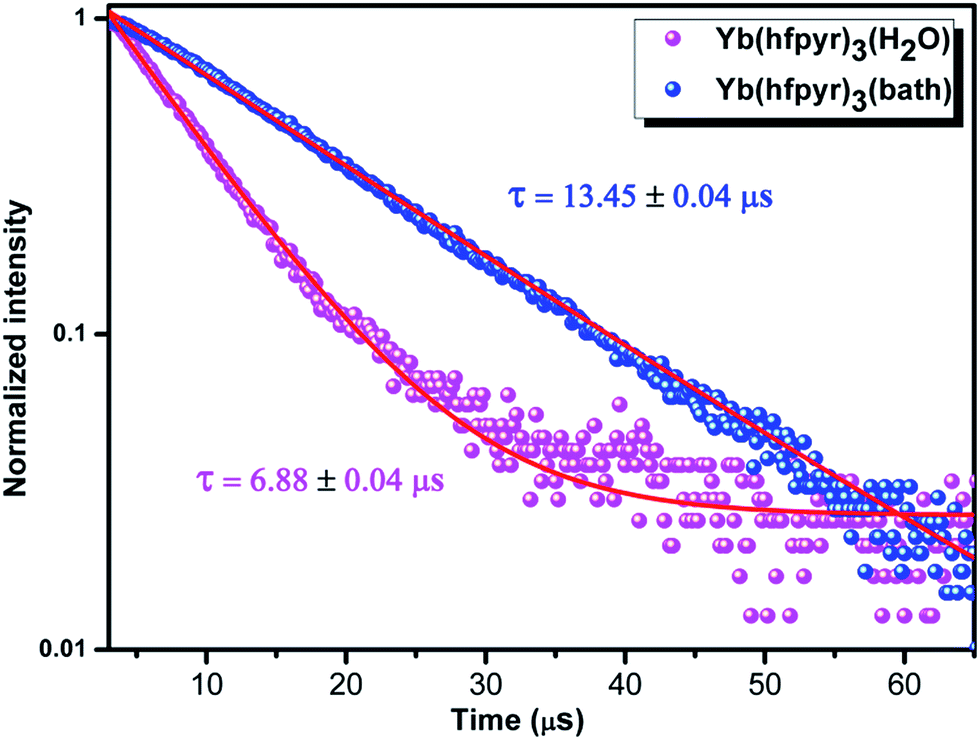 | ||
| Fig. 10 Experimental luminescence decay profiles for complexes 3 and 4 in solid state monitored at approximately 979 nm and excited 400 nm. | ||
| Complex | ARAD (s−1) | ANR (s−1) | τobs (μs) | ΦLn (%) | Φoverall (%) |
|---|---|---|---|---|---|
| Nd(hfpyr)3(H2O) 1 | 1607 | 3.65 × 105 | 2.80 ± 0.02 | 1.04 | 0.45 |
| Nd(hfpyr)3(bath) 2 | 1737 | 1.61 × 105 | 6.16 ± 0.03 | 2.28 | 1.07 |
| Yb(hfpyr)3(H2O) 3 | 2456 | 1.43 × 105 | 6.88 ± 0.04 | 0.34 | 1.69 |
| Yb(hfpyr)3(bath) 4 | 2389 | 7.20 × 104 | 13.45 ± 0.04 | 0.67 | 3.08 |
The overall quantum yields (Φoverall) of the developed NIR emitting Nd3+ and Yb3+ complexes (1–4) have been calculated intending to understand more about the photophysical properties. Therefore, it is appropriate to analyse the NIR emission behaviour of the Ln3+ complexes in terms of overall quantum yields (Φoverall). As it is well-known that, the overall quantum yield is generally regulated by the sensitization efficiency of the antenna molecule (Φsens) as well as the intrinsic luminescent quantum yield (ΦLn) of the Ln3+ ion [Φoverall = ΦsensΦLn]. The ΦLn of the complexes was determined by using the following eqn (1).2b,34
| ΦLn = (τobs/τrad) | (1) |
To calculate the efficiency of the sensitization process, it is necessary to know the radiative lifetime values (τrad), which are not easy to determine experimentally. It is clear from the literature that the τrad values for Nd3+ and Yb3+ vary widely and depend heavily on the solvent or the physical state of the sample. Taking into account 0.27 ms for Nd3+ as and a value of 2.0 ms commonly assumed for Yb3+, the data for sensitization Φsens = 0.45 for Nd3+, while a value >1 is noted for Yb3+, meaning that the actual radiative lifetime is larger than 2 ms in our system. Similar kind of results has also been reported by Bünzli and co-workers.7c
Synthesis, characterization and photophysical properties of PMMA doped hybrid materials
In view of the low cost, low optical absorbance and good mechanical properties,10 in the present study PMMA has been blended with the developed NIR emitting Yb3+ ternary complex (4) in proportions of 1, 3, 5, 7 and 9% (w/w) and corresponding polymeric films [PMMA@1% Yb, PMMA@3% Yb, PMMA@5% Yb, PMMA@7% Yb and PMMA@9% Yb] were isolated, characterized and evaluated their photophysical properties. The FT-IR spectra of precursor metal complex Yb(hfpyr)3(bath) 4 and the corresponding embedded Yb3+ complex in PMMA film (PMMA@7% Yb) were recorded in the 400–4000 cm−1 region and the results are shown in Fig. S29 in ESI.†The characteristic absorptions of the –O–CH2 asymmetric stretch, the –CH3 asymmetric stretch, the –CO stretch, the –O–CH3 deformation and the –C–O–C– symmetric stretch of pure PMMA was observed at 3000–3002, 2947–2950, 1730–1732, 1380–1385 and 989–993 cm−1 region, respectively.36 On the other hand, the weakening of vibrations of the ternary complex along with PMMA absorptions noted in the hybrid film indicate that the metal complex is embedded into PMMA matrix. The thermal stabilities of the precursor complex Yb(hfpyr)3(bath) 4 as well as the typical metal complex embedded into polymer film (PMMA@7% Yb) have been assessed by TGA and DTA analyses and the results are depicted in Fig. S24 and S30 respectively in ESI.† As can be clearly seen from the DTA results that the thermal stability of the parent Yb3+ ternary complex (366 °C decomposition temperature) was significantly improved after doping into the PMMA matrix (416 °C decomposition temperature). The TGA curve of PMMA@7% Yb film indicates that the decomposition starts at 290 °C. The polymer has been completely departed from the hybrid material when the temperature reaches at 427 °C.
The solid-state excitation and the emission profiles of a series of PMMA embedded Yb ternary metal complex [PMMA@1% Yb, PMMA@3% Yb, PMMA@5% Yb, PMMA@7% Yb and PMMA@9% Yb] polymeric films recorded at room temperature (298 K) are displayed in Fig. 11. A broad band noted in the wavelength region 250–500 nm of the excitation spectra can be attributed to the absorptions of the ligand systems. The emission spectra (λexc = 400 nm) clearly illustrates the presence of characteristic emission band of Yb3+ ion at 979 nm, which can be assigned to 2F5/2 → 2F7/2 transition.6c,34 Further, the luminescence intensity of the Yb3+ ion at 979 nm increases initially with the increase of dopant metal complex concentration in the polymer film to a maximum (PMMA@7% Yb) and thereafter decreases at higher concentration (PMMA@9% Yb). The energy transfer between the Yb3+ ions themselves is a non-radiative process, which accounts for the observed decrease in the Yb3+ emission, especially at high dopant metal complex concentration. It is noteworthy to mention that the emission intensity of the 7% Yb complex doped PMMA film at 979 nm has been markedly improved (about 1.4 fold) as compared to the precursor ternary Yb3+ complex. As a consequence, the overall quantum yields of the hybrid materials (3.40–3.62%) have also been moderately enhanced (Table 4). In conclusion, the above results disclose that Yb3+ ternary complex retains its original photophysical properties even after doping into the PMMA matrix. Further, the thermal stability and film forming properties have been significantly improved as compared to precursor complex.
 | ||
| Fig. 11 Excitation and emission spectra of PMMA films doped with 1, 3, 5, 7 and 9% (w/w) of Yb(hfpyr)3(bath). The data were recorded at 298 K. | ||
| Complex/film | ARAD (s−1) | ANR (s−1) | τobs (μs) | ΦLn (%) | Φoverall (%) |
|---|---|---|---|---|---|
| Yb(hfpyr)3(bath) | 2289 | 7.20 × 104 | 13.45 ± 0.02 | 0.743 | 3.08 |
| PMMA@1% Yb | 2259 | 6.50 × 104 | 14.87 ± 0.02 | 0.738 | 3.36 |
| PMMA@3% Yb | 2317 | 6.54 × 104 | 14.76 ± 0.02 | 0.728 | 3.42 |
| PMMA@5% Yb | 2443 | 6.61 × 104 | 14.57 ± 0.02 | 0.744 | 3.56 |
| PMMA@7% Yb | 2432 | 6.47 × 104 | 14.88 ± 0.02 | 0.741 | 3.62 |
| PMMA@9% Yb | 2388 | 6.50 × 104 | 14.82 ± 0.02 | 0.743 | 3.54 |
The luminescent decay profiles of the polymeric hybrid films were obtained by monitoring the emission at 979 nm corresponding to 2F5/2 → 2F7/2 transition and excited at 400 nm and the results are given in Fig. 12 and S31.† The life time values of the isolated hybrid films are listed in Table 2. All the τ values of the hybrid polymer films (14–15 μs) are found to be moderately higher than the precursor Yb3+ ternary complex 4 (13.45 μs), thus highlighting the radiative process are operative in the doped films due to the absence of multiphonon relaxation by coupling with the –OH oscillators.9 Furthermore, the excited state lifetime values are not influenced by the doping process into the PMMA matrix.
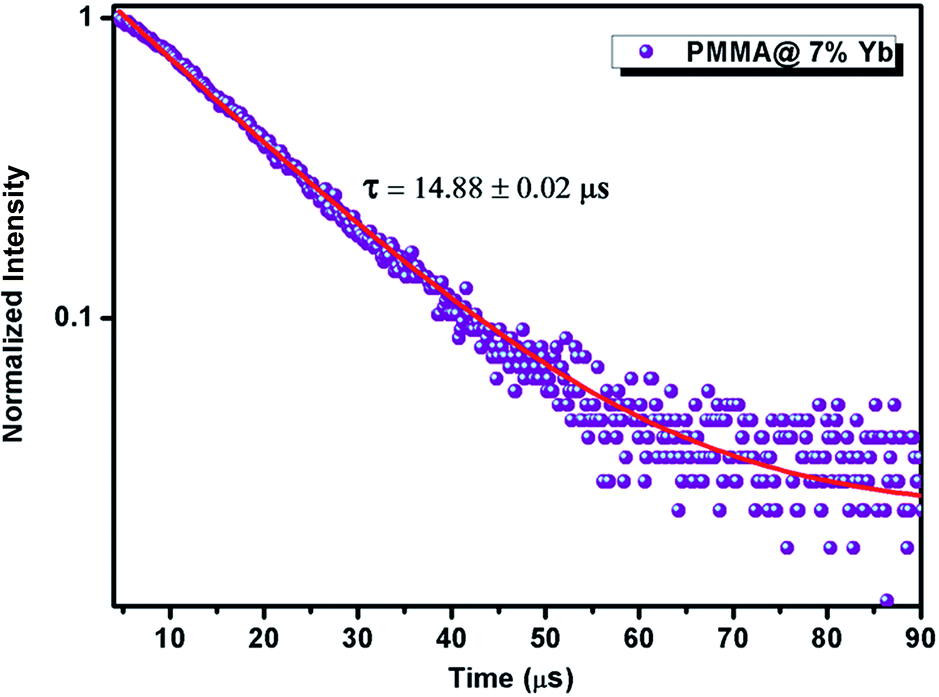 | ||
| Fig. 12 Experimental luminescence decay profiles of PMMA film@7% (w/w) of Yb(hfpyr)3(bath) monitored at approximately 975 nm and excited 400 nm. | ||
Conclusions
In summary, a novel β-diketonate ligand, 4,4,5,5,6,6,6-heptafluoro-3-hydroxy-1-(pyren-1-yl)hex-2-en-1-one has been successfully synthesized by incorporating highly conjugated pyrene moiety as a sensitizing unit and polyfluorinated alkyl group with low energy C–F oscillators, with an aim to develop near-infrared (NIR) emitting lanthanide complexes. Notably, the designed β-diketonate ligand has a triplet energy level of 16207 cm−1, which lies well above the energy of the main emitting level of Nd3+ (4F3/2 = 11257 cm−1) or Yb3+ (2F5/2 = 10400 cm−1), implying that it can act as an efficient antenna molecule for the sensitization of NIR emitting lanthanide ions. Further, the developed NIR emitting lanthanide complexes possess markedly high molar absorption coefficient values (about ε = 49000 to 50000 L mol−1 cm−1), indicating the adequate light-harvesting capacity of these compounds. The luminescent lifetimes and quantum yields values observed in the present study are found be significantly higher than many of the existing NIR emitting lanthanide β-diketonate complexes. Thus, the currently derived new Nd3+ and Yb3+ compounds may find potential applications as bioprobes in fluoroimmunoassay and new optical amplification materials for telecommunications. It is interesting to note that the thermal stability of the Yb3+ ternary complex incorporated PMMA film has been greatly enhanced as compared to parent compound, apart from exhibiting good film forming capacity.
Acknowledgements
One of the authors T. M. George thanks UGC, New Delhi for the award of Senior Research Fellowship.References
- (a) S. Comby and J.-C. G. Bünzli, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner, J.-C. G. Bünzli and K. P. Vitalij, Elsevier, 2007, vol. 37, pp. 217–470 Search PubMed; (b) J.-C. G. Bünzli and S. V. Eliseeva, Chem. Sci., 2013, 4, 1939 RSC; (c) S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC; (d) A. J. Amoroso and S. J. A. Pope, Chem. Soc. Rev., 2015, 44, 4723–4742 RSC; (e) J. C. G. Bünzli and S. V. Eliseeva, J. Rare Earths, 2010, 28, 824–842 CrossRef; (f) L. Prodi, E. Rampazzo, F. Rastrelli, A. Speghini and N. Zaccheroni, Chem. Soc. Rev., 2015, 44, 4922–4952 RSC.
- (a) P. A. Tanner and C.-K. Duan, Coord. Chem. Rev., 2010, 254, 3026–3029 CrossRef CAS; (b) J. Feng and H. Zhang, Chem. Soc. Rev., 2013, 42, 387–410 RSC; (c) L. Armelao, S. Quici, F. Barigelletti, G. Accorsi, G. Bottaro, M. Cavazzini and E. Tondello, Coord. Chem. Rev., 2010, 254, 487–505 CrossRef CAS; (d) J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- (a) X. Wang, H. Chang, J. Xie, B. Zhao, B. Liu, S. Xu, W. Pei, N. Ren, L. Huang and W. Huang, Coord. Chem. Rev., 2014, 273–274, 201–212 CrossRef CAS; (b) J.-C. G. Bünzli, Acc. Chem. Res., 2006, 39, 53–61 CrossRef PubMed; (c) M. L. P. Reddy and S. Sivakumar, Dalton Trans., 2013, 42, 2663–2678 RSC; (d) E. G. Moore, A. P. S. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542–552 CrossRef CAS PubMed.
- (a) A. D. Bettencourt-dias, P. S. Barber and S. Viswanathan, Coord. Chem. Rev., 2014, 273–274, 165–200 CrossRef; (b) D. V. Kazakov and F. E. Safarov, Photochem. Photobiol. Sci., 2014, 13, 1646–1649 RSC; (c) M. L. P. Reddy, V. Divya and R. Pavithran, Dalton Trans., 2013, 42, 15249–15262 RSC; (d) Y. Ma and Y. Wang, Coord. Chem. Rev., 2010, 254, 972–990 CrossRef CAS.
- (a) K. Binnemans, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. B. Karl, A. Gschneidner and K. P. Vitalij, Elsevier, 2005, vol. 35, pp. 107–272 Search PubMed; (b) D. B. A. Raj, B. Francis, M. L. P. Reddy, R. R. Butorac, V. M. Lynch and A. H. Cowley, Inorg. Chem., 2010, 49, 9055–9063 CrossRef CAS PubMed; (c) T. M. George, M. J. Sajan, N. Gopakumar and M. L. P. Reddy, J. Photochem. Photobiol., A, 2016, 317, 88–99 CrossRef CAS; (d) J. Sun, B. Song, Z. Ye and J. Yuan, Inorg. Chem., 2015, 54, 11660–11668 CrossRef CAS PubMed; (e) T. V. Usha Gangan and M. L. P. Reddy, Dalton Trans., 2015, 44, 15924–15937 RSC; (f) F. Cao, Z. Yuan, J. Liu and J. Ling, RSC Adv., 2015, 5, 102535–102541 RSC; (g) P. N. Remya, S. Biju, M. L. Reddy, A. H. Cowley and M. Findlater, Inorg. Chem., 2008, 47, 7396–7404 CrossRef CAS PubMed; (h) S. Biju, D. B. A. Raj, M. L. P. Reddy and B. M. Kariuki, Inorg. Chem., 2006, 45, 10651–10660 CrossRef CAS PubMed; (i) B. Francis, C. Heering, R. O. Freire, M. L. P. Reddy and C. Janiak, RSC Adv., 2015, 5, 90720–90730 RSC; (j) K. Miyata, Y. Konno, T. Nakanishi, A. Kobayashi, M. Kato, K. Fushimi and Y. Hasegawa, Angew. Chem., Int. Ed., 2013, 52, 6413–6416 CrossRef CAS PubMed; (k) J. Yuasa, T. Ohno, H. Tsumatori, R. Shiba, H. Kamikubo, M. Kataoka, Y. Hasegawa and T. Kawai, Chem. Commun., 2013, 49, 4604–4606 RSC; (l) Y. Hirai, T. Nakanishi, Y. Kitagawa, K. Fushimi, T. Seki, H. Ito, H. Fueno, K. Tanaka, T. Satoh and Y. Hasegawa, Inorg. Chem., 2015, 54, 4364–4370 CrossRef CAS PubMed; (m) P. P. Lima, M. M. Nolasco, F. A. A. Paz, R. A. S. Ferreira, R. L. Longo, O. L. Malta and L. D. Carlos, Chem. Mater., 2013, 25, 586–598 CrossRef CAS; (n) V. Divya, R. O. Freire and M. L. Reddy, Dalton Trans., 2011, 40, 3257–3268 RSC; (o) V. Divya, V. Sankar, K. G. Raghu and M. L. P. Reddy, Dalton Trans., 2013, 42, 12317–12323 RSC.
- (a) C. Yu, Z. Zhang, L. Liu, H. Li, Y. He, X. Lu, W.-K. Wong and R. A. Jones, New J. Chem., 2015, 39, 3698–3707 RSC; (b) Y. Hou, J. Shi, W. Chu and Z. Sun, Eur. J. Inorg. Chem., 2013, 7, 3063–3069 CrossRef; (c) Z. Zhang, C. Yu, L. Liu, H. Li, Y. He, X. Lü, W. K. Wong and R. A. Jones, J. Photochem. Photobiol., A, 2016, 314, 104–113 CrossRef CAS.
- (a) S. Comby, D. Imbert, C. Vandevyver and J. C. Bünzli, Chemistry, 2007, 13, 936–944 CrossRef CAS PubMed; (b) N. M. Shavaleev, R. Scopelliti, F. Gumy and J. C. Bünzli, Inorg. Chem., 2008, 47, 9055–9068 CrossRef CAS PubMed; (c) S. Comby, D. Imbert, A. S. Chauvin and J. C. G. Bünzli, Inorg. Chem., 2006, 45, 732–743 CrossRef CAS PubMed; (d) E. R. Trivedi, S. V. Eliseeva, J. Jankolovits, M. M. Olmstead, S. Petoud and V. L. Pecoraro, J. Am. Chem. Soc., 2014, 136, 1526–1534 CrossRef CAS PubMed; (e) A. Sanguineti, A. Monguzzi, G. Vaccaro, F. Meinardi, E. Ronchi, M. Moret, U. Cosentino, G. Moro, R. Simonutti, M. Mauri, R. Tubino and L. Beverina, Phys. Chem. Chem. Phys., 2012, 14, 6452 RSC; (f) S. Dang, J. B. Yu, X. F. Wang, Z. Y. Guo, L. N. Sun, R. P. Deng, J. Feng, W. Q. Fan and H. J. Zhang, J. Photochem. Photobiol., A, 2010, 214, 152–160 CrossRef CAS; (g) X. S. Ke, B. Y. Yang, X. Cheng, S. L. F. Chan and J. L. Zhang, Chem.–Eur. J., 2014, 20, 4324–4333 CrossRef CAS PubMed; (h) P. B. Glover, A. P. Bassett, P. Nockemann, B. M. Kariuki, R. Van Deun and Z. Pikramenou, Chem.–Eur. J., 2007, 13, 6308–6320 CrossRef CAS PubMed.
- (a) A. W. Woodward, A. Frazer, A. R. Morales, J. Yu, A. F. Moore, A. D. Campiglia, E. V. Jucov, T. V. Timofeeva and K. D. Belfield, Dalton Trans., 2014, 43, 16626–16639 RSC; (b) P. Martín-Ramos, P. S. Pereira da Silva, V. Lavín, I. R. Martín, F. Lahoz, P. Chamorro-Posada, M. Ramos Silva and J. Martín-Gil, Dalton Trans., 2013, 42, 13516–13526 RSC; (c) L. Yang, Z. Gong, D. Nie, B. Lou, Z. Bian, M. Guan, C. Huang, H. J. Lee and W. P. Baik, New J. Chem., 2006, 30, 791–796 RSC; (d) A. Monguzzi, R. Tubino, F. Meinardi, A. O. Biroli, M. Pizzotti, F. Demartin, F. Quochi, F. Cordella and M. A. Loi, Chem. Mater., 2009, 21, 128–135 CrossRef CAS; (e) X. Guo, H. Guo, L. Fu, L. D. Carlos, R. A. S. Ferreira, L. Sun, R. Deng and H. Zhang, J. Phys. Chem. C, 2009, 113, 12538–12545 CrossRef CAS; (f) T. S. Kang, B. S. Harrison, M. Bouguettaya, T. J. Foley, J. M. Boncella, K. S. Schanze and J. R. Reynolds, Adv. Funct. Mater., 2003, 13, 205–210 CrossRef CAS; (g) Z. Ahmed and K. Iftikhar, J. Phys. Chem. A, 2013, 117, 11183–11201 CrossRef CAS PubMed; (h) N. M. Shavaleev, R. Scopelliti, F. Gumy and J. C. G. Bünzli, Eur. J. Inorg. Chem., 2008, 9, 1523–1529 CrossRef; (i) B. L. Reid, S. Stagni, J. M. Malicka, M. Cocchi, A. N. Sobolev, B. W. Skelton, E. G. Moore, G. S. Hanan, M. I. Ogden and M. Massi, Chem.–Eur. J., 2015, 21, 18354–18363 CrossRef CAS PubMed.
- (a) G. M. Davies, R. J. Aarons, G. R. Motson, J. C. Jeffery, H. Adams, S. Faulkner and M. D. Ward, Dalton Trans., 2004, 1136–1144, 10.1039/b400992d; (b) J. Feng, J.-B. Yu, S.-Y. Song, L.-N. Sun, W.-Q. Fan, X.-M. Guo, S. Dang and H.-J. Zhang, Dalton Trans., 2009, 2406, 10.1039/b819644c; (c) N. M. Shavaleev, S. J. A. Pope, Z. R. Bell, S. Faulkner and M. D. Ward, Dalton Trans., 2003, 808–814, 10.1039/b300294b.
- S. Biju, Y. K. Eom, J.-C. G. Bünzli and H. K. Kim, J. Mater. Chem. C, 2013, 1, 6935–6944 RSC.
- B. Li, H. Li, P. Chen, W. Sun, C. Wang, T. Gao and P. Yan, Phys. Chem. Chem. Phys., 2015, 17, 30510–30517 RSC.
- (a) A. J. Howarth, M. B. Majewski and M. O. Wolf, Coord. Chem. Rev., 2015, 282–283, 139–149 CrossRef CAS; (b) A. G. Crawford, A. D. Dwyer, Z. Q. Liu, A. Steffen, A. Beeby, L. O. Palsson, D. J. Tozer and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 13349–13362 CrossRef CAS PubMed.
- (a) R. Haldar, K. Prasad, P. K. Samanta, S. Pati and T. K. Maji, Cryst. Growth Des., 2016, 16, 82–91 CrossRef CAS; (b) F. Liu, C. Tang, Q. Q. Chen, F. F. Shi, H. B. Wu, L. H. Xie, B. Peng, W. Wei, Y. Cao and W. Huang, J. Phys. Chem. C, 2009, 113, 4641–4647 CrossRef CAS; (c) C. Tang, F. Liu, Y. J. Xia, J. Lin, L. H. Xie, G. Y. Zhong, Q. L. Fan and W. Huang, Org. Electron., 2006, 7, 155–162 CrossRef CAS; (d) C. Tang, F. Liu, Y.-J. Xia, L.-H. Xie, A. Wei, S.-B. Li, Q.-L. Fan and W. Huang, J. Mater. Chem., 2006, 16, 4074 RSC.
- J. E. Sohna Sohna and F. Fages, Tetrahedron Lett., 1997, 38, 1381–1384 CrossRef CAS.
- S. Faulkner, M. C. Carrie, S. J. A. Pope, J. Squire, A. Beeby and P. G. Sammes, Dalton Trans., 2004, 1405–1409, 10.1039/b401302f.
- S. J. A. Pope, Polyhedron, 2007, 26, 4818–4824 CrossRef CAS.
- (a) Y. Hasegawa, T. Ohkubo, K. Sogabe, Y. Kawamura, Y. Wada, N. Nakashima and S. Yanagida, Angew. Chem., Int. Ed., 2000, 39, 357–360 CrossRef CAS; (b) A. Døssing, Eur. J. Inorg. Chem., 2005, 2005, 1425–1434 CrossRef; (c) A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. Gareth Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–504, 10.1039/a808692c.
- (a) J. Li, H. Li, P. Yan, P. Chen, G. Hou and G. Li, Inorg. Chem., 2012, 51, 5050–5057 CrossRef CAS PubMed; (b) D. B. A. Raj, S. Biju and M. L. P. Reddy, J. Mater. Chem., 2009, 19, 7976–7983 RSC.
- (a) D. B. Ambili Raj, S. Biju and M. L. Reddy, Dalton Trans., 2009, 36, 7519–7528 RSC; (b) V. Divya and M. L. P. Reddy, J. Mater. Chem. C, 2013, 1, 160–170 RSC; (c) D. B. A. Raj, S. Biju and M. L. P. Reddy, Inorg. Chem., 2008, 47, 8091–8100 CrossRef CAS PubMed.
- (a) W. Fan, J. Feng, S. Song, Y. Lei, L. Zhou, G. Zheng, S. Dang, S. Wang and H. Zhang, Nanoscale, 2010, 2, 2096–2103 RSC; (b) Z. Zhang, W. Feng, P. Su, X. Lü, J. Song, D. Fan, W. K. Wong, R. A. Jones and C. Su, Inorg. Chem., 2014, 53, 5950–5960 CrossRef CAS PubMed.
- W. Li, P. Yan, G. Hou, H. Li and G. Li, RSC Adv., 2013, 3, 18173–18180 RSC.
- (a) K. S. Bejoymohandas, A. Kumar, S. Sreenadh, E. Varathan, S. Varughese, V. Subramanian and M. L. P. Reddy, Inorg. Chem., 2016, 55, 3448–3461 CrossRef CAS PubMed; (b) K. S. Bejoymohandas, A. Kumar, S. Varughese, E. Varathan, V. Subramanian and M. L. P. Reddy, J. Mater. Chem. C, 2015, 3, 7405–7420 RSC.
- S. V. Eliseeva, D. N. Pleshkov, K. A. Lyssenko, L. S. Lepnev, J. C. Bünzli and N. P. Kuzmina, Inorg. Chem., 2010, 49, 9300–9311 CrossRef CAS PubMed.
- J. A. Cunningham, D. E. Sands, W. F. Wagner and M. F. Richardson, Inorg. Chem., 1969, 8, 22–28 CrossRef CAS.
- S. Biju, M. L. P. Reddy, A. H. Cowley and K. V. Vasudevan, Cryst. Growth Des., 2009, 9, 3562–3569 CAS.
- V. Divya, S. Biju, R. L. Varma and M. L. P. Reddy, J. Mater. Chem., 2010, 20, 5220–5227 RSC.
- A. R. Ramya, D. Sharma, S. Natarajan and M. L. P. Reddy, Inorg. Chem., 2012, 51, 8818–8826 CrossRef CAS PubMed.
- F. J. Steemers, W. Verboom, D. N. Reinhoudt, E. B. van der Tol and J. W. Verhoeven, J. Am. Chem. Soc., 1995, 117, 9408–9414 CrossRef CAS.
- L. Yang, Z. Gong, D. Nie, B. Lou, Z. Bian, M. Guan, C. Huang, H. J. Lee and W. P. Baik, New J. Chem., 2006, 30, 791–796 RSC.
- O. L. Malta, H. F. Brito, J. F. S. Menezes, F. R. Gonçalves e Silva, C. de Mello Donegá and S. Alves Jr, Chem. Phys. Lett., 1998, 282, 233–238 CrossRef CAS.
- (a) P. O. Alink, S. I. Klink, L. Grave, F. G. A. Peters, J. W. Hofstraat, F. Geurts and F. C. J. M. van Veggel, J. Chem. Soc., Perkin Trans. 2, 2001, 363–372 Search PubMed; (b) W.-T. Wong and W. P.-W. Lai, New J. Chem., 2000, 24, 943–944 RSC; (c) F. C. J. M. van Veggel and J. W. Stouwdam, Nano Lett., 2002, 2, 733–737 CrossRef; (d) L. N. Sun, J. B. Yu, G. L. Zheng, H. J. Zhang, Q. G. Meng, C. Y. Peng, L. S. Fu, F. Y. Liu and Y. N. Yu, Eur. J. Inorg. Chem., 2006, 19, 3962–3973 CrossRef; (e) J.-C. G. Bünzli and S. V. Eliseeva, Springer Ser. Fluoresc., 2011, 1–45, DOI:10.1007/4243.
- O. L. Malta, F. R. Goncalves e Silva, C. Reinhard, H.-U. Güdel, C. Piguet, J. E. Moser and J.-C. G. Bünzli, J. Phys. Chem. A, 2002, 106, 1670–1677 Search PubMed.
- R. J. Aarons, G. M. Davies, G. R. Motson, J. C. Jeffery, H. Adams, S. Faulkner and M. D. Ward, Dalton Trans., 2004, 1136–1144 Search PubMed.
- (a) L. D. Carlos, R. A. S. Ferreira, V. D. Bermudez and S. J. L. Ribeiro, Adv. Mater., 2009, 21, 509–534 CrossRef CAS PubMed; (b) A. de Bettencourt-Dias, Dalton Trans., 2007, 2229–2241, 10.1039/b702341c.
- (a) L. N. Puntus, K. J. Schenk and J.-C. G. Bünzli, Eur. J. Inorg. Chem., 2005, 2005, 4739–4744 CrossRef; (b) Y. F. Yuan, T. Cardinaels, K. Lunstroot, K. Van Hecke, L. Van Meervelt, C. Görller-Walrand, K. Binnemans and P. Nockemann, Inorg. Chem., 2007, 46, 5302–5309 CrossRef CAS PubMed.
- P. Y. Weizuo Li, G. Hou, H. Li and G. Li, Dalton Trans., 2013, 42, 11537–11547 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1473942. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra12220e |
| This journal is © The Royal Society of Chemistry 2016 |