
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Hydroxyphthalimide catalysts as bioactive pro-oxidants†
L.
Melone
ab,
P.
Tarsini
a,
G.
Candiani
*a and
C.
Punta
*a
aDepartment of Chemistry, Materials, and Chemical Engineering “Giulio Natta”. Politecnico di Milano, Piazza L. Da Vinci 32 and INSTM local unit, 20133 Milano, Italy. E-mail: gabriele.candiani@polimi.it; carlo.punta@polimi.it
bUniversità degli Studi e-Campus, Via Isimbardi 10, Como, 22060 Novedrate, Italy
First published on 17th February 2016
Abstract
The catalytic role of N-hydroxyphthalimide (NHPI) in promoting free-radical hydrogen atom transfer (HAT) reactions, well-documented for processes of industrial and synthetic interest, is here investigated for the first time in a biological environment. While NHPI by itself did not show any bioactivity, selected NHPI-derivatives (NHPIDs) revealed the ability to activate the intracellular formation of reactive oxygen species (ROS), causing the depletion of glutathione (GSH) levels and an increase in oxidative stress (OS). The evident bioactivity of some of these derivatives resulted in a significant reduction of the viability in osteosarcoma MG-63, suggesting a new, potential role of NHPIDs as pro-oxidant drugs. The key role of the N–OH group in promoting oxidative stress is demonstrated.
1. Introduction
NHPI is a cheap molecule, which is emerging as a valuable organocatalyst capable of promoting free-radical aerobic oxidation by activating hydrogen atom transfer (HAT) processes.1The efficiency and selectivity of this catalyst have been related to the favourable combination of enthalpic, polar, and entropic effects.2
As depicted in Scheme 1, the catalytic cycle implies the in situ generation of phthalimide-N-oxyl (PINO) radical by several metal1 and metal-free initiators.3
 | ||
| Scheme 1 General free-radical cycle for the NHPI-catalysed oxidation of organic substrates. | ||
PINO, whose formation was proved by Electron Spin Resonance (ESR) spectroscopy,4 is the real responsible of the catalytic activity of NHPI in oxidative processes, being directly involved in the free-radical propagation step by hydrogen atom abstraction from different activated organic substrates (Scheme 1, path i). The resulting carbon centred radicals undergo molecular oxygen addition, leading to the formation of the corresponding peroxyl radicals (Scheme 1, path ii).
The catalytic cycle is completed by the HAT reaction from a new NHPI molecule to the peroxyl radical (Scheme 1, path iii), favouring the concomitant formation of hydroperoxides and a new PINO unit, so that the radical chain is maintained.
Moreover, the possible presence of transition metal ions would favour the reduction of the hydroperoxide, leading to the formation of alkoxyl radicals which in turn are even faster in abstracting hydrogen from C–H bonds and from NHPI itself (Scheme 1, path iv).
Due to its unique catalytic role, in the last decade this derivative has attracted increasing attention, and its use has been also suggested for processes of industrial relevance.5,6
We also showed how NHPI and related derivatives (NHPIDs) are effective in catalysing the peroxidation of polyunsaturated fatty acids (PUFAs).7,8 In that case the final purpose was the selective synthesis of PUFAs' corresponding hydroperoxides. However, by considering that PUFAs are important components of the cellular membrane and environment, the manifested efficiency of this process prompted us to further investigate the possible catalytic action of NHPIDs in promoting oxidative stress (OS) in a biological environment in vitro.
The reasons of this interest rely on the still controversial role played by OS in regulating cancer development and apoptosis. OS occurs when redox homeostasis within the cell is altered.9 While experimental evidences show that an increase in the cellular concentrations of reactive oxygen species (ROS), like alkoxyl and peroxyl radicals, can promote DNA damage, angiogenesis, and metastasis,10 more recently researchers recognized the possible role of ROS in cancer therapy, leading to the conclusion that, to consider OS as outright toxic would be a mistake.11 In fact, it was reported that an increase of intracellular ROS content to cytotoxic levels would selectively induce apoptotic cell death in cancer.9 Thereby, besides the investigation of natural and synthetic agents with antioxidant activity,12,13 the development of pro-oxidant agents capable of inducing OS is nowadays emerging as an intriguing anticancer strategy.14–16
Herein, for the first time we demonstrate that the pro-oxidant catalytic activity of NHPIDs, originally developed for industrial applications, can be exerted also in vitro to address relevant biological issues. Striking outcomes of the study include (i) the key role that lipophilic tails on the aromatic ring play in favouring the interaction of NHPI with cells; (ii) the association between the cytotoxic action of NHPIDs and their catalytic activity as pro-oxidants, evidenced by the increase of OS, followed by the depletion of intracellular glutathione (L-γ-glutamyl-L-cysteinyl-glycine, GSH) levels and consequently apoptotic cell death; (iii) the importance of having free N–OH moieties in order to exhibit a pro-oxidant activity, suggesting that the catalytic action in vitro could still follow the free-radical HAT mechanism described in Scheme 1.
2. Results and discussion
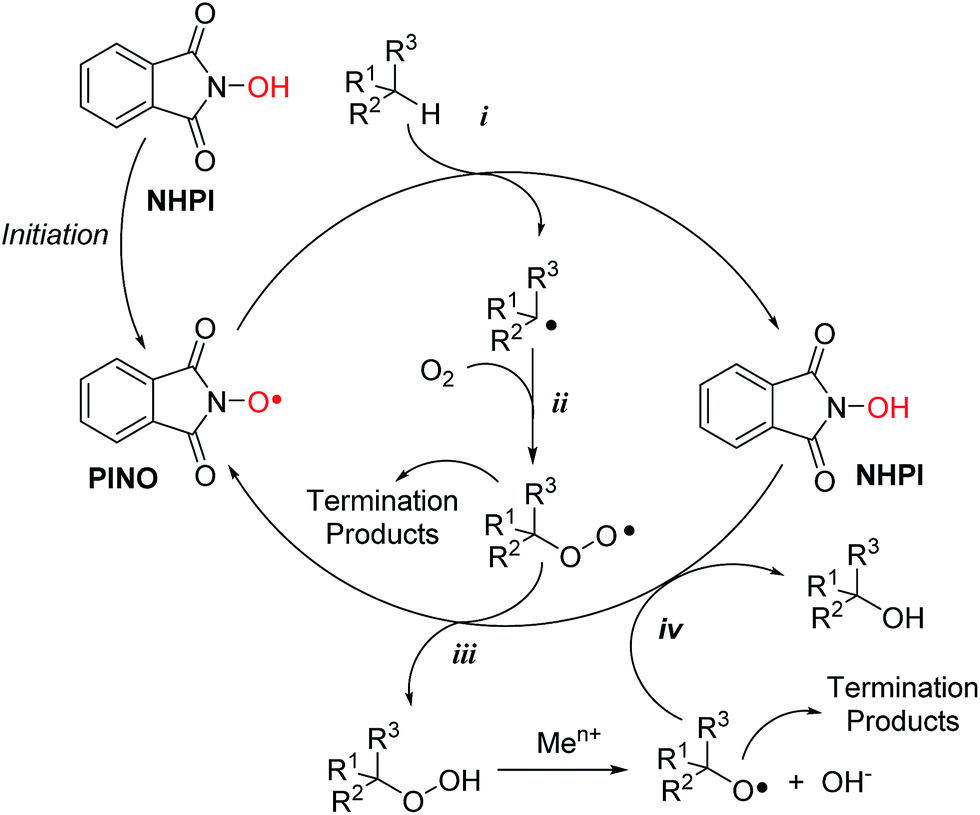
Human osteosarcoma MG-63 cells were chosen as target recipients because being responsive to OS insult.17,18Disappointingly, first experiments carried out in the presence of increasing amounts of NHPI for 24 h showed that it did induce significant cytotoxicity only at extremely high concentrations (EC50 = 7 mM), as determined by AlamarBlue® fluorimetric assay (Fig. 1A). We thus hypothesized that this behavior would be associated to the strong polar character of NHPI, a key factor already investigated for industrial applications.19,20 In biological systems, high polarity could drastically reduce the reactivity of NHPI towards the plasma membrane, preventing its effective interaction with the cell.
 | ||
| Fig. 1 Cytotoxicity of (A) NHPI, (B) 1-OH, (C) 2-OHvs.2-Me (full black cycles vs. empty squares, respectively), (D) 3-OHvs.3-Me (full black cycles vs. empty squares, respectively) and (E) 4-OHvs.4-Me (full black cycles vs. empty squares, respectively) after 24 h of exposure as a function of concentration in MG-63 human osteosarcoma cells. | ||
In order to ascertain this, we synthesized NHPIDs bearing different lipophilic tails (Fig. 2), reproducing the same synthetic solutions suggested to overcome the industrial limit of NHPI related to its solubility.20
 | ||
| Fig. 2 NHPIDs and corresponding N-methyl derivatives. | ||
Compounds 1-OH and 2-OH were obtained starting from trimellitic anhydride chloride, after optimization of the experimental procedure reported by Ishii for the synthesis of 2-OH.21
Similarly, compound 3-OH was synthesized in the presence of the purposely prepared (R)-2-(1-hydroxybutan-2-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione, following the approach recently reported by our group.22 Interestingly, specifically functionalized naphthalimides have already shown good anticancer activity upon a variety of human cancer cells due to their high intercalation affinity towards DNA.23 The cytotoxic properties of these derivatives were usually ascribed to the side chains present on the naphthalene moiety. However, data reported in the literature suggested that this target group would favor cellular internalization, rendering derivative 3-OH attractive for our purpose. Moreover, we recently reported how catalyst 3-OH could be photo-activated by UV radiation, further promoting aerobic oxidation.22
In addition, we also synthesized compound 4-OH, a bis-NHPI lipophilic catalyst, by reacting hydroxylamine hydrochloride with 4,4′-(4,4′-isopropylidenediphenoxy)bis (phthalic anhydride), a cheap and commercially available di-anhydride obtained from bisphenol A. The catalytic efficiency and the high lipophilic character of compound 4-OH have been already documented by our group, for the solvent-free selective aerobic oxidation of alkyl aromatics.20
NHPIDs were tested in MG-63 cancer cell line to check their potential, concentration-dependent, cytotoxicity. The results are reported in Fig. 1. Compounds 2-OH, 3-OH, and 4-OH displayed EC50 at 100, 200 and 110 μM, respectively (Fig. 1C–E, full black cycles), well below the values for the poorly cytotoxic compound 1-OH (Fig. 1B).
These observations prompted us to further investigate the reasons for such a bioactivity. If our original assumption was correct, the decrease in cell viability following challenging with compounds 2-OH, 3-OH, and 4-OH had to be due to a significant elevation of the OS. In fact, being NHPIDs 2-OH, 3-OH, and 4-OH more lipophilic than, and as pro-oxidant as 1-OH, they would be expected to interact with cells and exert potential pro-oxidant effects to a greater extent than the latter molecule. Moreover, onto consideration of the HAT mechanism according to which the NHPIDs promote oxidation (Scheme 1), we wanted to be sure that the eventual pro-oxidant action was specific to these compounds and relied on the presence of the N–OH group, rather than to the introduction of the lipophilic tails. To do so, we challenged osteosarcoma cells with compounds 2-OH, 3-OH, and 4-OH and the purposely synthesized N-methyl molecules 2-Me, 3-Me, and 4-Me (Fig. 1C–E, empty squares vs. full black cycles, respectively). In this way we could evaluate and compare the eventual cytotoxicity exclusively associated to the grafting of the phthalimide aromatic ring (Fig. SI8†). In line with our expectations, 2-Me to 4-Me derivatives were far less cytotoxic than parent N-OHs.
Following these observations, we moved to consider the possible occurrence of OS both directly, by evaluating the increase of ROS, and indirectly, by measuring GSH depletion. In fact, chemotherapy agents (e.g. cisplatin) are detoxified by antioxidant defense mechanisms, such as enzymatic conjugation to the tripeptide GSH.24 Abrogation of such drug-resistant mechanisms by redox modulation would thus have significant therapeutic implications.
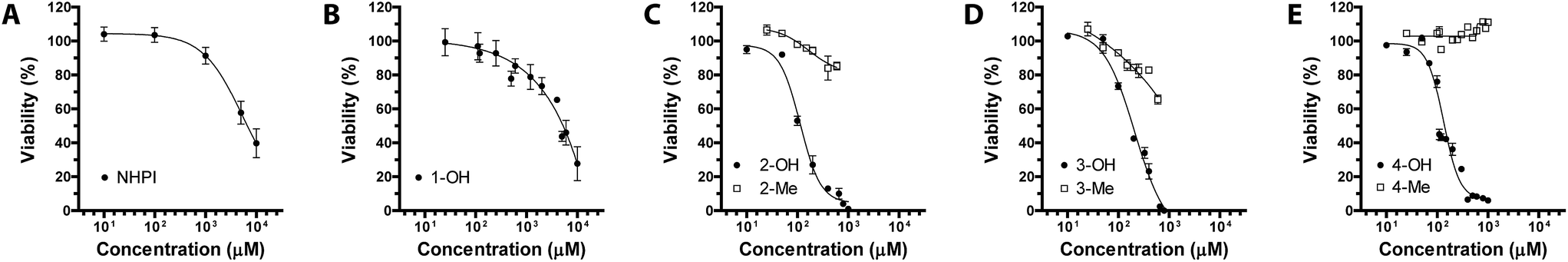
Interestingly, we herein show the generation of large quantities of ROS induced by the treatment with compounds 2-OH, 3-OH, and 4-OH used each at its respective EC50 (130 ± 5%, 294 ± 20%, 234 ± 23%, all p < 0.05 vs. CTRL cells). Furthermore, our results about OS induced by derivatives 2-Me, 3-Me, and 4-Me (all not statistically significant vs. CTRL cells) (Fig. 3) pointed out that the mechanism whereby NHPIDs 2-OH, 3-OH, and 4-OH induce cytotoxicity is associated to the N–OH moiety. Indeed, injury to cells occurs only when ROS overwhelm the biochemical defense of the cell.9 Moreover, GSH consumption that mirrors the depletion of cellular antioxidant defenses,25 is related to such OS insult, as confirmed by the significantly greater GSH depletion with the parent molecule 4-OH, if compared to 4-Me compound (71 ± 4% vs. 92 ± 7%, respectively, p < 0.05) (Fig. SI9†).
 | ||
| Fig. 3 Oxidative stress (OS) levels expressed in terms of reactive oxygen species (ROS) production in untreated MG-63 cells (CTRL group) as compared to 1-OH (200 μM), 2-OH and 2-Me (100 μM, both), 3-OH and 3-Me (200 μM, both), and 4-OH and 4-Me-treated human osteosarcoma cells (110 μM, both). | ||
We would like to point out that these results cannot be taken for granted. From one point of view, the aqueous environment could inhibit NHPIDs' catalytic properties by means of the hydrogen bonding involving N–OH moiety. On the other hand, we operated for the first time at really lower concentrations of the catalyst with respect to classical synthetic applications. It is in fact well known that peroxidation kinetics promoted by NHPI are significantly dependent on catalyst concentration.1
The difference noticed among the EC50 of 2-OH (100 μM), 3-OH (200 μM) and 4-OH (110 μM) highlights one more the key role played by the lipophilic tails, suggesting that a proper design of the nitroxide pro-oxidant would allow to improve its biological performance. Such a difference is emphasized by the inherent behavior of the resulting 2-OH to 4-OH but not 1-OH, to aggregate. In our experimental conditions, 2-OH, 3-OH, and 4-OH compounds did form monodisperse aggregates with hydrodynamic diameters comprised between 136 and 628 nm (Fig. SI10–SI12†).
It is worth noting that, although also 2-Me, 3-Me, and 4-Me do form aggregates in water in our experimental conditions (Fig. SI13–SI15†), sometimes (2-Me) with lower, sometimes (3-Me, 4-Me) with higher hydrodynamic diameters than the corresponding N-hydroxy derivatives, they are in any case much less cytotoxic (Fig. 1).
This confirms once again the key function of any N–OH moiety. Overall, the inherent tendency of pro-oxidants 2-OH to 4-OH to aggregate do have a twofold advantage over less lipophilic molecules as 1-OH. First, submicrometric aggregates can be easily endocytosed by cells. Moreover, apart from being pro-oxidant drugs per se, –OHs do form aggregates in water that might offer place to accommodate hydrophilic, lipophilic as well as amphiphilic drugs. This colloidal behavior would extend their use as chemotherapeutic carriers or drug delivery systems.26
Excess OS kills or damages cells mostly by apoptosis, and eventually by other mechanisms.27–29 In the former case, alterations in the redox status of the cell to a more oxidizing environment occurs prior to activation of the final phase of cysteine aspartic acid-specific protease known as caspases that orchestrate the execution phase of apoptosis by cleaving multiple structural and repair proteins.9 Between the fourteen caspases identified in mammals, a subset of downstream, or executioner caspases (Caspase-3, and -7) is actually responsible for the demolition phase of apoptosis.30 We thereby focused our attention on the primary effector caspases necessary for triggering the cleavage of the vast majority of subcellular proteins.
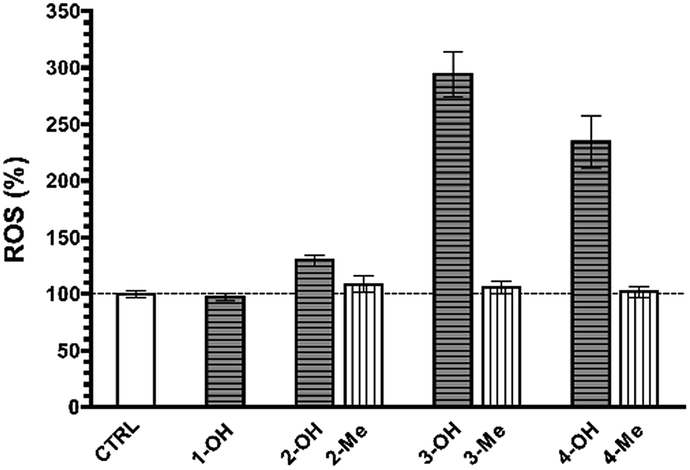
As shown in Fig. 4, Caspase-3 and -7 activity was significantly higher in cells treated with any of the NHPIDs as compared to each respective –Me derivative (597 ± 47% vs. 126 ± 7% for 2-OH and 2-Me, 679 ± 25% vs. 104 ± 6% for 3-OH and 3-Me, 6.664 ± 223% vs. 126 ± 7% for 4-OH and 4-Me, all p < 0.05), strengthening the idea that overt cytotoxicity specifically induced by 2-OH, 3-OH, and 4-OH was linked to OS insult. This, in turn, led to some GSH depletion and engages cells in caspase-mediated apoptotic cell death.
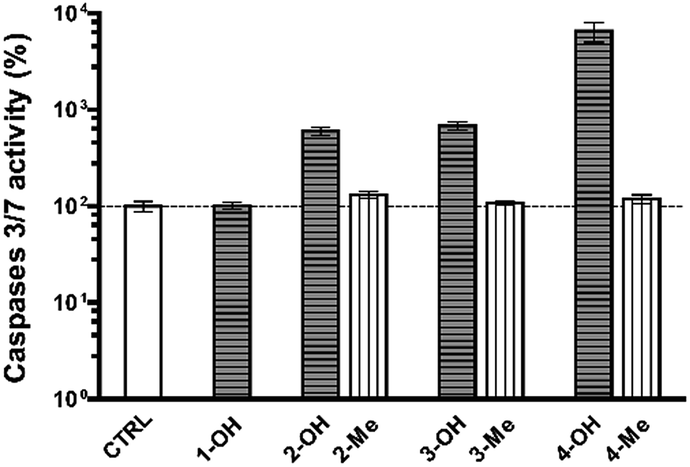 | ||
| Fig. 4 Caspase-3 and -7 activities in untreated MG-63 cells (CTRL group) as compared to 1-OH (200 μM), 2-OH and 2-Me (100 μM, both), 3-OH and 3-Me (200 μM, both), and 4-OH and 4-Me-treated human osteosarcoma cells (110 μM, both). | ||
3. Conclusions
In summary, lipophilic NHPIDs 2-OH, 3-OH, and 4-OH were synthesized and their activity as pro-oxidant catalysts was substantiated in vitro in osteosarcoma MG-63 cells.The overt cytotoxicity of these derivatives was associated to an increase of OS levels, and to a consequent depletion of intracellular GSH content, leading to caspase activation and apoptotic cell death. Interestingly, the markedly lower bioactivity of the N-methyl derivatives 2-Me, 3-Me, and 4-Me shed light on the key role played by N–OH group in promoting oxidation even in vitro, suggesting the occurrence of the classical HAT process.
Taken together, these observations provide unambiguous evidence that NHPI, an oxidation catalyst widely used in a plethora of abiotic industrial applications, once tethered to suitable lipophilic moieties is a promising pro-oxidant in vitro.
It is worth noting that, in this preliminary investigation, our aim was to verify an hypothesis, that is the potential bioactivity of NHPIDs, rather than to design the best drug solutions for in vitro and in vivo applications.
Even though further investigation is necessary to make these catalysts more effective and selective towards target cells, we believe that these preliminary results may open up the possibility of the development of novel anticancer chemicals and strategies based on NHPI catalysis.
4. Experimental
4.1. General
All reagents used in this work are commercially available (Sigma-Aldrich, Italy) and were used as received without further purification unless otherwise stated. 1H and 13C-NMR of the products were recorded at room temperature (r.t.) with a Bruker Avance-400 NMR spectrometer. Compounds 1–4 were stored dried at 4 °C and solubilized in dimethyl sulfoxide (DMSO) before use.4.2. Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v:v) CHCl3:MeOH as eluent (Rf: 0.60 – orange color spot if TLC is exposed to NH3). Yield: 90%. Mp: 115.0 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.91 (s, 1H), 8.49 (d, 2H), 8.42 (d, 2H), 8.18 (d, 1H), 8.01 (s, 1H), 7.84 (m, 1H + 2H), 5.43 (s, 1H), 4.95 (dd, 1H), 4.76 (dd, 1H), 2.21 (m, 1H), 2.018 (m, 1H), 0.97 (t, 3H). 1H-NMR and 13C-NMR spectra of this derivative is reported in Fig. SI4.†
:1 (v:v) CHCl3:MeOH as eluent (Rf: 0.60 – orange color spot if TLC is exposed to NH3). Yield: 90%. Mp: 115.0 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.91 (s, 1H), 8.49 (d, 2H), 8.42 (d, 2H), 8.18 (d, 1H), 8.01 (s, 1H), 7.84 (m, 1H + 2H), 5.43 (s, 1H), 4.95 (dd, 1H), 4.76 (dd, 1H), 2.21 (m, 1H), 2.018 (m, 1H), 0.97 (t, 3H). 1H-NMR and 13C-NMR spectra of this derivative is reported in Fig. SI4.†
Compounds 1-OH and 2-OH were synthetised similarly to 3-OH, following the procedures previously described,20 first preparing the ester from MeOH and dodecyl alcohol respectively and trimellitic anhydride chloride. The corresponding NHPIDs 1-OH and 2-OH were obtained using NH2OH·HCl and purified by chromatography on silica-gel using 9:1 (v:v) CHCl3:MeOH as eluent (Rf: 0.60 (3), 0.76 (4) – orange color spot if TLC is exposed to NH3). Yield: 94%. Mp: 172.0 °C (1-OH), 85.0 °C (2-OH). 1H NMR of 1-OH (400 MHz, acetone-d6) δ (ppm): 9.95 (broad s, 1H), 8.43 (d, 1H), 8.28 (s, 1H), 7.96 (d, 1H), 3.98 (s, 3H). 1H-NMR of 2-OH (400 MHz, DMSO-d6) δ (ppm): 8.44 (d, 1H), 8.31 (s, 1H), 7.96 (s, 1H), 4.40 (t, 2H), 1.83 (q, 2H), 1.6–1.2 (m, 18H), 0.88 (t, 3H). 1H-NMR and 13C-NMR spectra of these derivatives are reported in Fig. SI1 and 2,† respectively.
In a 50 mL two-necked flask, 2.60 g (5 mmol) of purified 4,4′-(4,4′-isopropylidenediphenoxy)bis(phthalic anhydride) were added to 30 mL of anhydrous pyridine. After adding an excess of hydroxylamine hydrochloride (30 mmol, 2.85 g), the mixture was heated under reflux (119 °C) for 1 h in a microwave reactor. After cooling the pyridine was evaporated under vacuum, obtaining a viscous orange product which was dissolved in ethylacetate (150 mL). The organic phase was extensively washed with 0.5 M HCl (100 mL, 3 times) and deionized water (dH2O, 100 mL, 3 times) in order to remove the residual pyridine and finally dried with anhydrous sodium sulphate. The evaporation of the solvent gave a pale yellow foam, which was purified by chromatography on silica-gel using 9:1 (v:v) CHCl3:MeOH as eluent (Rf: 0.54 – orange color spot if TLC is exposed to NH3). Yield: 93%. 1H-NMR of 4-OH (400 MHz, DMSO-d6): δ = 10.77 (s, 2H), 7.80 (d, 2H), 7.34 (d, 4H), 7.29 (d, 2H), 7.24 (s, 2H), 7.08 (d, 4H), 1.69 (s, 6H). 1H-NMR and 13C-NMR spectra of this derivative is reported in Fig. SI6.†
2-Me . Yield: 95%; 1H-NMR (400 MHz, CDCl3): δ (ppm): 8.44 (d, 1H), 8.37 (s, 1H), 7.88 (s, 1H), 4.44 (t, 2H), 3.19 (s, 1H), 1.77 (q, 2H), 1.45–1.10 (m, 18H), 0.85 (t, 3H).
3-Me . Yield: 94%; 1H-NMR (400 MHz, CDCl3) δ (ppm): 8.59 (d, 2H), 8.33 (s, 1H), 8.26 (d, 1H), 8.21 (d, 2H), 7.79 (d, 1H), 7.75 (t, 2H), 5.55 (m, 1H), 5.05 (dd, 1H), 4.85 (dd, 1H), 2.34 (m, 1H), 2.09 (m, 1H), 1.01 (t, 3H).
4-Me . Yield: 94%; 1H-NMR (400 MHz, CDCl3) δ (ppm): 7.77 (d, 2H), 7.36 (d, 4H), 7.29 (d, 2H), 7.24 (s, 2H), 7.00 (d, 4H), 3.15 (s, 6H), 1.73 (s, 6H).
1H-NMR and 13C-NMR spectra of these derivatives are reported in Fig. SI3, SI5 and SI7,† respectively.
4.3. Biological procedures
:v) of Fetal Bovine Serum (FBS), 1 mM sodium pyruvate, 10 mM Hepes buffer, 100 U mL−1 penicillin, 0.1 mg mL−1 streptomycin, and 2 mM glutamine.
:v) DMSO-containing compounds at the desired concentration and kept in contact with cells for 24 h before analysis. Cells grown in complete medium containing 1.5% (v:v) DMSO were considered as untreated controls (CTRL).
:v) Tween in 50 mM Tris–HCl buffer, pH 7.5 for at least 10 min at r.t. Cells were then scraped, collected and subjected to 3 consecutive freezing/thawing cycles (−80 °C/r.t.) and the fluorescence in cell lysates measured by means of a microplate reader (λex = 485 nm; λem = 530 nm), then normalized over the total protein content of each sample as determined by bicinchoninic acid (BCA) protein assay kit (Pierce, USA). Experiments were performed at least 3 times in quadruplicate and values expressed as percentage of CTRL cells.
:v) sulfosalicylic acid (SSA) in dH2O and subjected to 2 freezing/thawing cycles (−80 °C/r.t.). After an incubation of 10 min at 4 °C, lysates were finally deproteinized by centrifugation for 10 min at 6 × 103 g and GSH levels determined in supernatants with Glutathione assay kit according to the manufacturer's instructions. Experiments were performed at least twice in triplicate and values, normalized for protein content, were expressed as percentage of CTRL cells.
Acknowledgements
The authors gratefully acknowledge the financial support from MIUR – FIRB Futuro in Ricerca 2008 (RBFR08XH0H) and PRIN 2010–2011 (PROxi Project 2010PFLRJR_005).Notes and references
- (a) F. Recupero and C. Punta, Chem. Rev., 2007, 107, 3800–3842 CrossRef CAS PubMed; (b) C. Galli, P. Gentili and O. Lanzalunga, Angew. Chem., Int. Ed., 2008, 47, 4790–4796 CrossRef CAS PubMed; (c) S. Coseri, Catal. Rev., 2009, 51, 218–292 CrossRef CAS; (d) C. Punta and C. Gambarotti, in Ideas in Chemistry and Molecular Sciences: Advances in Synthetic Chemistry; ed. B. Pignataro, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010, pp. 3–24 Search PubMed; (e) S. Wertz and A. Studer, Green Chem., 2013, 15, 3116–3134 RSC.
- F. Minisci, C. Punta and F. Recupero, J. Mol. Catal. A: Chem., 2006, 251, 129–149 CrossRef CAS.
- L. Melone and C. Punta, Beilstein J. Org. Chem., 2013, 9, 1296–1310 CrossRef CAS PubMed.
- (a) Y. Ishii, K. Nakayama, M. Takeno, S. Sakaguchi, T. Iwahama and Y. Nishiyama, J. Org. Chem., 1995, 60, 3934–3935 CrossRef CAS; (b) R. Amorati, M. Lucarini, V. Mugnaini, G. F. Pedulli, F. Minisci, F. Recupero, F. Fontana, P. Astolfi and L. Greci, J. Org. Chem., 2003, 68, 1747–1754 CrossRef CAS PubMed.
- L. Melone, S. Prosperini, G. Ercole, N. Pastori and C. Punta, J. Chem. Technol. Biotechnol., 2014, 89, 1370–1378 CrossRef CAS.
- L. Melone and C. Punta, in Liquid Phase Aerobic Oxidation Catalysis, ed. P. Alsters and S. Sthal, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2015, in press Search PubMed.
- C. Punta, C. L. Rector and N. A. Porter, Chem. Res. Toxicol., 2005, 18, 349–356 CrossRef CAS PubMed.
- L. Melone, M. Petroselli, N. Pastori and C. Punta, Molecules, 2015, 20, 15881–15892 CrossRef CAS PubMed.
- K. Kannan and S. K. Jain, Pathophysiology, 2000, 7, 153–163 CrossRef CAS PubMed.
- (a) L. J. Marnett, Carcinogenesis, 2000, 21, 361–370 CrossRef CAS PubMed; (b) C. C. Benz and C. Yau, Nat. Rev. Cancer, 2008, 8, 875–879 CrossRef CAS PubMed; (c) J. P. Kehrer, J. D. Robertson and C. V. Smith, in Comprehensive Toxicology, ed. C. A. McQueen, Elsevier, Oxford, 2nd edn 2010, vol. 1.14, pp. 277–307 Search PubMed; (d) M. L. Circu and T. Y. Aw, Free Radical Biol. Med., 2010, 48, 749–864 CrossRef CAS PubMed; (e) J.-C. Lee, Y.-O. Son, P. Pratheeshkumar and X. Sci, Free Radical Biol. Med., 2012, 53, 742–757 CrossRef CAS PubMed; (f) N. A. Porter, J. Org. Chem., 2013, 78, 3511–3524 CrossRef CAS PubMed.
- C. Jacob and P. G. Winyard, in Redox Signaling and Regulation in Biology and Medicine, Wiley-VCH, Weinheim, 2009 Search PubMed.
- L. Valgimigli and D. A. Pratt, in Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, John Wiley & Sons, Chichester, 2012, pp. 1623–1677 Search PubMed.
- (a) R. Serwa, T.-G. Nam, L. Valgimigli, S. Culbertson, C. L. Rector, B.-S. Jeong, D. A. Pratt and N. A. Porter, Chem.–Eur. J., 2010, 16, 14106–14114 CrossRef CAS PubMed; (b) P. T. Lynett, K. Butts, V. Vaidya, G. E. Garrett and D. A. Pratt, Org. Biomol. Chem., 2011, 9, 3320–3330 RSC; (c) J. J. Hanthorn, L. Valgimigli and D. A. Pratt, J. Org. Chem., 2012, 77, 6908–6916 CrossRef CAS PubMed; (d) B. Li, J. R. Harjani, N. S. Cormier, H. Madarati, J. Atkinson, G. Cosa and D. A. Pratt, J. Am. Chem. Soc., 2013, 135, 1394–1405 CrossRef CAS PubMed.
- L. Raj, T. Ide, A. U. Gurkar, M. Foley, M. Schenone, X. Li, N. J. Tolliday, T. R. Golub, S. A. Carr, A. F. Shamji, A. M. Stern, A. Mandinova, S. L. Schreiber and S. W. Lee, Nature, 2011, 475, 231–234 CrossRef CAS PubMed.
- (a) L. Valgimigli and R. Iori, Environ. Mol. Mutagen., 2009, 50, 222–237 CrossRef CAS PubMed; (b) C. Martín-Cordero, A. J. León-González, J. M. Calderón-Montaño, E. Burgos-Morón and M. López-Lázaro, Curr. Drug Targets, 2012, 13, 1006–1028 CrossRef.
- (a) R. Amorati, L. Valgimigli, P. Dinér, K. Bakhtiari, M. Saeedi and L. Engman, Chem.–Eur. J., 2013, 19, 7510–7522 CrossRef CAS PubMed; (b) S. Suy, J. B. Mitchell, A. Samuni, S. Mueller and U. Kasid, Cancer, 2005, 103, 1302–1313 CrossRef CAS PubMed.
- (a) D. Pezzoli, M. Zanda, R. Chiesa and G. Candiani, J. Controlled Release, 2013, 165, 44–53 CrossRef CAS PubMed; (b) G. Candiani, D. Pezzoli, L. Ciani, R. Chiesa and S. Ristori, PLoS One, 2010, 5, e13430 Search PubMed.
- Z. Chang, J. Xing and X. Yu, Tumour Biol., 2014, 35(1), 753–758 CrossRef CAS PubMed.
- (a) L. Melone, C. Gambarotti, S. Prosperini, N. Pastori, F. Recupero and C. Punta, Adv. Synth. Catal., 2011, 353, 147–154 CrossRef CAS; (b) L. Melone, S. Prosperini, C. Gambarotti, N. Pastori, F. Recupero and C. Punta, J. Mol. Catal. A: Chem., 2012, 355, 155–160 CrossRef CAS.
- M. Petroselli, P. Franchi, M. Lucarini, C. Punta and L. Melone, ChemSusChem, 2014, 7, 2695–2703 CrossRef CAS PubMed.
- N. Sawatari, T. Yokota, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2001, 66, 7889–7891 CrossRef CAS PubMed.
- L. Melone, P. Franchi, M. Lucarini and C. Punta, Adv. Synth. Catal., 2013, 355, 3210–3220 CrossRef CAS.
- (a) M. F. Braña, M. Cacho, A. Gradillas, B. Pascual-Teresa and A. Ramos, Curr. Pharm. Des., 2001, 7, 1745–1780 CrossRef; (b) M. F. Braña and A. Ramos, Curr. Med. Chem.: Anti-Cancer Agents, 2001, 1, 237–255 CrossRef; (c) I. Czerwinska, S. Sato and S. Takenaka, Bioorg. Med. Chem., 2012, 20, 6416–6422 CrossRef CAS PubMed.
- S. Komiya, M. C. Gebhardt, D. C. Mangham and A. Inoue, J. Orthop. Res., 1998, 16, 15–22 CrossRef CAS PubMed.
- (a) D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS PubMed; (b) X. Guo, R. A. Mittelstaedt, L. Guo, J. G. Shaddock, R. H. Heflich, A. H. Bigger, M. M. Moore and N. Mei, Toxicol. In Vitro, 2013, 27, 1496–1502 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- N. Zanzami, P. Marchetti, M. Castedo, D. Decaudin, A. Macho, T. Hirsch, S. A. Susin, P. X. Petit, B. Mignotte and G. Kroemer, J. Exp. Med., 1995, 182, 367–377 CrossRef.
- N. Zanzami, S. A. Susin, P. Marchetti, T. Hirsch, M. Castedo, I. Gómez-Monterrey and G. Kroemer, J. Exp. Med., 1996, 183, 1533–1544 CrossRef.
- Y. Chen, E. McMillan-Ward, J. Kong, S. J. Israels and S. B. Gibson, Cell Death Differ., 2008, 15, 171–182 CrossRef CAS PubMed.
- E. A. Slee, C. Adrain and S. J. Martin, J. Biol. Chem., 2001, 276, 7320–7326 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra26556h |
| This journal is © The Royal Society of Chemistry 2016 |