
Acid catalyzed synthesis of 2-(2-aminophenyl)quinazoline-4-amine and reaction with aromatic aldehydes†
Elina Marinho and
M. Fernanda Proença*
Chemistry Department, University of Minho, Campus de Gualtar, 4710-057 Braga, Portugal. E-mail: fproenca@quimica.uminho.pt
First published on 8th January 2016
Abstract
The hydrochloride salt of 2-(2-aminophenyl)quinazoline-4-amine, prepared from a quinazolino[3,4-a]quinazoline, was reacted with aromatic aldehydes under conventional heating or microwave irradiation, leading to high yields of tetracyclic dihydroquinazolines.
Introduction
The quinazoline motif is widely present in naturally occurring alkaloids isolated from plants, in commercial drugs and in a diversity of bioactive compounds. The synthetic strategies to prepare different quinazoline derivatives and their biological properties have been extensively documented in reviews and monographs.1 The reported activities that include anticancer, antimicrobial, anti-inflammatory, antiviral, antitubercular, anticonvulsant or anti-depressant, highly depend on the substitution pattern and/or on the fused heterocyclic system associated to the quinazoline skeleton. This observation continues to inspire researchers in the development of new and eco-friendly methods to create diversity-oriented libraries of highly functionalized compound. Conventional heating methods have been generally applied,2 as well as other strategies that include the use of efficient and greener catalysts,3 multicomponent reactions4 or microwave irradiation.5The importance of 1,2-dihydroquinazoline derivatives has been documented and different methods have been reported for their synthesis, involving heating conditions or microwave irradiation. These compounds have been previously prepared from the reaction of 2-aminobenzamidine with aldehydes or ketones in refluxing ethanol6 or from 2-aminobenzonitrile and Grignard reagents, followed by condensation with an aldehyde.7 1,2-Dihydroquinazolines were also prepared from the reaction of 2-aminobenzophenone with aldehydes in urea, under microwave irradiation8 or in the presence of ammonium acetate catalyzed by DMAP, in ethanol at 40 °C.9 A different strategy used the microwave-promoted reaction of 2-aminoarylalkanone O-phenyl oximes with aldehydes in toluene.10
The presence of reactive functional groups in the quinazoline core has been widely used to explore the preparation of structurally novel types of derivatives and/or the combination with recognized pharmacophores, to modify traditional drugs. The amino group, in particular, can be regarded as an important synthon and a recent publication reports the selective amidation of 2,4-diaryl quinazolines.11 The reaction uses sulfonyl azides as the amine source and proceeds with exceptional regioselectivity in the ortho position of the 2-aryl group, induced by a rhodium catalyst and assisted by a meta substituent on the ring.
As a continuation of our ongoing study on the synthesis of quinazolino[3,4-a]quinazolines11 herein we report their transformation into 2-aminophenyl quinazolines. These compounds were used in the reaction with aromatic aldehydes to generate novel and highly stable dihydro-quinazolino[3,4-a]quinazolines by two different eco-friendly methods.
Results and discussion
Quinazolino[3,4-a]quinazolines 1, used as starting material for the preparation of 2-(2-amino)quinazolines 3, were previously prepared from the reaction of anthranilonitrile and triethylorthoformate (TEOF). The product was isolated in 80% yield from ethanol and 1 molar equivalent of nitric acid, after 3 h (1a) or 3 days (1b) at 40 °C.12In the present work, the reactivity of the dimeric quinazoline 1 was studied in ethanol/methanol or aqueous solution, in the presence of inorganic acid or base (Table 1).
|
|||
|---|---|---|---|
| Entry | Reagents | Reaction conditions | Product (yield) |
| 1 | 1a·HNO3 | H2O, r.t., 18 h | 2a, X = NO3 (70%) |
| 2 | 1a·HNO3 | NaOH (aq.) 3 M, r.t., 18 h | 2a (96%) |
| 3 | 1a | H2O, 60 °C, 18 h | 2a (92%) |
| 4 | 1a | NaOH (aq.) 3 M, r.t., 18 h | 2a (92%) |
| 5 | 1a·HNO3 | MeOH, HCl, r.t., 4 h | 3a, X = Cl (93%) |
| 6 | 1a·HNO3 | H2O, EtOH, HNO3, r.t., 6 days | 3a, X = NO3 (91%) |
| 7 | 1b·HNO3 | MeOH, HCl, r.t., 16 h | 3b, X = Cl (93%) |
| 8 | 2a·HNO3 | H2O, HNO3, 110 °C, 1 day | 3a, X = NO3 (70%) |
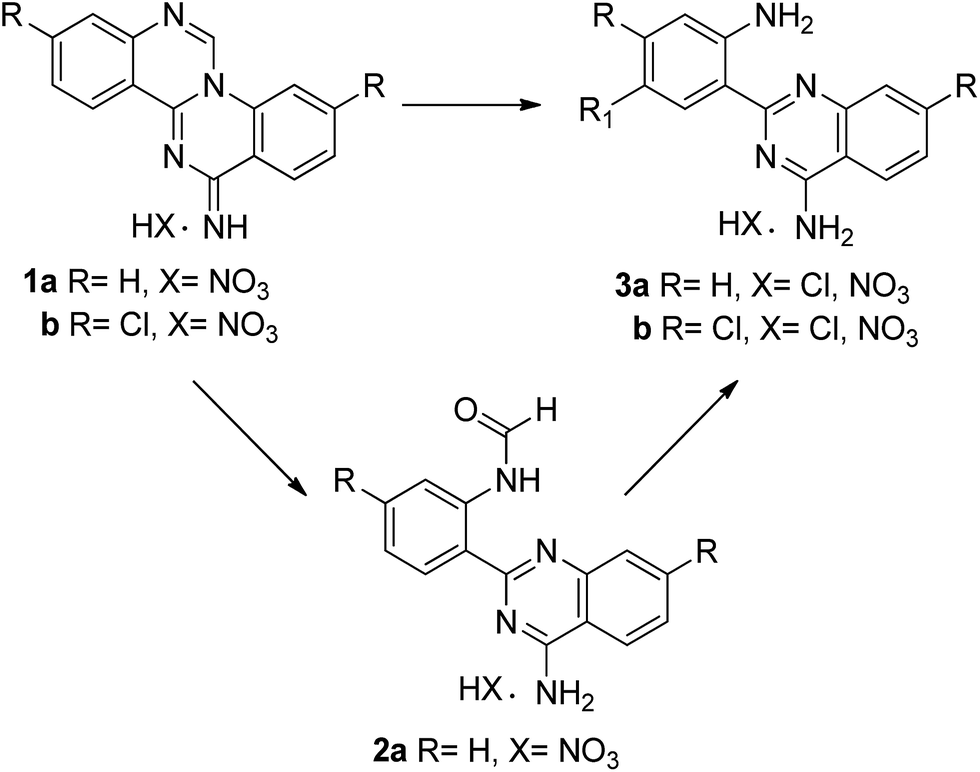
The reaction of 1a, used as the nitrate salt, was initially performed in water, at room temperature, for 18 h. The formamide unit was maintained in the product (2a), isolated in 70% yield upon hydrolysis (entry 1). The yield was considerably improved using 3 M NaOH under similar experimental conditions (entry 2, 96%). The synthesis was equally successful from the neutralized quinazolino[3,4-a]quinazolines 1a, leading to the same product 2a in 92% yield either upon heating at 60 °C in water or in aqueous base at room temperature (entries 3 and 4). Compounds 1a and 1b were also combined with conc. hydrochloric acid in methanol, at room temperature (entries 5 and 7). The hydrochloride salt of the corresponding products 3a and 3b precipitated from solution and were isolated as yellow solids by simple filtration after 4 h (3a) or 16 h (3b), both in 93% yield. The use of nitric acid in ethanol/water (Table 1, entry 6) led to the nitrate salt of 3a, isolated in 91% yield after 6 days, at room temperature. These results indicate that, in acidic medium, hydrolysis is followed by cleavage of the formyl group, leading to the diamine 3 in excellent yield. This was confirmed by the isolation of 3a when 2a was heated in aqueous nitric acid (entry 8). The hydrochloride salt of 3a was neutralized in acetone and 3 M NaOH solution and the product was isolated in 77% yield.
The synthesis of 2-(2-aminophenyl)quinazolin-4-amine 3a was previously reported from the reaction of anthranilonitrile with sodium hydride in dimethyl sulphoxide, at 0 °C (3 h) followed by 21 h at 25 °C.13 The product was isolated in almost quantitative yield after addition of aqueous HCl. This reaction was equally successful using sodium methoxide in dioxane, but the product was isolated in 75% yield.14 Combining anthranilonitrile with 10 mol% of potassium t-butoxide15 under microwave irradiation (700 W) for 1 minute led also to compound 3a, isolated in 82% yield.
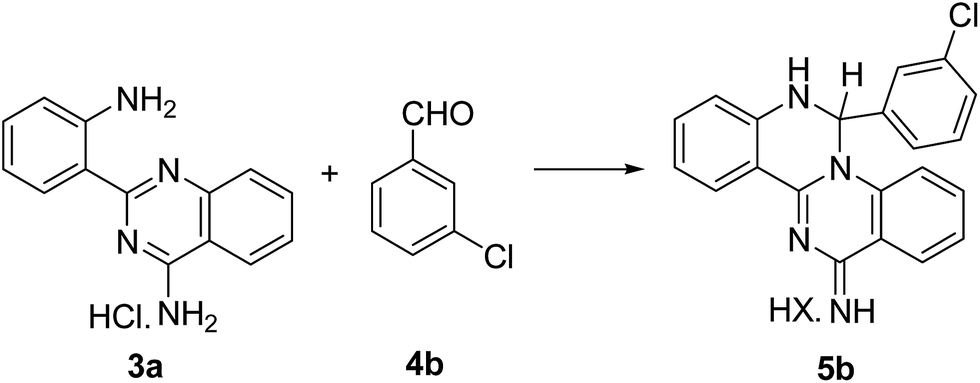
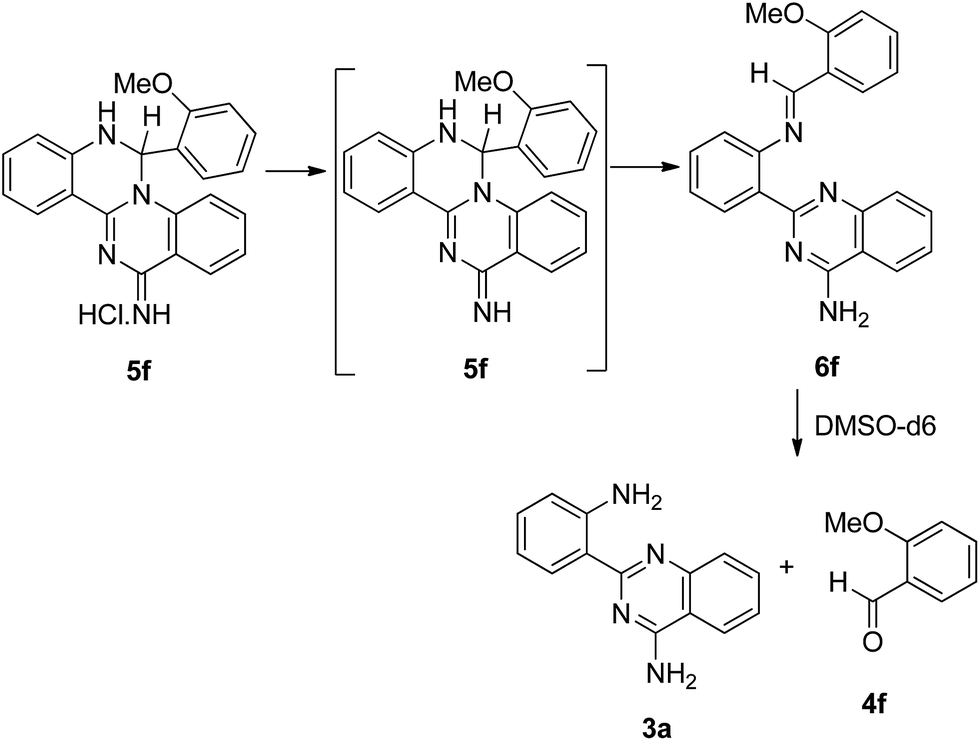
The reaction between the neutral pyrimidine 3a and 2-methoxybenzaldehyde 4f was initially followed by 1H NMR, in deuterated DMSO (600 μL; 3a, 10 mg; 4f, 1.1 equiv.). No reaction was observed after 7 h at 60 °C and addition of a catalytic amount of TFA was equally unsuccessful, after 20 h at 60 °C. The experiment was reproduced in ethanol and the reagents (3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4f, 1:1.1 molar ratio) were refluxed for 30 h. TLC showed no evidence for product formation and 5f was only quantitatively generated after addition of HCl (1 molar equivalent) and reflux for 30 min. This result indicates that the presence of one equivalent of acid is essential for a smooth reaction to occur and all the experiments were performed from the hydrochloride salt of 3a. The quinazolino[3,4-a]quinazolines 5 were generated by nucleophilic attack of the most reactive amino group to the carbonyl function, followed by intramolecular cyclization to the neighbouring quinazoline nitrogen, with elimination of water.
:4f, 1:1.1 molar ratio) were refluxed for 30 h. TLC showed no evidence for product formation and 5f was only quantitatively generated after addition of HCl (1 molar equivalent) and reflux for 30 min. This result indicates that the presence of one equivalent of acid is essential for a smooth reaction to occur and all the experiments were performed from the hydrochloride salt of 3a. The quinazolino[3,4-a]quinazolines 5 were generated by nucleophilic attack of the most reactive amino group to the carbonyl function, followed by intramolecular cyclization to the neighbouring quinazoline nitrogen, with elimination of water.
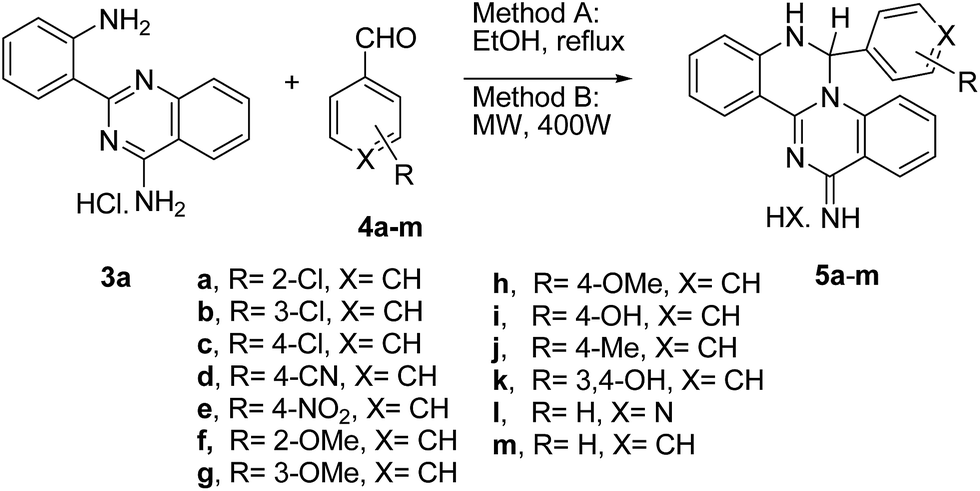
A preliminary study on the effect of solvent, temperature and acid/base catalysis on the product yield was performed using the reaction of 3a with 4b (Table 2). When 3a and 4b were combined in a 1:2 molar ratio, using THF as solvent and nitric acid catalysis, the quinazoline 3a was still present after 6 days at room temperature (entry 1). Changing the solvent to ethanol under similar reaction conditions led to the isolation of 5b in 85% yield after 3 days at room temperature (entry 2). Ethanol was definitely a better solvent than THF and was selected for the remaining experiments where trimethylamine or DBU were used as base catalysts (entries 3 and 4 respectively). After 1.5 days at room temperature, experiment 3 resulted in a complex mixture while experiment 4 allowed the isolation of a solid product that proved to be a combination of 3a and 5b in a 1:2.7 molar ratio, by 1H NMR. Refluxing the reagents in ethanol proved to be the most convenient approach, resulting in 86% isolated yield of 5b after 2 h (entry 5). The use of microwave irradiation (400 W for 5 min) was equally successful leading to 94% yield of the product (entry 6).
|
|||
|---|---|---|---|
| Entry | Reagents (equiv.) | Reaction conditions | Product (yield) |
| a By 1H NMR.b By TLC. | |||
| 1 | 3a + 4b (1:2) |
THF, HNO3, r.t., 6 days | 3a + 5b (1:1.4)a |
| 2 | 3a + 4b (1:2) |
EtOH, HNO3, r.t., 3 days | 5b (85%) |
| 3 | 3a + 4b (1:2) |
EtOH, NEt3, r.t., 1.5 days | Complex mixtureb |
| 4 | 3a + 4b (1:2) |
EtOH, DBU, r.t., 1.5 days | 3a + 5b (1:2.7)b |
| 5 | 3a + 4b (1:1.1) |
EtOH, reflux, 2 h | 5b (86%) |
| 6 | 3a + 4b (1:1.2) |
EtOH, MW, 400 W, 5 min | 5b (94%) |
Experiments 5 and 6 were considered the optimal reaction conditions and compound 3a was reacted with a variety of aromatic aldehydes (4a–m) either under conventional heating conditions in ethanol (method A) or under microwave irradiation at a constant power of 400 W (method B). Table 3 summarizes the individual experimental conditions that were used and the overall yield of product 5, isolated as a yellow solid. Products 5a–m were collected by simple filtration in excellent yield from both methods A or B.
|
|||
|---|---|---|---|
| Entry | Reagents (equiv.) | Reaction conditions | Product (yield) |
| 1 | 3a + 4a (1:1.2) |
EtOH, reflux, 2 h | 5a (84%) |
| 2 | 3a + 4a (1:1.2) |
EtOH, MW, 400 W, 5 min | 5a (85%) |
| 3 | 3a + 4b (1:1.1) |
EtOH, reflux, 2 h | 5b (86%) |
| 4 | 3a + 4b (1:1.2) |
EtOH, MW, 400 W, 5 min | 5b (94%) |
| 5 | 3a + 4c (1:1.2) |
EtOH, reflux, 12 h | 5c (91%) |
| 6 | 3a + 4c (1:1.2) |
EtOH, MW, 400 W, 5 min | 5c (94%) |
| 7 | 3a + 4d (1:1.1) |
EtOH, reflux, 1.5 h | 5d (86%) |
| 8 | 3a + 4d (1:1.2) |
EtOH, MW, 400 W, 5 min | 5d (84%) |
| 9 | 3a + 4e (1:1.32) |
EtOH, reflux, 5 h | 5e (92%) |
| 10 | 3a + 4e (1:1.2) |
EtOH, MW, 400 W, 5 min | 5e (90%) |
| 11 | 3a + 4f (1:1.1) |
EtOH, reflux, 15 min | 5f (97%) |
| 12 | 3a + 4f (1:1.2) |
EtOH, MW, 400 W, 5 min | 5f (96%) |
| 13 | 3a + 4g (1:1.1) |
EtOH, reflux, 25 min | 5g (94%) |
| 14 | 3a + 4g (1:1.2) |
EtOH, MW, 400 W, 5 min | 5g (96%) |
| 15 | 3a + 4h (1:1.1) |
EtOH, reflux, 50 min | 5h (86%) |
| 16 | 3a + 4h (1:1.2) |
EtOH, MW, 400 W, 5 min | 5h (90%) |
| 17 | 3a + 4i (1:1.2) |
EtOH, reflux, 4 h | 5i (83%) |
| 18 | 3a + 4i (1:1.2) |
EtOH, MW, 400 W, 5 min | 5i (84%) |
| 19 | 3a + 4j (1:1.2) |
EtOH, reflux, 12.5 h | 5j (86%) |
| 20 | 3a + 4j (1:1.2) |
EtOH, MW, 400 W, 5 min | 5j (90%) |
| 21 | 3a + 4k (1:1.1) |
EtOH, reflux, 1.5 h | 5k (91%) |
| 22 | 3a + 4k (1:1.2) |
EtOH, MW, 400 W, 5 min | 5k (93%) |
| 23 | 3a + 4l (1:1.1) |
EtOH, reflux, 4 h | 5l (92%) |
| 24 | 3a + 4j (1:1.2) |
EtOH, MW, 400 W, 5 min | 5l (95%) |
| 25 | 3a + 4m (1:1.1) |
EtOH, reflux, 4 h | 5m (74%) |
| 26 | 3a + 4m (1:1.1) |
EtOH, MW, 400 W, 5 min | 5m (76%) |
The quinazolines 5 proved to be highly stable in air and heat. A solution of 5b in deuterated DMSO was heated to the boiling point of the solvent with no detected degradation of the product, by 1H NMR performed when the solution reached room temperature. Products 5 clearly showed the presence of a sp3 carbon and two characteristic protons on NH and CH in adjacent positions (by 1H NMR, δ 8.65 and 7.72 ppm, J 4 Hz). The feasibility to generate the aromatic tetracyclic skeleton in the presence of an oxidizing agent was also tested. Studies on the evolution of 5b were conducted by 1H NMR spectroscopy in DMSO-d6 solution and in the presence of DDQ. A catalytic amount of DDQ was added to a solution of 5b (3 mg) in deuterated DMSO (650 μL) and the NMR tube was allowed to stand at 20 °C. The spectrum was registered after 1 and 5 days, evidencing that the composition of the solution remained unchanged. The sample was placed at 60 °C and after 7 days, the two doublets were still clearly present. The mixture was heated to the boiling point of the NMR solvent during 1 to 5 minutes. Again no evolution was observed in the 1H NMR spectrum and the coupling between the NH and CH protons was undoubtedly present.
Compound 5b demonstrated a high stability to temperature and to oxidation but in the 1H NMR spectrum, some of the compounds 5 consistently showed the presence of trace amounts of aldehyde and quinazoline 3a in a 1:1 molar ratio. This observation led us to study the effect of temperature on the chemical composition of the NMR solution of a selection of these compounds in DMSO-d6. The solutions were prepared using 3 mg of solid material in 650 μL of deuterated solvent and the 1H NMR spectra were registered at 20 °C and 80 °C. Table 4 summarizes the relative percentage of tetracyclic compound 5 and starting material 3a, calculated from the integration of the triplet at δ 7.47–7.50 ppm corresponding to C9–H (5b, 5h, 5m) of the doublet at δ 6.98–7.03 ppm corresponding to C8–H (5c, 5d, 5j) or the doublet at δ 6.56 ppm for C6′–H of 5g. Compound 3a was quantified from the triplet at δ 6.71 ppm for C5–H. For each compound, the relative percentage of the tetracyclic structure and of starting material (5:3a), present in solution at 80 °C and 20 °C, was calculated.
| Comp | R (aldehyde) | σ*a | 20 °C | 80 °C | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 5 (%) | 3a (%) | Kb | logK |
5 (%) | 3a (%) | Kb | logK |
|||
| a Calculated using the “ACD Lab Sigma Predictor” program.b K = [5]/[3a]. | ||||||||||
| 5h | p-OMe | 0.36 | 76 | 24 | 3.17 | 0.50 | 50 | 50 | 1 | 0 |
| 5j | p-Me | 0.46 | 80 | 20 | 4.00 | 0.60 | 53 | 47 | 1.12 | 0.05 |
| 5m | H | 0.60 | 84 | 16 | 5.25 | 0.72 | 55 | 45 | 1.27 | 0.10 |
| 5g | m-OMe | 0.66 | 87 | 13 | 6.69 | 0.83 | 59 | 41 | 1.44 | 0.16 |
| 5c | p-Cl | 0.75 | 90 | 10 | 9.00 | 0.95 | 69 | 31 | 2.23 | 0.35 |
| 5b | m-Cl | 0.85 | 93 | 7 | 13.29 | 1.12 | 72 | 28 | 2.57 | 0.41 |
| 5d | p-CN | 1.05 | 96 | 4 | 24.00 | 1.38 | 77 | 23 | 3.35 | 0.52 |
In an attempt to understand if the nature and position of the substituent on the aromatic ring of the aldehyde influenced the relative amounts of the species in solution at 80 °C and 20 °C we used the Hammett–Brown parameter (σ*), calculated for the aryl group using the “ACD Lab Sigma Predictor” program. The relationship of this parameter with logK, defined as the ratio K = [5]/[3a], is depicted in Scheme 1. A linear relationship was observed between these values, either at 80 °C or at 20 °C, confirming that the polar effect of the aromatic substituent influences this equilibrium.
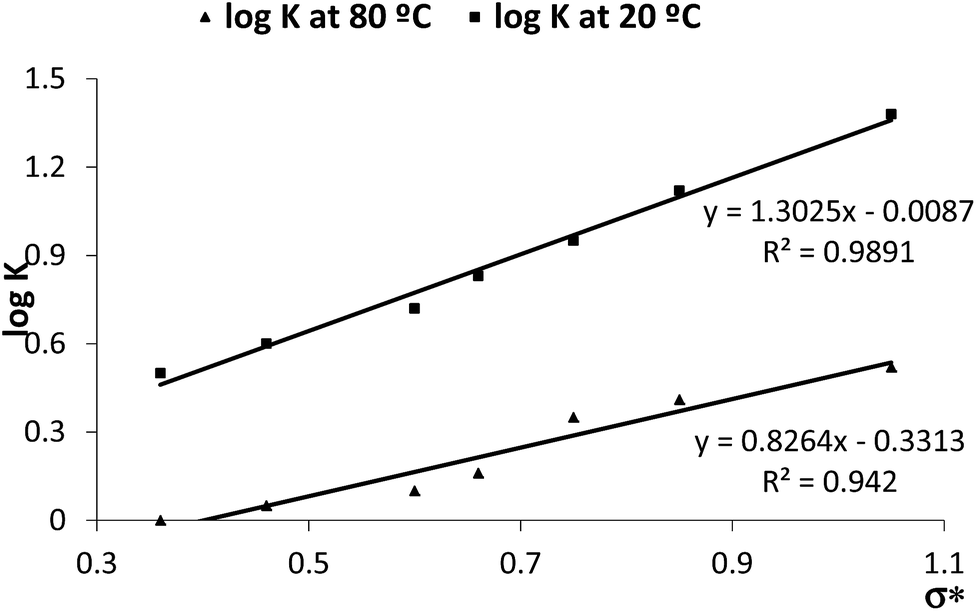 | ||
| Scheme 1 Plot of logK (K = [5]/[3]) in DMSO-d6 versus the Hammett–Brown parameters σ*. | ||
Considering that the Hammett–Brown parameter (σ*) reflects the polar effect of the aromatic substituent incorporating the R group, the analysis of these results suggests that as the electron-withdrawing character of the substituent increases, the ratio of the tetracyclic structure 5 increases when compared to the precursor 3a. This effect is more evident at 20 °C than at 80 °C.
The equilibrium may be due to a strong interaction between the two nitrogen atoms of the dihydropyrimidine unit in compound 5 with a water molecule, as is represented in Scheme 2. In this association, the inter-conversion boat-chair must be accelerated as the temperature is raised. At 80 °C, a higher contribution of the boat conformation is expected, favouring the intramolecular nucleophilic attack of the oxygen atom to the sp3 carbon, regenerating the reagents 3a and 4.
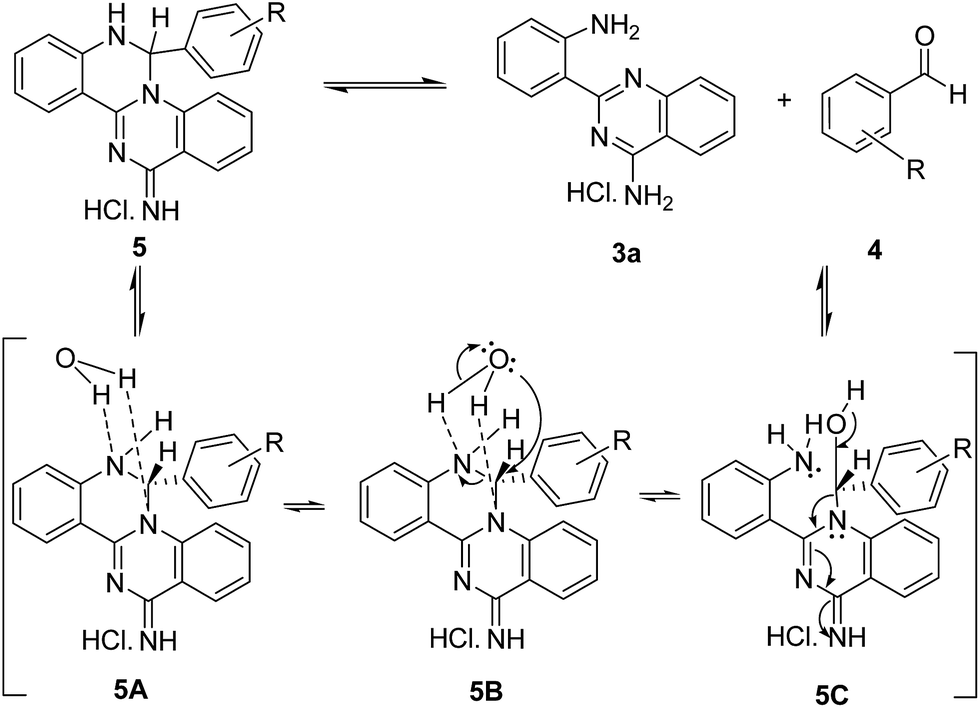 | ||
| Scheme 2 Mechanistic proposal for the equilibrium between the reagents 3a and 4 and the tetracyclic structure 5, induced by heating. | ||
The presence of electron withdrawing substituents in the aromatic ring decreases the basicity of the adjacent amino groups, and the stability of the association with the water molecule is consequently reduced, leading to an enhanced concentration of the tetracyclic structure 5. When the sample studied at 80 °C was allowed to cool to 20 °C, the compound ratio reproduced the values initially registered by 1H NMR at this temperature, clearly evidencing an equilibrium that favours the tetracyclic structure.
All compounds were characterized by the usual analytical and spectroscopic techniques and a selection was also studied by 15N/1H HMBC correlation spectra (Table 5).
| Compound | δ N1 | δ N2 | δ N3 | |
|---|---|---|---|---|
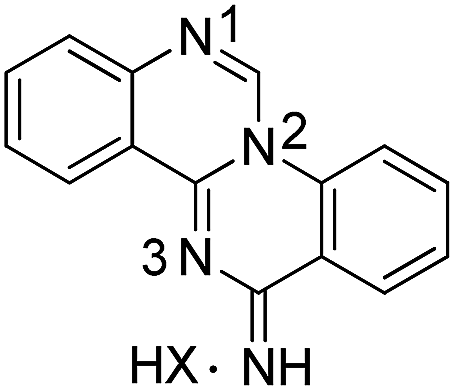 |
1a | |||
| HX = HNO3 | 262.81 | 169.57 | 252.82 | |
| Neutral | 249.81 | 160.30 | — | |
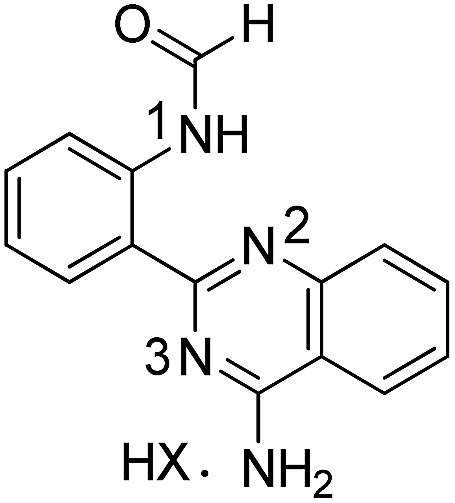 |
2a | |||
| HX = TFA | 136.00 | 240.10 | — | |
| Neutral | 133.81 | — | — | |
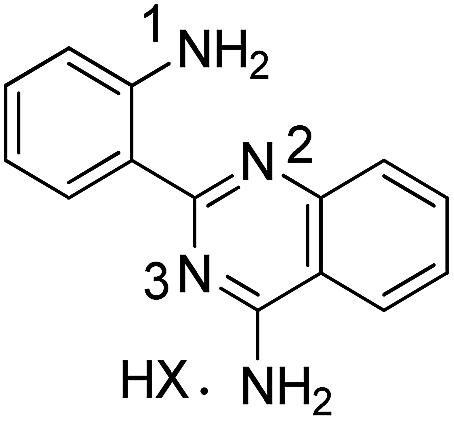 |
3a | |||
| HX = HNO3 | 74.06 | 146.74 | — | |
| HX = TFA | 73.47 | 145.45 | 220.58 | |
 |
5c | |||
| R = Cl | 75.05 | 145.32 | 216.31 | |
| 5h | ||||
| R = OMe | 76.20 | 146.30 | 216.40 |
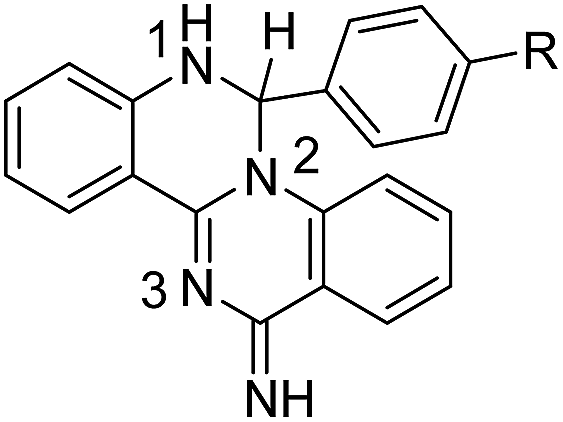
The N1 and N3 nitrogen atoms in the protonated form of 1a exhibit high chemical shifts (δ 252.82 and 262.81 ppm) while a considerably lower chemical shift was assigned to N2 (δ 169.59 ppm). The specific assignments were based on the correlations of N1 and N2 with the neighbouring ring protons and the remaining signal was assigned to N3. The signal for the exocyclic amino group on the quinazoline ring could never be detected. Protonation affects all the nitrogen atoms of the tetracyclic ring of 1a, with a decrease of 10–13 ppm observed for the chemical shift of N1 and N2. For compound 2a, protonation has only a minor effect on the signal for N1, suggesting that the proton must be centrally located on the quinazoline ring. Compound 3a (protonated), 5b and 5h all share comparable chemical shifts for N1 (the sp3 nitrogen of an amino group), N2 (a sp3 nitrogen partially involved in the aromatic π system) and N3 (a sp2 ring nitrogen). This observation suggests that protonation of 3a is predominantly occurring on N2.
Attempts to neutralize compound 5f in 3 M NaOH (Table 6, entry 1) led to an oily solid material identified by TLC as a mixture of 3a and 4f. In order to avoid the use of aqueous base, the neutralization was performed by DBU in acetonitrile (entry 2). A white solid was immediately formed and identified by NMR as the imine 6f. The imine proton (δH 8.68 ppm and δC 155.59 ppm) correlates with the neighbouring carbon atoms in both aromatic rings. The product was always contaminated with the hydrochloride salt of DBU together with traces of the neutral species 5f, as evidenced by a doublet at δH 7.56 ppm (J 4 Hz) and δC 59.29 ppm assigned to the sp3 carbon. Keeping the NMR solution at room temperature for one week, resulted in complete evolution to 3a and 4f due to prompt imine hydrolysis induced by traces of water in the solvent.
Experimental
General methods
All compounds were fully characterized by elemental analysis and spectroscopic data. The NMR spectra were recorded at room temperature, on a Varian Unity Plus (1H: 300 MHz, 13C: 75 MHz), or Bruker Avance III 400 (1H: 400 MHz, 13C: 100 MHz) including the 1H–13C and 1H–15N correlation spectra (HMQC and HMBC). Deuterated DMSO was used as solvent. The peak patterns are indicated as follows: s, singlet; d, doublet; t, triplet; m, multiplet; q, quartet and br, broad. The coupling constants, J, are reported in hertz (Hz). IR spectra were recorded on a FT-IR Bomem MB 104 using Nujol mulls and NaCl cells. The reactions under microwave irradiation were performed on a CEM microwave reactor, model Mars 5, using a quartz open vessel.All reactions were monitored by thin layer chromatography (TLC) using silica gel 60 F254 (Merck). The melting points were determined on a Stuart SMP3 melting point apparatus and are uncorrected. Elemental analyses were performed on a LECO CHNS-932 instrument. High resolution mass spectra (HRMS) were obtained from the C.A.C.T.I. – Universidade de Vigo (Spain).
Synthesis of [2-(4-aminoquinazolin-2-yl)phenyl]formamide (2a)
A solution of the nitrate salt of 13H-quinazolino[3,4-a]quinazolin-13-imine nitrate 1a (0.08 g, 0.26 mmol) in 3 M NaOH (1 mL) was stirred at room temperature for 18 h, leading to a yellow solid suspension. The solid was filtered, washed with water and identified as 2-(4-aminoquinazolin-2-yl)phenyl]formamide 2aGeneral procedure for the synthesis of the hydrochloride salt of 2-(2-aminophenyl) quinazolin-4-amine (3)
Concentrated HCl (5 μL) was added to a pale yellow suspension of the nitrate salt of 13H-quinazolino[3,4-a]quinazolin-13-imine 1 in methanol (2 mL). The reaction mixture was stirred at room temperature leading to a homogeneous yellow solution after 5–15 min. A yellow solid started to precipitate after 25–45 min and the suspension was stirred for a further 4–15 h. The solid was filtered and washed with diethyl ether, leading to the pure product 3.General procedure for the synthesis of the hydrochloride salt of 6-aryl-6,7-dihydro-13H-quinazolino[3,4-a]quinazolin-13-imine (5)
The aldehyde 4 (1.1–1.2 equiv.) was added to a yellow suspension of the hydrochloride salt of 2-(2-aminophenyl)quinazolin-4-amine 2a in ethanol (2–3 mL). The suspension was refluxed for 15 min to 12.5 h (method A) or irradiated at a constant power of 400 W for 5 min (method B). The solid was filtered and washed with diethyl ether, leading to the pure product 5.Conclusions
A simple and eco-friendly approach was developed for the synthesis of quinazolines 3, prepared from the corresponding quinazolino[3,4-a]quinazolines 1, in methanol and hydrochloric or nitric acid, at room temperature. The hydrochloride salt of the unsubstituted quinazoline 3a was reacted with a slight excess of a selection of aromatic aldehydes, leading to a novel and highly stable tetracyclic structure 5. The reaction was performed under reflux in ethanol (15 min to 12.5 h) or under microwave irradiation at a constant power of 400 W (5 min). All the products were isolated in a high purity form by simple filtration and in 74 to 97% yield. The stability of compounds 5 is associated with protonation, as neutralization results in the imine precursor that can be easily hydrolysed.Acknowledgements
We gratefully acknowledge the financial support by the University of Minho and FCT through the Portuguese NMR network (RNRMN), the Project F-COMP-01-00124-FEDER-022716 (ref. FCT PEst-C/QUI/UI0686/2011) FEDER-COMPETE, and a PhD grant awarded to Elina Marinho (SFRH/BD/73659/2010).Notes and references
- For recent reviews see: I. Khan, A. Ibrar, N. Abbas and A. Saeed, Eur. J. Med. Chem., 2014, 76, 193 CrossRef CAS PubMed; V. G. Ugale and S. B. Bari, Eur. J. Med. Chem., 2014, 80, 447 CrossRef PubMed; I. Khan, A. Ibrar, N. Abbas and A. Saeed, Eur. J. Med. Chem., 2015, 90, 124 CrossRef PubMed; M. Garg, M. Chauhan, P. K. Singh, J. M. Alex and R. Kumar, Eur. J. Med. Chem., 2015, 97, 444 CrossRef PubMed.
- For recent publications see: C. Mendoza-Martínez, N. Galindo-Sevilla, J. Correa-Basurto, V. M. Ugalde-Saldivar, R. G. Rodríguez-Delgado, J. Hernandez-Pineda, C. Padierna-Mota, M. Flores-Alamo and F. Hernandez-Luis, Eur. J. Med. Chem., 2015, 92, 314 CrossRef CAS PubMed; M. Mahdavi, K. Pedrood, M. Safavi, M. Saeedi, M. Pordeli, S. K. Ardestani, S. Emami, M. Adib, A. Foroumadi and A. Shafiee, Eur. J. Med. Chem., 2015, 95, 492 CrossRef PubMed; Y. Zhang, Y.-J. Huang, H.-M. Xiang, P.-Y. Wang, D.-Y. Hu, W. Xue, B.-A. Song and S. Yang, Eur. J. Med. Chem., 2014, 78, 23 CrossRef PubMed; S. Ravez, A. Barczyk, P. Six, A. Cagnon, A. Garofalo, L. Goossens and P. Depreux, Eur. J. Med. Chem., 2014, 79, 369 CrossRef PubMed; A. M. Alanazi, A. A.-M. Abdel-Aziz, I. A. Al-Suwaidan, S. G. Abdel-Hamide, T. Z. Shawer and A. S. El-Azab, Eur. J. Med. Chem., 2014, 79, 446 CrossRef PubMed; A. Elkamhawy, J. Lee, B.-G. Park, I. Park, A. N. Pae and E. J. Roh, Eur. J. Med. Chem., 2014, 84, 466 CrossRef PubMed; H. Luo, S. Yang, Y. Cai, Z. Peng and T. Liu, Eur. J. Med. Chem., 2014, 84, 746 CrossRef PubMed; C. Mendoza-Martínez, J. Correa-Basurto, R. Nieto-Meneses, A. Marquez-Navarro, R. Aguilar-Suarez, M. D. Montero-Cortes, B. Nogueda-Torres, E. Suarez-Contreras, N. Galindo-Sevilla, A. Rojas-Rojas, A. Rodriguez-Lezama and F. Hernandez-Luis, Eur. J. Med. Chem., 2015, 96, 296 CrossRef PubMed; K. S. Van Horn, W. N. Burda, R. Fleeman, L. N. Shaw and R. Manetsch, J. Med. Chem., 2014, 57, 3075 CrossRef PubMed; H. Lechuga-Eduardo, H. F. Olivo and M. Romero-Ortega, Eur. J. Org. Chem., 2014, 5910 CrossRef; O. E. Alawode, V. K. Naganaboina, T. Liyanage, J. Desper and S. Rayat, Org. Lett., 2014, 16, 1494 CrossRef PubMed.
- For recent publications see: Y. Yan, Y. Xu, B. Niu, H. Xie and Y. Liu, J. Org. Chem., 2015, 80, 5581 CrossRef CAS PubMed; W. Lu, J. Ma, J. Hu, J. Song, Z. Zhang, G. Yang and B. Han, Green Chem., 2014, 16, 221 RSC; R. Deshidi, S. Devari and B. A. Shah, Eur. J. Org. Chem., 2015, 1428 CrossRef; C. Li, W.-T. Zhang and X.-S. Wang, J. Org. Chem., 2014, 79, 5847 CrossRef PubMed.
- For a recent publication see: Z. Zhang, Y.-S. Zhang, Z.-J. Quan, Y.-X. Da and X.-C. Wang, Tetrahedron, 2014, 70, 9093 CrossRef CAS.
- For recent publications see: A. Gellis, C. Kieffer, N. Primas, G. Lanzada, M. Giorgi, P. Verhaeghe and P. Vanelle, Tetrahedron, 2014, 70, 8257 CrossRef CAS; Y. Kabri, M. D. Crozet, T. Terme and P. Vanelle, Eur. J. Org. Chem., 2015, 3806 CrossRef.
- N. Finch and H. W. Gschwend, J. Org. Chem., 1971, 36, 1463 CrossRef CAS.
- J. Bergman, A. Brynolf, B. Elman and E. Vuorinen, Tetrahedron, 1986, 42, 3697 CrossRef CAS; L. Strekowski, M. T. Cegla, D. B. Harden, J. L. Mokrosz and M. J. Mokrosz, Tetrahedron Lett., 1988, 29, 4265 CrossRef.
- R. Sarma and D. Prajapati, Green Chem., 2011, 13, 718 RSC.
- C. Derabli, R. Boulcina, G. Kirsch, B. Carboni and A. Debache, Tetrahedron Lett., 2014, 55, 200 CrossRef CAS.
- F. Portela-Cubillo, J. S. Scott and J. C. Walton, J. Org. Chem., 2009, 74, 4934 CrossRef CAS PubMed.
- C. Zhang, Y. Zhou, Z. Deng, X. Chen and Y. Peng, Eur. J. Org. Chem., 2015, 8, 1735 CrossRef.
- E. Marinho, R. Araújo and F. Proença, Tetrahedron, 2010, 66, 8681 CrossRef.
- A. Gescher, M. F. G. Stevens and C. P. Turnbull, J. Chem. Soc., Perkin Trans. 1, 1977, 107 RSC.
- J. E. van Muijlwijk-Koezen, H. Timmerman, H. van der Goot, W. M. P. B. Menge, J. F. D. Künzel, M. Groote and A. P. Ijzerman, J. Med. Chem., 2000, 43, 2227 CrossRef CAS PubMed and references therein. P. G. Baraldi, M. A. Tabrizi, S. Gessi and P. A. Borea, Chem. Rev., 2008, 108, 238 CrossRef PubMed.
- J. A. Seijas, M. P. Vásquez-Tato and M. M. Martínez, Tetrahedron Lett., 2000, 41, 2215 CrossRef and references therein.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra19785f |
| This journal is © The Royal Society of Chemistry 2016 |