
LED and visible light-induced metal free ATRP using reducible dyes in the presence of amines†
Ceren
Kutahya
a,
F. Simal
Aykac
a,
Gorkem
Yilmaz
*a and
Yusuf
Yagci
*ab
aIstanbul Technical University, Department of Chemistry, Maslak, Istanbul 34469, Turkey
bCenter of Excellence for Advanced Materials Research (CEAMR) and Department of Chemistry, Faculty of Science, King Abdulaziz University, P. O. Box 80203, Jeddah 21589, Saudi Arabia. E-mail: gorkemyilmaz@itu.edu.tr; yusuf@itu.edu.tr
First published on 5th September 2016
Abstract
A new photoinitiating system involving electron acceptor dyes, namely, eosin Y and erythrosin B, in conjunction with alkyl halides and amines for photoinduced ATRP of (meth)acrylates and vinyl monomers in the absence of inorganic catalysts is reported. The polymerizations were efficiently activated by the photomediated redox processes to produce polymers with controlled chain end functionality and narrow molecular weight distribution. The dye/amine system was shown to be efficient under various colors of LED and industrially available visible light irradiation. The livingness nature of the polymerization was proved by GC analyses and the irradiation dependency of polymerization was confirmed by light on/off experiments.
Introduction
Among the most common controlled/living radical polymerization methods, ATRP became the most widely used strategy because of its applicability to a wide range of monomer formulations and initiators.1,2 However, the requirement of low oxidation state transition metal complexes as catalysts (i.e., CuX/L, X: halide and L: ligand) make the reaction conditions extremely sensitive to air as the corresponding metal oxides can easily be formed. Therefore, several approaches were proposed to generate the catalysts starting from their higher oxidation state analogs. Either using organic reductants3,4 or applying photochemical strategies,5–11 convenient conditions for ATRP can be provided. Among these strategies, photochemical approaches have been the most-widely investigated and applied as they provide temporal and spatial control over the processes.12–20 Either by direct initiation of the Cu(II)/L via UV irradiation or indirect initiation at higher wavelengths using free radical initiators, photosensitizers, nanoparticles and porous structures in conjunction with Cu(II)/L illustrated the feasibility of conducting ATRP at ambient temperatures.21–25 Notably, similar strategies were also applied to copper catalyzed azide–alkyne cycloaddition reactions where the presence of CuX salts is neccessary.26,27 For example, the use of polynuclear aromatic compounds (PACs) such as phenothiazine, perylene and anthracene were shown to simultaneously generate Cu(I)/L from the stable Cu(II)/L complexes, which catalyzed CuAAC for macromolecular syntheses. The mechanism considers an electron transfer from the excited state PACs (sensitizers) to Cu(II) to yield the required Cu(I) catalyst.28Although these strategies bring enormous advantages, the necessity of inorganic catalysts in such processes could not be dealt with until recently. Recent studies showed that the utilization of PACs, such as phenothiazine29–31 and perylene,32 together with alkyl halides can achieve ATRP even in the absence of inorganic catalysts to produce monodisperse polymers with controlled chain-end functionalities. Previously, fluorescein was also shown to mediate ATRP in the presence of amines.33 The mechanism involves an electron transfer from the amine to the excited state fluorescein, which reduces alkyl halides to generate radicals responsible for initiation. The reversibility of the electron transfer steps allows for the living nature of the process as well as control over the chain-end functionality. In addition to these advantages, the photoinduced, metal-free ATRP systems are expected to be applicable for the modification of various surfaces.34
Eosin Y and erythrosin B are well-known reducible dyes with light absorption up to around 600 nm. The halide substituents on the core chromophoric structure make them even more suitable for reduction upon photochemical excitation. Herein, we present the use of two electron-acceptor dyes, eosin Y and erythrosin B, with electron donor amines and various alkyl halide sources to mediate the metal-free photo-ATRP of commercially available monomers. We performed detailed mechanistic, kinetic and spectroscopic studies, and in the light of the detailed experimental evidence, we proposed a plausible polymerization mechanism.
Experimental
Materials
Methyl methacrylate (MMA, 99%; Aldrich), styrene (St, 99%, Aldrich), and 2-hydroxyethyl methacrylate (HEMA, 99%; Aldrich) were passed through a basic alumina column to remove the inhibitor. N,N,N′,N′′,N′-Pentamethyldiethylenetriamine (PMDETA, 99%; Aldrich) was distilled before use. Ethyl α-bromoisobutyrate (EBI, 98%, Sigma Aldrich), ethyl 2-bromopropionate (EBP, 99%, Sigma Aldrich), (1-bromoethyl)benzene (BEB, 97%, Sigma Aldrich), N,N-dimethylformamide (DMF, Aldrich, 99.5%), methanol (99.9%; Merck), toluene (Aldrich, 99.5%), tetrahydrofuran (Aldrich, 99.5%), eosin Y (Merck, 99%), and erythrosin B (Merck, 99%) were used as received.Polymerizations
Measurements
1H NMR and 13C NMR of the intermediates and final polymers were recorded at room temperature at 500 MHz on an Agilent VNMRS 500 spectrometer. UV spectra were recorded on a Shimadzu UV-1601 spectrometer. The resolution was 4 cm−1, and 24 scans were performed with a 0.2 cm s−1 scan speed. Gel permeation chromatography (GPC) measurements were obtained from a Viscotek GPCmax autosampler system consisting of a pump, a Viscotek UV detector, and Viscotek a differential refractive index (RI) detector. Three ViscoGEL GPC columns (G2000H HR, G3000H HR, and G4000H HR, 7.8 mm internal diameter, 300 mm length) were used in series. THF was used as an eluent at a flow rate of 1.0 mL min−1 at 30 °C. Both detectors were calibrated using polystyrene standards with a narrow molecular weight distribution. Data were analyzed using Viscotek OmniSEC Omni-01 software.Results and discussion
The absorption regimes of eosin Y, erythrosin B and fluorescein were tested by UV-vis spectral analysis in N,N′-dimethylformamide (DMF) (Fig. 1). The presence of an amine (i.e., N,N,N′,N′′,N′-pentamethyldiethylenetriamine (PMDETA)) in the solutions had a significant impact on the photophysical properties of the dyes as the five membered lactone functionality in their structures is prone to undergo a ring opening reaction at high pH levels.33 Both eosin Y and erythrosin B display photoactivity up to 600 nm, which enables their application as photocatalysts at lower energies in comparison to fluorescein (Fig. 1). | ||
| Fig. 1 UV-vis spectra of fluorescein, eosin y and erythrosin b in the presence of PMDETA. | ||
Steady state fluorescence studies were performed to investigate the nature of the interactions with the dyes and alkyl halides in the presence of amines. Results showed that the emission spectra of both dye/amine systems decreased with a non-linear regime upon addition of ethyl 2-bromo propionate (EBP) (ESI, SF2†). Previous studies showed that structurally similar chromophores undergo a series of electron transfer reactions with the additive alkyl halide to form the corresponding alkyl radicals through a reductive quenching mechanism upon light irradiation. These reports suggested that eosin Y and erythrosin B can be used for the photopolymerization of various monomers.
To test the ability of eosin Y and erythrosin B to mediate ATRP, methyl methacrylate (MMA) was polymerized under reduced pressure using EBP and PMDETA under visible light irradiation. For comparison, fluorescein was also used as a photosensitizer since it was previously shown to be efficient in the photo-ATRP process. The results are tabulated in Table 1.
| Runs | Dyeb | [MMA]/[EBP]/[PMDETA]/[Dye] | Conv.c (%) |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d (g mol−1) d (g mol−1) |
M
w/Mnd |
|---|---|---|---|---|---|
| a V MMA = 2.0 mL, VDMF = 1.0 mL, λ ∼ 400–500 nm, time = 120 min. b E-Y: Eosin Y, E-B: Erythrosin B, F: Fluorescein. c Determined gravimetrically. d Determined by gel permeation chromatography using polystyrene standards. | |||||
| 1 | E-Y | 200/1/1/0.1 | 8.7 | 15100 |
1.33 |
| 2 | E-B | 200/1/1/0.1 | 20 | 90000 |
1.20 |
| 3 | F | 200/1/1/0.1 | 9.9 | 17500 |
1.52 |
| 4 | E-Y | 200/1/0/0.1 | — | — | — |
| 5 | E-B | 200/1/0/0.1 | — | — | — |
| 6 | E-Y | 200/1/1/0.5 | 2.1 | 14500 |
1.71 |
| 7 | E-B | 200/1/1/0.5 | — | — | — |
| 8 | F | 200/1/1/0.5 | 22.5 | 11100 |
1.76 |
The results showed that both dyes displayed a higher sensitization efficiency in comparison to fluorescein, which might be attributed to their better absorption characteristics in the irradiation region. In addition, the polymers obtained with eosin Y and erythrosin B show narrower molecular weight characteristics (runs 1–3). To gain more insight into the polymerization mechanism, similar experiments were conducted in the absence of PMDETA as the electron donor source (runs 4 and 5). However, no polymer was attained, which proves that the presence of an electron donor source is a prerequisite for the polymerization to occur. When the concentrations of the dyes were increased, the polymerization conversions observed with both eosin Y and erythrosin B showed a dramatic decrease, but this had no effect on the conversion of the polymerization performed with fluorescein (runs 6–8). This can be accredited to the excimer (excited state dimer) formation of both eosin Y and erythrosin B, which is a well-known phenomenon promoted by high monomer density in some specific chromophores.35
The effect of the light source on polymerization was also investigated by replacing the visible light source with an appropriate LED light. As can be seen from Table 2, green, white and blue LEDs lead to almost similar conversions in the polymerizations. However, higher polydispersities were observed in the polymers obtained, which might be attributed to the lower light intensities in comparison to the visible light source.
| Dyeb | LED colour | [MMA]/[EBP]/[PMDETA]/[Dye] | Conv.c (%) |
M
nd (g mol−1) |
M
w/Mnd |
|---|---|---|---|---|---|
| a [MMA]/[EBP]/[PMDETA]/[Dye]: 200/1/1/0.1, VMMA = 2.0 mL, VDMF = 1.0 mL, samples were irradiated by LED light at room temperature. b E-Y: Eosin Y, E-B: Erythrosin B. c Determined gravimetrically. d Determined by gel permeation chromatography using polystyrene standards. | |||||
| E-Y | Green | 200/0.1/1/0.1 | 28 | 41200 |
1.85 |
| E-B | Green | 200/0.1/1/0.1 | 15.9 | 57600 |
1.42 |
| E-Y | White | 200/1/1/0.1 | 20.1 | 8700 | 1.48 |
| E-B | White | 200/1/1/0.1 | 20.0 | 13700 |
2.47 |
| E-Y | Blue | 200/1/1/0.1 | 20.2 | 31600 |
1.62 |
| E-B | Blue | 200/1/1/0.1 | 19.5 | 22500 |
1.74 |
The effect of the alkyl halide structure on polymerization was also investigated. Using either secondary and tertiary alkyl halides with neigh boring ester functions was shown to yield successful polymerization as well as the secondary benzyl halides, as can be seen in Table S1 (see ESI†).
The effect of the polarity of the reaction media on metal-free controlled radical polymerization was also tested. For this purpose, the typical polymerization procedures were applied with changing the polymerization solvent (Table 3). Even though satisfying conversions were attained in both solvents, a decrease in the polarity of the solvent yielded polymers with broader molecular weight distributions. This might be due to the better stabilization of the ionic species, which are generated during the electron transfer steps, in more polar solvents.
| Dyeb | Solvent | Conv.c (%) |
M
nd (g mol−1) |
M
w/Mnd |
|---|---|---|---|---|
| a [M]/[EBP]/[PMDETA]/[Dye]: 200/1/1/0.1, VMMA = 2.0 mL, VDMF = 1.0 mL; λ ∼ 400–500 nm, time = 120 min. b E-Y: Eosin Y, E-B: Erythrosin B. c Determined gravimetrically. d Determined by gel permeation chromatography using polystyrene standards. | ||||
| E-Y | DMF | 8.7 | 15100 |
1.33 |
| E-B | DMF | 20.0 | 90000 |
1.20 |
| E-Y | THF | 19 | 17100 |
1.41 |
| E-B | THF | 21.4 | 15000 |
1.40 |
| E-Y | Toluene | 13.6 | 22100 |
1.80 |
| E-B | Toluene | 7.5 | 16600 |
1.42 |
To investigate the efficiency of eosin Y in mediating the metal-free photo-ATRP of structurally different monomers, similar polymerizations were performed using styrene (S), hydroxyethyl methacrylate (HEMA) and tert-butyl acrylate (t-BA) (Table 4). All of the monomers yielded polymers with reasonable molecular weight distributions with varying conversions. The variations can be undoubtedly attributed to the propagating rate constants of the monomers.
| Monomer (M) |
k
pb (L mol−1 s−1) |
Conv.c (%) |
M
nd (g mol− 1) |
M
w/Mnd |
|---|---|---|---|---|
| a [M]/[EBP]/[PMDETA]/[Dye]: 200/1/1/0.1, VMMA = 2.0 mL, VDMF = 1.0 mL, λ ∼ 400–500 nm, time = 120 min. b Free radical propagation rate constants at 50 °C. c Determined gravimetrically. d Determined by gel permeation chromotography using polystyrene standards. e Could not be determined as the Mn value exceeds the limits of GPC. | ||||
| MMA | 6.4 × 102 | 8.7 | 15100 |
1.37 |
| S | 2.4 × 102 | 2.6 | 4500 | 1.55 |
| HEMA | 2.6 × 103 | 25.1 | ∞e | — |
| t-BA | 2.8 × 104 | 42.3 | 47300 |
1.68 |
The light dependency of the polymerization kinetics was investigated by light on/off experiments. For this purpose, the polymerization mixtures were placed in a Schlenk tube under a nitrogen atmosphere, irradiated at λ ∼ 350 nm, and kept in dark for repeated cycles. At certain time intervals, equivalent volumes of samples were syringed out from the system and precipitated into excess methanol to gravimetrically determine the conversion and analyze the molecular weight characteristics of the polymers obtained at each step by GPC measurements. The results demonstrate the polymerization is ultimately irradiation dependent, and almost no polymerization occurred when the solutions were kept in the dark (Fig. 2).
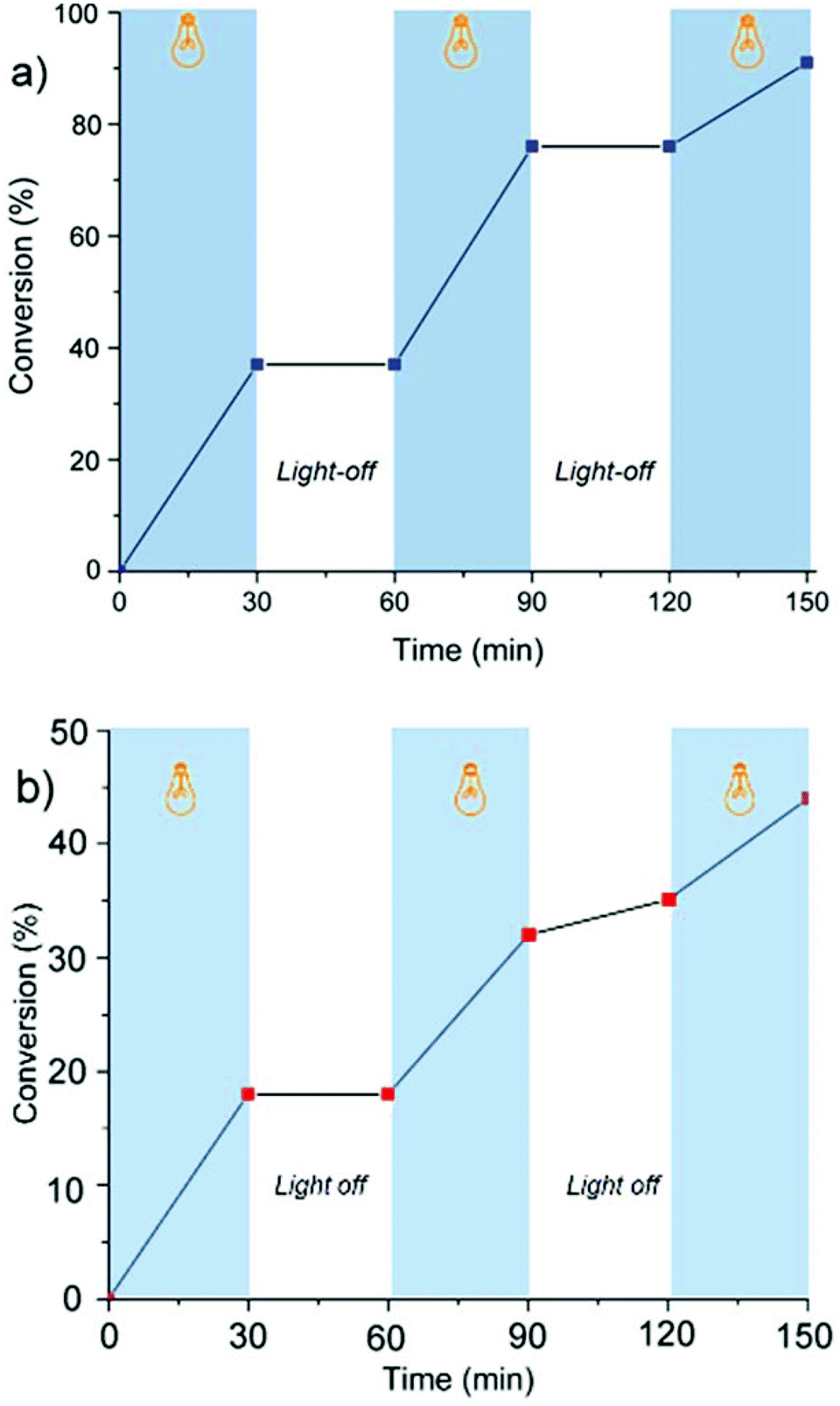 | ||
| Fig. 2 Monomer conversion (%) vs. time using erythrosin B (a) and eosin Y (b) to determine the dependency of propagation on irradiation: light on (blue regions) light off (white regions). | ||
To give further insight into the polymerization kinetics, several experiments were conducted to confirm the linear increase in the conversion during the irradiation time. A linear relationship between ln([M]0/[M]) and time indicated the living nature of the polymerization in the example of eosin Y (Fig. 3).
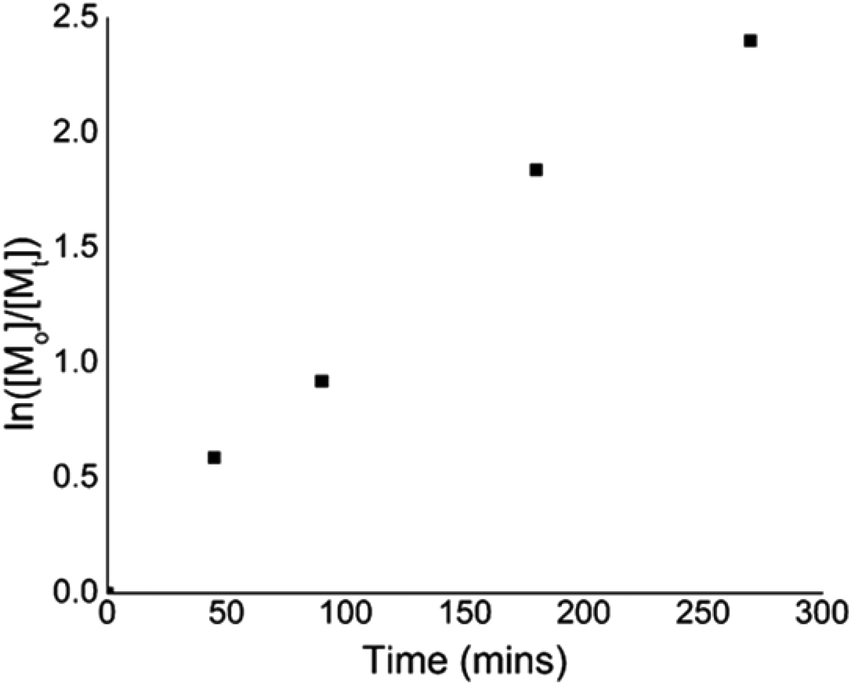 | ||
| Fig. 3 Kinetic plot for the polymerization of metal-free ATRP using eosin Y. | ||
In order to examine the chain-end fidelity of the polymers obtained, chain extension and block copolymerization experiments were performed. For this purpose, PMMA was used as the halide source, and identical polymerization conditions were applied as described in Table 4. GPC analyses showed that there are clear shifts to lower retention volumes, which indicated the success of the polymerizations from the chain ends of the precursor PMMA in either case (Fig. 4).
 | ||
| Fig. 4 Comparison of the GPC traces of precursor PMMA with (a) chain extended PMMA and (b) PMMA-b-PS before. | ||
In the light of these studies and the general photoexcited state behavior of the dyes, the following mechanism can be proposed for the polymerizations (Scheme 1).36,37 The excited state dyes undergo an electron transfer with electron donor amines. The formed radical anion dyes reduce the initiator alkyl halide to yield radicals responsible for the initiation. A back electron transfer from the halide anion to the amine radical cation concludes the formation of the dormant macroalkyl halide that returns to the polymerization cycle.
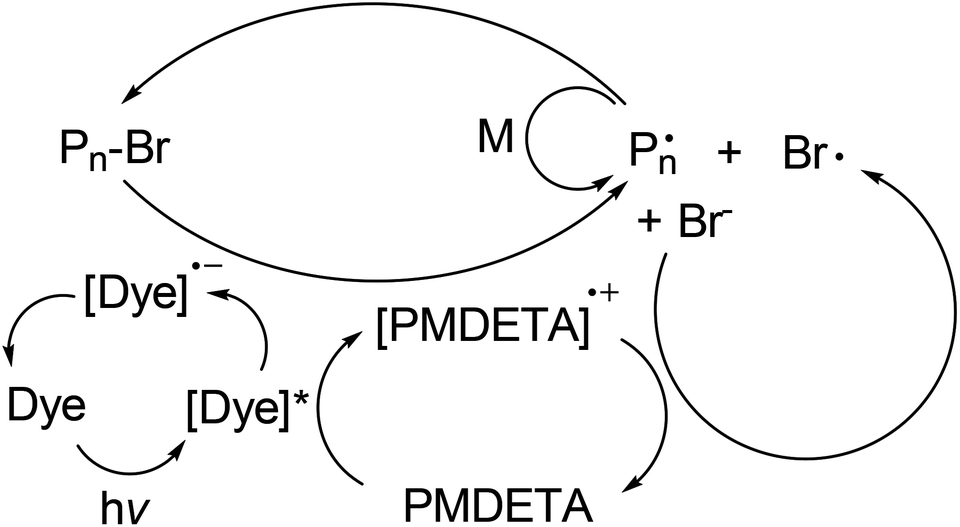 | ||
| Scheme 1 Proposed mechanism of photoinduced, metal-free ATRP using dye/amine initiating system. | ||
Conclusions
In conclusion, a successful photoinduced, metal-free ATRP by using reducible dyes in the presence of amine and alkyl halides was achieved. The results of the polymerization kinetics and controlled light on/off switching polymerizations together with the chain extension and block copolymerization experiments further proved the controlled nature of the polymerization system. The possibility of using commercially available dyes and working in the visible range of the electromagnetic spectrum, specifically with newly emerging light sources like LED, contribute to the efficiency and lower cost of controlled/living radical polymerization processes.References
- K. Matyjaszewski, Macromolecules, 2012, 45(10), 4015–4039 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101(9), 2921–2990 CrossRef CAS PubMed.
- Y. Gnanou and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2004, 42(2), 351–359 CrossRef CAS.
- K. Matyjaszewski, W. Jakubowski, K. Min, W. Tang, J. Y. Huang, W. A. Braunecker and N. V. Tsarevsky, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(42), 15309–15314 CrossRef CAS PubMed.
- D. Konkolewicz, K. Schroder, J. Buback, S. Bernhard and K. Matyjaszewski, ACS Macro Lett., 2012, 1(10), 1219–1223 CrossRef CAS.
- M. Ciftci, M. A. Tasdelen, W. W. Li, K. Matyjaszewski and Y. Yagci, Macromolecules, 2013, 46(24), 9537–9543 CrossRef CAS.
- M. Ciftci, M. A. Tasdelen and Y. Yagci, Polym. Chem., 2014, 5(2), 600–606 RSC.
- M. A. Tasdelen, M. Ciftci and Y. Yagci, Macromol. Chem. Phys., 2012, 213(13), 1391–1396 CrossRef CAS.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Chem. Phys., 2010, 211(21), 2271–2275 CrossRef CAS.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Rapid Commun., 2011, 32(1), 58–62 CrossRef CAS PubMed.
- F. Nzulu, S. Telitel, F. Stoffelbach, B. Graff, F. Morlet-Savary, J. Lalevee, L. Fensterbank, J. P. Goddard and C. Ollivier, Polym. Chem., 2015, 6(25), 4605–4611 RSC.
- J. T. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136(14), 5508–5519 CrossRef CAS PubMed.
- S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2015, 137(31), 9988–9999 CrossRef CAS PubMed.
- S. Shanmugam and C. Boyer, Science, 2016, 352(6289), 1053–1054 CrossRef CAS PubMed.
- S. Shanmugam, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2016, 55(3), 1036–1040 CrossRef CAS PubMed.
- M. A. Tasdelen and Y. Yagci, Angew. Chem., Int. Ed., 2013, 52(23), 5930–5938 CrossRef CAS PubMed.
- M.-A. Tehfe, J. Lalevée, S. Telitel, E. Contal, F. Dumur, D. Gigmes, D. Bertin, M. Nechab, B. Graff, F. Morlet-Savary and J.-P. Fouassier, Macromolecules, 2012, 45(11), 4454–4460 CrossRef CAS.
- M.-A. Tehfe, J. Lalevée, F. Morlet-Savary, B. Graff, N. Blanchard and J.-P. Fouassier, ACS Macro Lett., 2012, 1(1), 198–203 CrossRef CAS.
- T. Zhang, T. Chen, I. Amin and R. Jordan, Polym. Chem., 2014, 5(16), 4790–4796 RSC.
- T. Zhang, D. Gieseler and R. Jordan, Polym. Chem., 2016, 7(4), 775–779 RSC.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Chem. Phys., 2011, 212(18), 2036–2042 CrossRef CAS.
- S. Dadashi-Silab, M. A. Tasdelen, A. M. Asiri, S. B. Khan and Y. Yagci, Macromol. Rapid Commun., 2014, 35(4), 454–459 CrossRef CAS PubMed.
- S. Dadashi-Silab, M. A. Tasdelen, B. Kiskan, X. C. Wang, M. Antonietti and Y. Yagci, Macromol. Chem. Phys., 2014, 215(7), 675–681 CrossRef CAS.
- S. Dadashi-Silab, M. A. Tasdelen and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2014, 52(20), 2878–2888 CrossRef CAS.
- O. S. Taskin, G. Yilmaz, M. A. Tasdelen and Y. Yagci, Polym. Int., 2014, 63(5), 902–907 CrossRef CAS.
- M. A. Tasdelen, G. Yilmaz, B. Iskin and Y. Yagci, Macromolecules, 2012, 45(1), 56–61 CrossRef CAS.
- B. Sandmann, B. Happ, M. D. Hager, J. Vitz, E. Rettler, P. Burtscher, N. Moszner and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2014, 52(2), 239–247 CrossRef CAS.
- G. Yilmaz, B. Iskin and Y. Yagci, Macromol. Chem. Phys., 2014, 215(7), 662–668 CrossRef CAS.
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. R. de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136(45), 16096–16101 CrossRef CAS PubMed.
- X. Pan, C. Fang, M. Fantin, N. Malhotra, W. Y. So, L. A. Peteanu, A. A. Isse, A. Gennaro, P. Liu and K. Matyjaszewskit, J. Am. Chem. Soc., 2016, 138(7), 2411–2425 CrossRef CAS PubMed.
- X. C. Pan, N. Malhotra, A. Simakova, Z. Y. Wang, D. Konkolewicz and K. Matyjaszewski, J. Am. Chem. Soc., 2015, 137(49), 15430–15433 CrossRef CAS PubMed.
- G. M. Miyake and J. C. Theriot, Macromolecules, 2014, 47(23), 8255–8261 CrossRef CAS.
- X. Liu, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2016, 7(3), 689–700 RSC.
- S. Telitel, F. Dumur, D. Campolo, J. Poly, D. Gigmes, J. Pierre Fouassier and J. Lalevée, J. Polym. Sci., Part A: Polym. Chem., 2016, 54(5), 702–713 CrossRef CAS.
- E. Zipfel, J. R. Grezes, W. Seiffert and H. W. Zimmermann, Histochemistry, 1982, 75(4), 539–555 CAS.
- J.-P. Fouassier, in Photoinitiation, Photopolymerization, and Photocuring: Fundamentals and Applications, Hanser Gardner Publications, Munich, Germany, 1995 Search PubMed.
- J. V. Crivello, K. Dietliker, G. Bradley and S. T. Limited, in Chemistry & Technology of UV & EB Formulation for Coatings, Inks & Paints, John Wiley, Chichester, UK, 1998, vol. 3 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py01417h |
| This journal is © The Royal Society of Chemistry 2016 |