
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tunable dynamic hydrophobic attachment of guest molecules in amphiphilic core–shell polymers†
Jörg
Reichenwallner
a,
Anja
Thomas
b,
Lutz
Nuhn
bc,
Tobias
Johann
b,
Annette
Meister
ad,
Holger
Frey
b and
Dariush
Hinderberger
*a
aInstitute of Chemistry, Martin-Luther-Universität Halle-Wittenberg, Von-Danckelmann-Platz 4, 06120 Halle, Germany. E-mail: dariush.hinderberger@chemie.uni-halle.de
bInstitute of Organic Chemistry, Johannes Gutenberg-University, Duesbergweg 10-14, 55128 Mainz, Germany
cDepartment of Pharmaceutics, Ghent University, Ottergemsesteenweg 460, 9000 Ghent, Belgium
dInstitute of Biochemistry and Biotechnology, Martin-Luther-Universität Halle-Wittenberg, Kurt-Mothes-Straße 3, 06120 Halle, Germany
First published on 24th August 2016
Abstract
In this study, synthesis and dynamic properties of amphiphilic core–shell polymers are reported as monitored through their interaction with small amphiphilic molecules. Brush-like structures are formed with a hydrophobic core surrounded by a hydrophilic shell utilizing controlled radical addition–fragmentation chain transfer (RAFT) polymerization of macromonomers consisting of linear polyglycerol chains attached to alkylene methacrylate. Continuous wave electron paramagnetic resonance (CW EPR) spectroscopy is employed to study how the amphiphilic, paramagnetic spin probe 16-DSA (16-doxyl stearic acid) interacts with polymers of different alkylene chain lengths in their hydrophobic cores and different polyglycerol chain lengths in their hydrophilic shells. The spin probe exhibits dynamic hydrophobic attachment to the polymers and reveals an indirect, dynamics-based view of polymer effects such as temperature response, aggregation and ligand binding properties. Increasing the hydrophobic alkylene chain length in the polymers alters the physical properties of the core region significantly. A large set of controllable functional polymer properties can be adjusted by the degree of polymerization and alkylene spacer length. Partial aggregation of the polymers further modifies the binding properties. Applying dynamic light scattering (DLS), transmission electron microscopy (TEM) and molecular dynamic (MD) simulations, the complex dynamic behavior found with EPR spectroscopy was further complemented and verified.
1. Introduction
In recent years, synthetic pathways have been established to construct graft copolymers from amphiphilic macromonomers1–8 with high molecular weights of up to 470 kDa and narrow and monomodal weight distributions (PDI < 1.20).9 Therefore, these polymers are especially well suited for the design of specialized and reproducible functional properties in the realm of smart materials,10–13e.g., employing their capabilities to host small ligand molecules in their hydrophobic core.14–17One method to monitor ligand uptake is to chemically attach paramagnetic nitroxide moieties as spin labels to the ligands, which makes them amenable to electron paramagnetic resonance (EPR) spectroscopic investigations. A standard biological example for the case of fatty acid uptake is the major blood transport protein albumin, whose ligand uptake capabilities are mainly driven by thermodynamics, by electrostatic and hydrophobic (entropic) forces.18 So far, EPR spectroscopy has contributed substantial information about the mutual interactions of this biomacromolecule with spin-labeled fatty acids, specifically doxyl stearic acids (see Fig. 1),19–24 and has indirectly provided a profound basis for functional analysis that will be applied in this study. Furthermore, it has been shown that albumin fatty acid binding capacities can be modulated artificially by post-translational surface modifications, aiming for both, an increased25 or a decreased ligand uptake capacity.26
 | ||
| Fig. 1 Composition of all six core–shell polymers CnSm. (a) Generalized chemical structure of a core–shell polymer with N macromonomer repeating units consisting of m glycerol and n methylene components. (b) Propylene spacer polymers C3S32 (blue) and C3S16 (pale blue). (c) Hexylene spacer polymers C6S32 (orange) and C6S16 (pale blue). (d) Undecanoylene spacer polymers C11S14 (green) and C11S16 (pale blue). On the left hand scheme in (b), (c), and (d), the individual polymers are schematically color coded as they will appear in our subsequent analysis. A brush-like representation of the polymers is also given on the right hand side of (b)–(d) highlighting the hydrophobic core (red) and shell region (blue). The pale blue polymers are all equipped with a S16 shell. (e) Chemical structure of the 16-DSA spin probe. | ||
From a synthetic point of view, charged dendronized polymers have already been reported as fatty acid transporters whose uptake capabilities scale exponentially with the dendrimer generation.27 In principle, this shows that it is possible to tune ligand uptake capabilities by chemical modification of polymeric building blocks. Additionally, upon application of external stimuli such as pH,28 ionic liquids, salt,27 light16 or temperature the bound fatty acid ligands can also be released selectively, e.g. in Poloxamers29 (= Pluronics) or elastin-like polypeptides28 (ELPs).
Spectroscopically, we can study paramagnetic fatty acids by continuous wave (CW) EPR to determine their distribution in different environments based on polarity and their individual binding affinities as well as partition coefficients. The two major reference points are the free (f) and strongly bound and immobilized (b) spectral fractions that can easily be separated by their spectral shape and therefore facilitate extraction of valuable dynamic information (see Fig. 2). The basis for this spectral separation is the lineshape broadening upon decelerated diffusion.30–34 Freely tumbling fatty acid ligands exhibit typical isotropic three line EPR spectra, with well-defined solvent-specific line splittings (aiso) as it is commonly encountered for fast rotating nitroxide radicals with rotational correlation times τc of about 10–500 ps. Upon binding to a macromolecular substrate such as a polymer or a protein, the rotational motion is slowed down about three orders of magnitude to the ns-timescale, as it then reflects the slower rotational motion of the corresponding macromolecular structure (τc > 0.5 ns).34,35 Then, broad anisotropic EPR lineshapes are observed that are dominated by the shape, size and polarity of the macromolecular environment. As a nonpolar environment leads to a local shift of the electron spin density towards the oxygen atom in nitroxide radicals, the hyperfine interactions between electron and 14N nucleus and hence the aiso-value is reduced linearly with decreasing polarity.36,37 Thus, the spectral parameters, i.e., the individual dynamic fractions f and b, as well as τc and aiso, are the key to dissecting multicomponent EPR spectra. This forms the foundation of our approach of detecting structural and dynamic changes of the polymer itself, extending the established phenomenon of merely observing ligand binding towards polymers.14,16,17
 | ||
| Fig. 2 Collection of CW EPR spectra of different amphiphilic core–shell polymers as probed with 16-DSA at T = 25 °C. (a) Summary of EPR spectra Sk(B) from polymers with propylene (C3), hexylene (C6) and undecanoylene (C11) spacers. (b) Representative simulation of polymer C6S32,sim (orange) composed of an overlay of three exactly balanced (see orange numbers) fractions ϕi,k(B) of dynamic components Fi,k(B) in this case being b1, b2 and f1 (compare to a). The components a1 and g1 are also shown for completeness. All simulation traces are shown in grey. Measurements were conducted in H2O at concentrations of 1 wt% (C3) and 4 wt% (C6 and C11) and a 16-DSA concentration of about 200 μM. | ||
Furthermore, structural and dynamical aspects are complemented by dynamic light scattering (DLS), transmission electron microscopy (TEM) and molecular dynamics (MD) simulations, giving additional resolution of different dynamic processes on the nanoscopic scale. We will in the following show that our deliberate physico-chemical, EPR-based strategic approach can lead to an almost holistic view of how macromolecular structure and dynamic responses can be tuned.
2. Results and discussion
2.1 Polymer synthesis and screening by EPR spectroscopy
In this study, we characterize different amphiphilic core–shell polymers, in which parameters like the degree of polymerization (N) of the polymethacrylate backbone, but particularly the length of hydrophilic linear polyglycerol shells (linPPGm, in short denoted as Sm) and alkylene core spacer lengths (in short denoted as Cn) are varied. As a simplification all polymers will be denominated with a distinctive abbreviation CnSm according to their structural composition. The chemical rationale of all macromonomers is given in Fig. 1a with respect to its most important tunable entities (m, n), also highlighting the chemical structure of the polymethacrylate backbone comprising N subunits. In Fig. 1b–d a schematic representation of all core–shell polymers is given in a cylindrical and brush-like representation. All polymers were synthesized by radical addition–fragmentation chain transfer (RAFT) polymerization, a controlled polymerization technique, that provides well-defined (block co-)polymer architectures with narrow molecular weight distributions and precisely defined α- and ω-end-functionalities.38–40 To the best of our knowledge, the grafting through polymerization of polyglycerol-based macromonomers by RAFT leading to well-defined end-functional graft copolymers has not been described in the literature to date. We succeeded in synthesizing the amphiphilic polyglycerol-based macromonomers by clicking azido alkyl methacrylates of different spacer lengths (propylene = C3, hexylene = C6, undecanoylene = C11) to the propargyl-functional linPPGm in a copper-catalyzed azide–alkyne cycloaddition (CuAAC). While the synthesis of C3 and C6-based core–shell polymers has been reported before,9 the longer undecanoylene spacer has not been employed up to now. Using (4-cyanopentanoic acid)dithiobenzoate as charge transfer agent for the RAFT polymerization, well-defined core–shell polymers of narrow polydispersity are accessible that can even further be modified towards heterotelechelic graft copolymers with reductive cleavable or non-cleavable fluorescent end groups. Details on synthesis and characterization summarized in the Experimental section and in the ESI† demonstrate the versatility of RAFT polymerization conditions for amphiphilic polyglycerol-based macromonomers towards well-defined end-functional core–shell polymers.For analysis of the dynamic properties of the polymers, we chose the amphiphilic spin probe 16-DSA41,42 to monitor the microenvironment of the polymer solutions and potential binding to the polymers by means of EPR spectroscopy.28,43 The chemical structure of 16-DSA is given in Fig. 1e.
The advantage of using this spin probe is the rigid attachment of the nitroxide moiety to the alkyl chain, so that the change in motional freedom of this long chain (C18) is directly and accurately reflected in an EPR spectrum.41,44 In Fig. 2a, CW EPR spectra of 16-DSA co-dissolved in solutions of all available core–shell polymers are shown. Interestingly, the overall spectral shape evidently only changes significantly with alkylene spacer length Cn, clearly indicating an affinity-based variation in the ratio of free (f) to bound (b) ligand due to the hydrophobic core. In the following we present data for one representative polymer from each group of similar alkylene spacer lengths: C3S32, C6S32, and C11S14. This selection allows studying the effect of short alkylene spacers shielded by a thick hydrophilic shell (C3S32 and C6S32) and long alkylene spacers shielded by a much thinner hydrophilic layer (C11S14), so that we can observe the maximum number of individual effects of structural properties (spacer length, shell thickness and degree of polymerization) on the ligand binding and conformational changes while keeping the number of datasets at a manageable level.
The studied polymers are color coded as in Fig. 1b–d throughout the whole manuscript. All important polymer data such as molecular weight (MW), macromonomer weight (MWMM), polydispersity index of the macromonomer (PDIMM), and degree of polymerization (N) can be found in Table 1.
From an EPR spectroscopic point of view, we encounter experimental spectra that are composed of a set of five distinguishable sub-spectra Fi,k(B) resembling different spin probe dynamic regimes and environments as seen in Fig. 2b. We will later show that these different rotational and environmental regimes correspond to different topologies and mobilities in the three different core–shell polymer solutions. The index i denotes the type of sub-spectrum and k denotes the individual polymer sample. The according spectral components can be extracted from rigorous simulations of experimental EPR spectra. Upon addition of the 16-DSA spin probe to the polymer solution, we find 16-DSA as a freely tumbling species (f1), a species aggregated in micelles (a1, as the stearic acid spin probes have a critical micellar concentration of about 0.3 mM in phosphate buffered saline at pH 7.4),45 a slowly tumbling species (b1), an intermediately slow tumbling species (b2) and it also appears in a gel phase (g1) very similar to the hydrogels described in Junk et al.46 The respective spectral fractions of all species are given by ϕi,k. In principle, any occurring experimental spectrum Sk(B) can be reconstructed by the formal relation  where ∑ϕi,k = 1 (see Fig. 2b). Further explicit information about the simulations and reconstruction technicalities of experimental spectra can be found in the ESI.† All spectra from C6- and C11-polymer-solutions show distinct, broad spectral lines stemming from strong spin probe immobilization upon binding and overlapping of different dynamical regimes. While C6- and C11-type polymers show 16-DSA spectra exclusively composed of b1, b2, and f1, we show that C3S32 and C3S16 exhibit three line spectra resembling freely tumbling 16-DSA and C3S32 may comprise small fractions of f1, g1 and a1. The shape and relative fractions of these spectral components will be used for subsequent analysis.
where ∑ϕi,k = 1 (see Fig. 2b). Further explicit information about the simulations and reconstruction technicalities of experimental spectra can be found in the ESI.† All spectra from C6- and C11-polymer-solutions show distinct, broad spectral lines stemming from strong spin probe immobilization upon binding and overlapping of different dynamical regimes. While C6- and C11-type polymers show 16-DSA spectra exclusively composed of b1, b2, and f1, we show that C3S32 and C3S16 exhibit three line spectra resembling freely tumbling 16-DSA and C3S32 may comprise small fractions of f1, g1 and a1. The shape and relative fractions of these spectral components will be used for subsequent analysis.
2.2. Temperature dependence of ligand binding
The so-called hydrophobic effect is a driving force of ligand binding to macromolecules and is strongly dependent on temperature.47,48 Hence, we conducted temperature-dependent EPR measurements of the solutions to obtain the dynamic response of the polymers via their modulated interaction with the 16-DSA spin probe. As a reference, we investigated the temperature response of 16-DSA alone without polymer and found that neither micellization nor any phase transitions occur in the whole temperature range (see Fig. S1 and S2†). As the evaluation of a multicomponent EPR spectrum at a single temperature always suffers from a potential inseparability of the individual spectral components, measuring a temperature series may unravel all dynamic regimes, as the individual fractions have different temperature dependences and can thus be individually discerned and investigated in detail. In case of the polymers available, this strategy proved successful yet exceptionally complicated, as sub-spectra separation had to be conducted iteratively in a manual global fit. Ultimately, the results from spectral simulations of temperature-dependent measurements are displayed in Fig. 3a–d, where only the spectral fractions ϕi,k of the respective components are plotted as a function of temperature. Simulation parameters and all simulated spectra as well as the deconvolution routine can be found in the ESI in Table S1, Fig. S1 and S3–S6.† The individual 16-DSA concentrations were determined by (relative) spin counting and are shown as grey insets in Fig. 3. | ||
| Fig. 3 Temperature response from CnSm core–shell polymers in the temperature range from 5–95 °C loaded with 16-DSA depicted as fractions ϕi,j,k. (a) 1 wt% C3S32 spin probed with 105 μM 16-DSA. (b) 4 wt% C6S32 spin probed with 160 μM 16-DSA. (c) 4 wt% C11S14 spin probed with 204 μM 16-DSA and (d) a comparison of the slow (b1) and intermediately slow (b2) rotating components of C6S32- and C11S14-polymers. Micellar fractions a1 are shown in grey, gel fractions g1 in blue and free fractions f1 in black throughout. The responses of polymers C6S32 (orange) and C11S14 (green) can be subdivided in slow rotating (● = b1) and intermediately slow rotating (○ = b2) regimes. Error margins have been determined from spectral simulations. | ||
As can be seen in Fig. 3a, the bulk of the 16-DSA molecules in the C3S32 polymer solution resides in micelles or tumbles freely in solution. In the temperature range from 5–40 °C the CW EPR spectra reveal only free (f1) and micellar (a1) 16-DSA, with the sum of fractions ϕi,j,k adding up to 100%. The subscript j now extends the specificity of our spectral fractions from type of sub-spectrum (i) and polymer (k) and denotes the temperature T. The micelle fraction decreases above 40 °C giving rise to a simultaneous increase of free 16-DSA in solution, most probably by a temperature-based shift in equilibrium between micelles and free 16-DSA. Above 45 °C a third component emerges with a strongly hydrophobic/non-polar aiso-value of 40.24 MHz (14.36 G) and a rotational correlation time of τc = 0.4–0.7 ns. This is indicative of the formation of a hydrogel-like environment by the C3S32 polymer as it has been shown for thermoresponsive polymers49–51 and is therefore denoted as the sub-spectrum g1. A comparison with 16-DSA alone in DPBS52 buffer shows a clear deviation in the high-field region of the EPR spectra (Fig. S4†).
The steep increase of the gel fraction is nonlinear in the observable temperature range and can be expected to be time-dependent as well. Our EPR data show that the C3S32-core–shell polymer most probably forms small hydrogel-like entities above 40 °C by forming water depleted regions which are detected by the amphiphilic spin probes, while there is no spin probe uptake at temperatures close to room temperature. Loading studies with the smaller and more hydrophilic spin probe TEMPO did not indicate any immobilization or binding in the C3S32 polymer either (data not shown). The reasons for this uptake behavior are discussed in subsequent sections. One can further assume a complicated thermodynamic equilibrium in terms of competition for fatty acids between micelles and C3S32 polymer.53,54 Another interesting fact is that the emergence of large micelle fractions at such low fatty acid concentrations of 100–150 μM may indicate a polymer-induced decrease of the fatty acid solubility in aqueous solution (see Fig. S4†). In this context we have to mention that also the ionic strength of the DPBS buffer of the pure 16-DSA solution may alter the CMC towards higher values. Finally, our data show that for the C3S32 polymer effective binding and transport of amphiphilic ligands like our stearic acid derivatives can be excluded at least for individual polymers at ambient conditions.
The spin probes together with the C6S32-polymer in Fig. 3b display a completely different development of dynamic regime occupations. There are three overlapping spectral components Fi,j,k(B) in the whole temperature range (f1, b1, b2). The b1 and b2 fractions of 16-DSA strongly interact with the polymer and hence are significantly hampered in their rotational motion (shown in orange). The sum of bound fractions ∑ϕb,j,k (b1 and b2) constantly amounts to over 92% for all temperatures without any micellar aggregates (a1) or gels (g1) appearing. The respective aiso-value of 42.80 MHz (15.27 G) for both bound fractions bi of C6S32 indicates a much more hydrophobic environment than 44.26 MHz (15.79 G) for freely tumbling 16-DSA, giving rise to the assumption, that the spin probe is in fact in contact with the much better accessible hydrophobic and water-depleted core compared to the C3S32 polymer.
The fundamental difference in fractions b1 and b2 is given by the rotational correlation time τc. At room temperature b1 has a rotational correlation time of τc = 6.6 ns and b2 has an about three times lower value of τc = 2.0 ns. There is a minimum free 16-DSA fraction of 2.4% around 35 °C, exactly coinciding with the crossing of the curves of slow and intermediately slow tumbling fractions, both being occupied equally at this point. Additionally, from 5 °C onwards the free fraction f decreases from 4.2% to 2.4% at 35 °C, so the fatty acid uptake can be regarded to be initially enhanced by temperature and the temperature dependent ligand uptake attains a plateau-like behavior between 35 °C and 50 °C. For temperatures exceeding 50 °C the free fraction again increases to 7.8% at 95 °C, allowing for an optimum fatty acid uptake capability in the range from 35 °C to about 50 °C, which interestingly includes the range of mammalian body temperature. Fatty acid ligand uptake of the C6S32-polymer thus clearly occurs throughout the whole temperature range.
A slightly simpler dynamic temperature response can be found for the C11S14-polymer as shown in Fig. 3c, although also exhibiting an initial simultaneous occurrence of three spectral components Fi,j,k(B) as seen for the C6S32-polymer (f1, b1, b2). The C11S14-polymer features the longest alkylene spacers in the core and we observe an almost gradual decrease of free and slow bound subspectra f1 and b1, both almost completely vanishing above 45 °C and leaving a single spectral component b2 behind that can be tracked to temperatures as high as 95 °C. The corresponding aiso-value of 42.44 MHz (15.14 G) for both bound fractions of the C11S14-polymer is even more hydrophobic than for the C6S32-polymer, also strongly implying contact to the hydrophobic core region. Unlike in the C6S32-polymer, here the free fraction of 16-DSA is about one order of magnitude lower, decreasing from 0.45% at 5 °C to 0.17% at 45 °C, which is a clear indication of a higher affinity of the spin probe towards the hydrophobic undecanoylene-containing core as compared to the hexylene core. The C11S14-polymer is therefore considered to have the best 16-DSA accessibility of all three polymers as the thinner hydrophilic shell reduces spatial restriction regarding ligand entry to the hydrophobic core. The rotational correlation times of the two bound fractions b1 and b2 at room temperature are 6.6 ns and 2.6 ns, respectively. A direct comparison with the C6S32-polymer reveals that these values are almost identical at room temperature, so the fatty acid rotational dynamics seem not to be greatly affected by the molecular weight of the polymers C6S32 (470 kDa) and C11S14 (64.3 kDa). In Fig. 3d the occupation changes of both dynamic regimes b1 and b2 of C6S32- and C11S14 polymers are compared. This graph shows that the length of the Cn-spacer might operate as shifting the sigmoidally shaped curves (○ and ●) along the y-axis. We therefore suggest this interconversion of b1 to b2 with temperature to be the first tunable dynamic property of the core shell polymers, although, as it will be shown later in this manuscript, a polymer structural correlation cannot be made from EPR data alone. The ligand uptake curves, shown in Fig. 3 in black throughout indirectly detect the optimum functional state of the polymers. The temperature dependent motional dynamics of the spin probes and polymers are summarized in Fig. 4. The rotational correlation times τc of all immobilized fractions b1, b2 and g1 emerging from being in close contact with the polymers are depicted in Fig. 4a for the whole temperature range. In contrast to the C11S14-polymer which shows an almost linear growth of −log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) τc,j,k in the semilogarithmic diagram (logarithmic decrease of τc) in both bound fractions b1 and b2, the C6S32-polymer appears to have a kinked, more sigmoidal temperature response of its rotational dynamics as probed by 16-DSA. This is indicative for a two-state phase transition of the polymer leading to a slightly more restricted rotational motion of the spin probe. For both, C6S32 and C11S14 polymers, τc of the slow component b1 decreases from 11 ns to about 2 ns from 5–95 °C, and τc of the intermediately slow component b2 changes from about 3.6–5.2 ns at 5 °C to 0.40–0.47 ns at 95 °C, so the values are roughly similar and of course in the same rotational dynamic regime. As physically expected, the aiso-values for f1, b1 and b2 have also been found to be slightly temperature dependent and in addition are graphically summarized in Fig. S7.†
τc,j,k in the semilogarithmic diagram (logarithmic decrease of τc) in both bound fractions b1 and b2, the C6S32-polymer appears to have a kinked, more sigmoidal temperature response of its rotational dynamics as probed by 16-DSA. This is indicative for a two-state phase transition of the polymer leading to a slightly more restricted rotational motion of the spin probe. For both, C6S32 and C11S14 polymers, τc of the slow component b1 decreases from 11 ns to about 2 ns from 5–95 °C, and τc of the intermediately slow component b2 changes from about 3.6–5.2 ns at 5 °C to 0.40–0.47 ns at 95 °C, so the values are roughly similar and of course in the same rotational dynamic regime. As physically expected, the aiso-values for f1, b1 and b2 have also been found to be slightly temperature dependent and in addition are graphically summarized in Fig. S7.†
 | ||
| Fig. 4 Temperature-induced dynamics of spin probes and polymers. (a) Semilogarithmic plot of all emerging temperature-dependent immobilized fractions (g1, b1, b2) of 16-DSA in solutions of polymers C3S32, C6S32 and C11S14 as monitored by τc. The gel phase g1 of the C3S32-polymer is shown in blue, the immobilized fractions bi of the C6S32-polymer are shown in orange and C11S14-polymer fractions bi in green. Error margins for τc were estimated from simulations to range about 8%. (b) Temperature dependence of the main hydrodynamic radii RH,j,k as detected by DLS. Error bars were calculated according to the fluctuation of individual measurements at constant temperature. (c) Scattered light intensity (SLI) given as count rates (in kHz) from temperature dependent DLS measurements of all three polymers. (d) A dynamic model for the temperature dependent formation of aggregates due to SLI of (c) in the scheme of “dynamic hydrophobic aggregation” in all three polymers based on (a)–(c) and Fig. 3. The polymer concentrations for DLS measurements were 0.13 wt% for C3S32 (blue), 0.2 wt% for C6S32 (orange) and 0.13 wt% for C11S14 (green). | ||
Complementing the EPR-spectroscopic temperature series, we have performed temperature-dependent DLS measurements on the samples including 16-DSA. These measurements reveal the main hydrodynamic polymer size as a function of temperature. The actual size distributions from the regularization process55 appeared to be partly polydisperse depending on temperature and are given in Fig. S8.† While C11S14 seems to retain its main hydrodynamic size throughout the whole temperature range at about RH,j,11 = 4 nm, the shorter alkylene chain polymers C3S32 and C6S32 exhibit a slightly nonlinear size behavior.
The most prominent feature of this nonlinearity is a size decrease above 25 °C from 3.5 nm to 2.6 nm for C3S32 and 6.1 nm to 5.4 nm for C6S32 with the minimum appearing between 33 °C and 42 °C for both polymers. This decrease in size of ΔRH of 0.7–0.9 nm is assumed to be related to an intramolecular structural collapse most probably caused by the hydrophilic shell, as the short shell length polymer C11S14 does not show such a transition behavior. In turn, this complements our view from the EPR-derived motional restriction of the spin probe of the C6S32 polymer with the sigmoidal −logτc,j,6 curve above 25 °C in Fig. 4a. As it can be seen from Fig. 3a and 3b, the occurrence of this minimum in solution size in Fig. 4b is correlated with a newly initiated (C3) or increased (C6) accessibility of the hydrophobic core for both S32 polymers. For the hydrophilic shell the solvent quality of water is modulated (from being a good to being a poor solvent) in this temperature range, similar to what is found for ethylene-oxide based oligomers and polymers. This in fact generates a structural breathing that will be further on denoted as hydrophilic collapse. This hydrophilic collapse is apparently accompanied by a modified and improved ligand uptake that can be triggered by temperature. The spin probes in the C6S32 polymers experience a motional slowdown due to collapse of the formerly more water-swollen polyglycerol shell onto the hydrophobic core. As this stealth effect of the core is obviously based on the shell properties that can be modulated by synthesis, this is defined as the second dynamic tunable property.
Another clearly observable effect in the DLS measurements is the change of scattering intensity over temperature for each polymer solution (Fig. 4c). An increase in scattering intensity can generally considered to be correlated to the formation of larger particles in solution as it has been applied e.g. in Majhi et al.56 Thus, for C3S32 and C11S14 we observe dissolving low order aggregates or oligomers with increasing temperature and for C6S32 aggregates form with increasing temperature (see also Fig. S8†). When inspecting the individual polymer characteristics as summarized in Table 1, it appears very likely that the temperature induced formation and depletion of aggregates is connected with the degree of polymerization N (C6S32 by far has the highest N) and the aggregation probably obeys complicated thermodynamic phase transitions that are beyond the scope of this work. This behavior is considered to be the third tunable dynamic property of the core–shell polymers and will be termed as “dynamic hydrophobic aggregation”. A graphical sketch of these findings is given in Fig. 4d. All results will be later rationalized in a strategic overview in section 2.7.
Another observation that can be made and analyzed upon temperature increase is that when quantitatively analyzing the absolute number of spins in the EPR data shown in Fig. 3. Besides the described extraordinary temperature behavior of different sub-spectra, these polymers seem to exhibit a mild radical scavenger property, as the double integral of the first-derivative EPR spectra (number of spin-bearing 16-DSA molecules) decreases with temperature. The scavenging mechanism apparently gets stronger as the alkylene chain length Cn increases. Therefore, it can be tentatively linked to the increased residence time τr with higher ligand binding affinity of the longer alkylene chains (decreasing equilibrium dissociation constant KD ∼ 1/τr).57 In principle, the reduction of the nitroxide moiety is assumed to follow a disproportionation reaction58 for 16-DSA most probably similar to the thermal or acidic disproportionation reaction of the TEMPO spin probe.43,49,59,60 This circumstance proves the realization of studies comprising polymer heating as irreversible and reliable heating-cooling cycles are therefore impossible by EPR as, e.g., the C11S14 polymer depletes the spin-concentration by 80% in one heating procedure alone (2 hours). All results concerning the absolute number of spins during heating are summarized in Fig. S9,† but since we only analyze the data within the first, fast heating, for which the signal loss does not play a significant role, they are not investigated in more detail and are considered as a side effect.
2.3. Molecular dynamics (MD) simulations
Molecular dynamics simulations have the potential to add further insight to the solution behavior of the polymers. Although being purely phenomenological here a global analysis of the simulated structures gave a strong decrease of compactness of the polymers with increasing alkylene chain length. Therefore, we normalized the model radius of gyration RG,M to the model molecular weight MWM giving values from 0.0362 nm kDa−1 for C3S32, 0.0440 nm kDa−1 for C6S32 to 0.0580 nm kDa−1 for C11S14 coinciding with a slightly decreasing macromolecular density with alkylene spacer length on an empirical level. Generally, as the structure for the C3S32 polymer appears to be almost globular, the C11S14 structure is more sheet-like accompanied with a qualitatively decreasing diameter. The polyglycerol chains in the periphery of the core–shell polymers appear to be rather curled and irregular (Fig. S10†). From this viewpoint the polymer brushes can therefore only be seen as strictly globular (C3) or cylindrical in shape (C6 and C11) on short length scales only, of course depending on the degree of polymerization N and alkylene spacer length Cn. Further details from MD simulations are given in Table S2.†2.4. Ligand uptake capabilities
Going beyond the temperature dependence of ligand uptake of the core–shell polymers, we conducted a loading study to determine the ligand binding capacities and corresponding binding affinities of all polymers at room temperature. As only C6S32 and C11S14 polymers bind 16-DSA at ambient conditions, the C3S32 polymer is excluded from this study. To this end, we utilize the common strategy to increase the amount of ligand in about equidistant concentration steps while keeping the polymer concentration constant. The essential parameters (f and b) can be directly assessed from simulations of EPR spectra. If the ratio of bound to free ligand varies, we can construct Scatchard plots61–64 as a linear transformation of loading data, commonly conducted for proteins but also suitable for polymers.65 Particularly in case of EPR spectroscopy utilizing 16-DSA as a spin probe there are abundant loading studies of the blood transport protein albumin.19,21,23 Applying the Scatchard formula: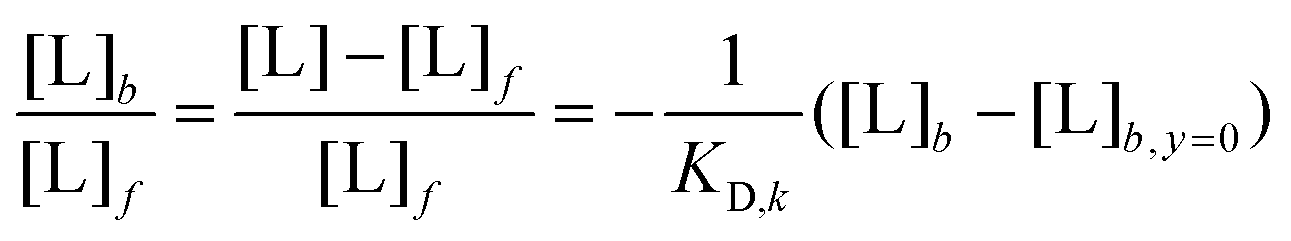 | (1) |
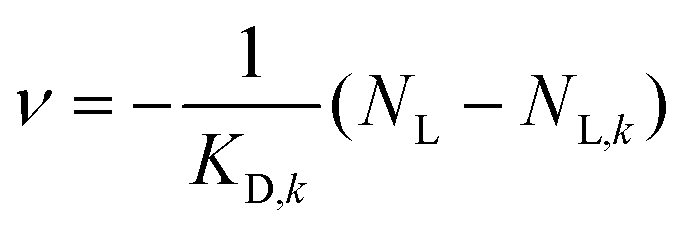 | (2) |
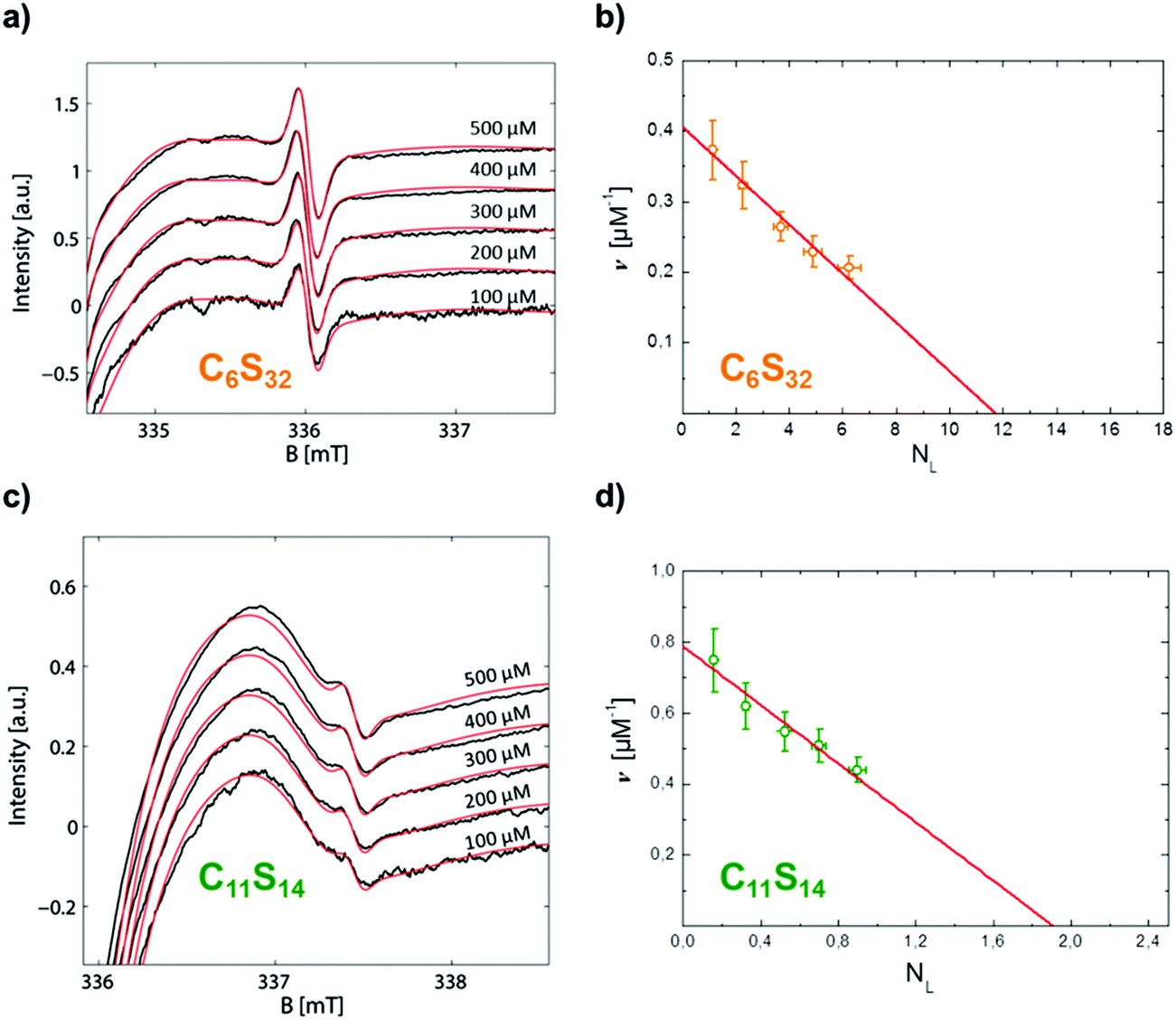 | ||
| Fig. 5 Scatchard plot construction of C6S32 and C11S14 loaded with 16-DSA in the nominal concentration range of 100–500 μM. The polymer concentration was 4 wt% each, i.e. 85 μM (C6S32) and 622 μM (C11S14). The most sensitive spectral parts of CW EPR spectra in the high-field range (335–338 mT) are highlighted. (a) All experimental CW EPR spectra (black) from 16-DSA loading of C6S32 with simulations (red). (b) Scatchard plot of C6S32 from (a) with linear fit according to eqn (2) (red). (c) All experimental CW EPR spectra (black) from 16-DSA loading of C11S14 with simulations (red). (d) Scatchard plot of C11S14 from (c) with linear fit (red). | ||
| k | C6S32 | C11S14 |
|---|---|---|
| K D,k [μM] | 28.82 ± 2.57 | 2.42 ± 0.35 |
| N L,MM,k | 0.0684 ± 0.0079 | 0.0445 ± 0.0080 |
| N L,k | 11.82 ± 1.37 | 1.96 ± 0.35 |
The linearity observed in both Scatchard plots is generally correlated with unspecific binding to equivalent binding sites.61 The change in the KD,k values upon increasing the hydrophobic alkylene chain length from C6 to C11 is accompanied by a decrease of more than one order of magnitude from KD,6 = 28.82 μM to KD,11 = 2.42 μM, so the ligand binding is much stronger for C11S14 due to the better accessibility of the hydrophobic core. The strong decrease of KD,k upon increased alkylene spacer length is the fourth dynamical tunable quantity. An astonishing finding is that regarding the maximum number of binding sites NL,MM,k per macromonomer the C6 polymer surpasses the C11 polymer slightly: a maximum number of macromonomer ligand binding sites of NL,MM,6 = 0.0684 is found for C6S32 compared to NL,MM,11 = 0.0445 for C11S14. The reason for this fact may be connected to the partial aggregation of the C11S14 polymer at room temperature and subsequent spatial restrictions to binding sites in the hydrophobic core, whereas C6S32 is mainly monomeric as obtained from our DLS data (Fig. 4c). The actual fatty acid capacity per polymer chain is clearly much higher for C6S32 with NL,6 = 11.82 than for C11S14 with NL,11 = 1.96 as derived from Fig. 5 (see also Table 2). The straightforward reason for this is the about 4-fold higher degree of polymerization of C6S32 (N = 172.8) compared to C11S14 (N = 44.0).
Thus, the creation of a single unspecific binding site in a core–shell polymer requires a minimum degree of polymerization of about 1/NL,MM,k = 14–22 macromonomers containing C6 or C11-spacers, relatively unaffected by spacer length. The fifth tunable functional parameter of the core–shell polymers is therefore the number of equivalent unspecific longitudinal ligand binding sites NL,k per polymer, controlled by the degree of polymerization N.
2.5. Transmission electron microscopy (TEM)
For a larger-scale, yet nanoscopic, view on the polymers we applied transmission electron microscopy (TEM). Although being of a size close to the resolution limit, all types of alkylene spacer-based core–shell polymers (C3S16, C6S32, C11S14) could be visualized after staining with uranyl acetate. Fig. 6 shows soft worm-like fiber (aggregates) with diameters between 1.5 and 2.5 nm for C3S16 (a), C6S32 (b), and C11S14 (c) assumed to be correspondent to the approximate hydrophobic core diameter. Due to the high polymer concentration and the flexibility of the fiber (aggregates), individual fibers are difficult to resolve in full length since they may partly lie on top of each other. | ||
| Fig. 6 TEM images of negatively stained core–shell polymers (a) C3S16 , (b) C6S32 and (c) C11S14. Yellow arrows point to fiber (aggregates) with diameters of a single polymer chain. The concentration for all polymers was set to 2 mg ml−1. Scale bars: 50 nm. | ||
Aggregated fibers with diameters of about 5 nm could be visualized by cryoEM for C6S32 (Fig. S12†). However, individual fibers are too small in size and too low in contrast to be visualized in detail by the cryoEM technique. Despite the low resolution of the TEM images, we can conclude from the shapes in the images that we encounter arbitrary, chain-like structures unlike the proposed unimolecular micelle picture for such polymers given in similar studies.16,66 We have to assume that for each core–shell polymer there is an ensemble of lower order aggregates, unimolecular micelles and chains at all temperatures with varying fractional occupations as also our DLS data exhibit partial polydisperse features especially for C3S32 and C11S14 at low temperatures and C6S32 at high temperatures (Fig. S8†).
2.6. Duality principle of fatty acid immobilization – proposal of the physical origin
The bimodal hydrophobic ligand immobilization we observed for C6S32 and C11S14 has also been detected for the physiological transporter molecule albumin67 and throughout literature it is considered as a ligand activation process in the substrate transporter molecule or a dynamic activation based on mutual interference and cooperation. The exact mechanism of fatty acid binding to a hydrophobic transport protein still remains somewhat enigmatic,18,68 but is thought to be of a stepwise nature69 as e.g. for cell entry.70–72 A vast amount of EPR spectroscopic studies have elucidated lipid, steroid and fatty acid dynamics in model membranes exhibiting lateral diffusion73–76 appearing among the principal physical phenomenon of jump diffusion first detected in pure water and lead.77–79 Several statistical mechanic considerations regarding rotational reorientation using the solution of the angular diffusion equation80–82 lead to practical mathematical relationships to discern between Brownian diffusion, free diffusion, and jump diffusion.34,81,83 We observed two bound spectral fractions Fi,j,k(B), namely b1 and b2, for polymers C6S32 and C11S14 (Fig. 3). The rotational correlation times τc of b1 and b2 differ by a factor of 2.41 ± 0.21 (C11) and 3.42 ± 0.50 (C6) in the whole observable temperature range. Jump diffusion can be excluded to appear in our polymer system, as we deal with spatially more separated fatty acid molecules appearing neither in crowded systems as micelles, nor in a membrane bilayer. The two remaining diffusion types, Brownian diffusion and free diffusion, are considered to be most suitable to fit into polymer bound fatty acid species, as the isotropic hyperfine coupling constant aiso was determined to be the same for b1 and b2. If 16-DSA, being attached to the polymer with the alkylene tail, underwent jump diffusion, displacements out of the polymer inside bulk water would give rise to a detectable polar spin probe environment as monitored by an increase of aiso. So, we assume a spontaneous displacement of the arbitrary molecular rotation angle ψ for free diffusion, while 16-DSA still remains attached to the polymer backbone. Especially, the reorientational model parameter B2 for slow motional EPR spectra introduced by Freed and coworkers81 separates Brownian diffusion from free diffusion by a factor of 7½ ≈ 2.65.34,84 This is surprisingly close to the observed differences in τc from subspectra b1 and b2. Moreover, the reorientational model parameter B2 is only explicitly determined for spherically symmetric rotation. On a very fundamental level of argumentation the intrinsic axial anisotropy in 16-DSA rotation might be responsible for small deviations from the theoretical value of 2.65 in polymers C6S32 and C11S14. A further hint indicating free diffusion is given in Fig. 4a and b, as the hydrophilic collapse of the polymer shell has the shape of a sigmoidal decrease in the –log τc curve, but only in subspectrum b2 of polymer C6S32. The contraction or collapse of the linear polyglycerol chains leads to a decreased local fluidity and therefore an increased local viscosity of the 16-DSA environment. This is only possible if 16-DSA in the rotational regime b2 intercalates at the interface of hydrophobic core and hydrophilic shell, which allows the spin probe dynamics to be influenced by the shell due to its close contact. In summary, the most reasonable physical basis of this dynamic hydrophobic interconversion process is a temperature induced switching of Brownian rotational diffusion towards free diffusion of bound 16-DSA indirectly reflecting the structural integrity and self-assembling features of the individual fatty acid-bearing polymer chains by the varying spectral fractions ϕb,j,k, while the temperature development of this process in C6S32 and C11S14 is almost identical (Fig. 3d, with a shift along y-axis). An elementary model for this dynamic hydrophobic interconversion of fatty acids is given in the conclusion.2.7. Tunable structure and dynamics of the core-shell polymers
We have revealed a wide variety of dynamic features based on the temperature dependence of the structure-based polymer–ligand and polymer–polymer interactions. In the combined picture of EPR and DLS we find that an increased ligand uptake takes place for the collapsed shell of the S32 polymers (Fig. 7a), C3S32 and C6S32, as binding gets stronger or is facilitated with increasing temperature, most probably due to a truncated <r2> diffusion distance from bulk water to the hydrophobic core. This can be seen as a decreasing stealth property of the hydrophilic shell with temperature. In case of high temperature aggregation (C6S32), the hydrophobic cores are additionally shielded and ligand binding is less probable, or ligands might even be expelled from the polymeric interior for sterical reasons. To highlight the polymer-based inherent dynamics of ligand binding, we characterize this complex behavior in general as “dynamic hydrophobic interconversion” (KAB) for the interconversion of rotational regimes A (b1) to B (b2) with the rate constants k1 and k−1 and the process of ligand binding as “dynamic hydrophobic binding” (KD) with the rate constants k2 and k−2 (Fig. 7b). Together with the process of “dynamic hydrophobic aggregation” as an intermolecular feature for all polymers (Fig. 4c), we summarize these processes in terms of “dynamic hydrophobic attachment”. A mathematical treatment of the resulting thermodynamics is beyond the scope of this work and is currently work in progress in our group. | ||
| Fig. 7 Physical basis for the dynamic hydrophobic attachment principle. (a) Sketch of the hydrophilic shell collapse as monitored for S32 polymers. (b) Depiction of dynamic hydrophobic binding and interconversion by means of the kinetic models for KD and KAB. (c) Polarity plot of the alkylene spacer length Cnversus hydrophobicity by means of aiso at 25 °C as probed by 16-DSA. | ||
The bigger such a hydrophobic core is designed relative to the hydrophilic shell, the smaller the dissociation constant KD and the stronger the ligand binding. In Fig. 7c we depict the aiso-values from our rigorous simulations of EPR spectra at 25 °C (see also Fig. S7†).
A clear decrease of aiso compared to the water-exposed spin probe 16-DSA (added as C0) is achieved upon binding to C6 and C11 polymers, and the lower aiso, the more nonpolar and water-depleted the hydrophobic core appears. The aiso-value of the 16-DSA spin probe freely tumbling in the C3 polymer solution is also depicted for completeness. Only at high temperatures the spin probe tends to evade exposition to the solvent by encapsulating into the small collapsed hydrophobic cores within a gel-like polymeric state, but only after the hydrophilic collapse has occurred and the hydrophobic core together with the collapsed shell forms a polymer-rich, water-separated microenvironment. Especially, the C6S32 polymer displays interesting functional aspects, which can be described in the framework of a temperature induced functional activation as a ligand uptake optimum is emerging between 35 and 50 °C therefore encompassing mammalian body temperature. Furthermore, we claim that a minimum hydrophobic spacer length is necessary to facilitate uptake of fatty acid molecules in the core–shell structure at room temperature, otherwise the core is invisible for the ligand, cloaked by the hydrophilic polyglycerol shell. This critical uptake spacer length is somewhere between the C3 and C6 alkylene subunits in the chains. The overall uptake capability per chain can simply be adjusted by the degree of polymerization (N).
We have not tested how functionality might also depend on concentration, for both ligand and polymer. Instead, for EPR studies we restricted ourselves to polymer and fatty acid ligand concentration ranges (several hundred μM) that are in the approximate range of the native concentration of the transport protein albumin in mammalian blood.18 This is an approach that is justified when considering such core–shell polymeric systems as protein-analogous transporters for drug delivery purposes. In similarity to often used polyethylene glycol (PEG) chains, the hydrophilic polyglycerol shell appears similar to water from a spectroscopic point of view, but reveals an indirect intricate effect upon chain and structural dynamics which might induce stealth properties to the hydrophobic cores of the polymers. This proposed stealth effect of the hydrophilic shell may even be too strong for the smaller hydrophobic cores of the C3S32 polymer so that no ligand uptake may occur regularly. This is also important for their potential use as drug transporting vehicles, as the stealth effect on the one hand is promising to enhance circulation times as a polysaccharide analogue9 while on the other hand the water-depleted core regions need to be intact and ligand migration from the core to the shell and the outside must be possible (and vice versa for, e.g., sensor-based applications). All this is excellently fulfilled in the modular core–shell type polymers presented in this study.
One of the hallmarks of the hydrophobic effect is the linear dependence of the increased surface area with transfer free energies when dissolving linear alkylene chains in solvents.85,86 In contrast, from our data it is apparent that the studied systems consisting of the polymerized macromonomers display a much more complicated cooperative interplay than single alkylene chains of varying length. For these albumin-analoguous polymers, the structural and dynamic parameters facilitating the imitation of protein-like behavior, as it has been shown in this study, are therefore much more difficult to assess.
From a physical methods point of view, an outstanding feature of using the nitroxide probing the dynamic molecular environment is the possibility to obtain an in depth functional view of a protein or polymer and the ligand itself without any other influences that might alter the results.64 A few other aspects could be clarified by complementary investigations regarding dynamic self-association and shape by TEM, DLS and molecular modeling. Especially TEM showed that we have rather elongated structures than spherical collapsed micellar structures (unimolecular micelles). The clear disadvantage of our EPR approach is the incapability of reversibility studies for increasing alkylene spacer length and the complexity of the spectral simulations, which should be partly overcome with the detailed guideline to rigorous spectroscopic analysis given in this paper and corresponding ESI.†
Although many of the functional principles may be intuitive, we here establish a CW EPR-based method for a reliable and complete quantitative dynamic molecular analysis of the core–shell polymers and their ligands and found remarkable implications and conclusions for several design principles. Our findings suggest a sum of tunable, functional parameters for the core–shell polymers:
(i) We have observed three structural tuning parameters namely alkylene spacer length (Cn), the length of the hydrophilic shell (Sm) and the degree of polymerization (N) of the macromonomers. These can largely be controlled by thorough and careful synthesis and subsequent polymer characterization (see also Experimental section and ESI†).7,9 This opens an elegant way to tailor-make polymers for delivery and uptake purposes by design and prior knowledge. Moreover, we found several dynamic properties that can be adjusted by chemical alteration of the core–shell structure.
(ii) The dynamic hydrophobic interconversion process of fatty acid species A (b1) and B (b2) can be designed by adjustment of Cn spacer length. This interconversion process (Fig. 3d) apparently introduces an optimum working temperature by a dynamic population crossing or transition of A and B for the polymer with optimum ligand uptake. It is influenced by intermolecular interactions like polymeric self-association as well as by intramolecular interactions as the accessibility of the hydrophobic core determined by the dynamics and size of the hydrophilic shell. This functional principle can be regarded as being the first dynamic tuning parameter. However, a thermodynamic model for this interconversion process has yet to be developed.
(iii) A long polyglycerol chain may lead to a hydrophilic collapse of the outer shell of the full polymer chain. This is the only dynamic effect that can be conditionally associated with the Sm chain but not directly to its length alone but rather towards the ratio of shell size (Sm) and core size (Cn). We assume a slight stealth effect of the hydrophilic shell to shield the hydrophobic core. This can be regarded as the second dynamic tuning parameter of the core–shell structures, and may also comprise a phase transition from a loose to a more compact structure of the polymer as also seen from DLS data. This hydrophilic collapse and stealth effect may further impose the onset of an aggregation process (C6S32) or the onset of ligand binding in a gel-like state (C3S32) to the system.
(iv) The intermolecular temperature-induced dynamic hydrophobic aggregation behavior of the polymers is found to be strongly connected to the degree of polymerization. Although implications are weak from EPR itself this is considered to be the third dynamic tuning parameter as confirmed by DLS data.
(v) The affinity of a ligand molecule to its binding substrate is determined by its macroscopic KD,k value as it can be extracted from e.g. Scatchard plots. By increasing alkylene spacer length (Cn) the KD,k value can be adjusted within orders of magnitude. The adjustment of the dynamic hydrophobic binding affinity towards a ligand molecule is the fourth dynamic tuning parameter of the core–shell polymers.
(vi) The degree of polymerization (N) is the decisive parameter for how many ligands can be bound to unspecific ligand binding sites per polymer (NL,k), as an increase of the alkylene spacer length (Cn) does not necessarily improve the number of bound ligands per macromonomer (NL,MM,k). This is also indicative for a longitudinal alignment of the bound ligands and is regarded as the fifth dynamic tuning parameter affected by the structure of the core–shell polymer. Partial aggregation behavior may furthermore alter the individual ligand binding capacity of a polymer. A single unspecific binding site is found to be constructed by polymerization of 14–22 macromonomers.
3. Conclusions
The overall characteristics of the mutual interactions of amphiphilic ligands and amphiphilic transporters is summarized as the concept of “dynamic hydrophobic attachment” as this term is to the best of our knowledge not used for any particular mechanism elsewhere. This term shall comprise an intermolecular polymeric “dynamic hydrophobic aggregation”, a ligand based “dynamic hydrophobic binding” and “dynamic hydrophobic interconversion” that have been shown to react sensitively on structural changes of the core–shell substrate. Astonishingly, as we have seen from C6S32 and C11S14 polymers the global rotational dynamics as monitored by τc do not seem to depend strongly on molecular weight of the polymer and therefore the degree of polymerization N, which simplifies comparative studies decisively. Nonetheless, the strong temperature dependence of both polymer dynamics and ligand affinity indicates suitable applications in minimally invasive surgery in terms of controlled ligand uptake and release in combination with e.g. LASER- or electro-hyperthermia treatment.87 This ligand uptake is not restricted to stearic acids only, but may be extended to any amphiphilic or hydrophobic medical ingredient that can be finally subject to a targeted drug release.64,88 In addition, the RAFT polymerization derived core–shell structures may further provide access to covalently modified telechelic structures with cleavable and non-cleavable release properties, as demonstrated in the ESI,† that enhance their applicability towards smart drug delivery purposes as well.Although being far from complete, this study gives a blueprint for the further design and the characterization of amphiphilic core–shell structures to systematically screen ligands and hydrophobic core and hydrophilic shell structures to obtain suitable synthetic drug transporter molecules working in the scheme of dynamic hydrophobic attachment.
4. Experimental section
4.1 Materials
All C3- and C6-based core–shell polymers were synthesized as described in Thomas et al.,9 while the C11-based core–shell polymers were prepared as described in the following. Solvents and reagents were purchased from Sigma-Aldrich or Acros Organics and used without further purification, unless stated otherwise. The spin-labeled fatty acid 16-DSA (Sigma-Aldrich, Taufkirchen) was used without further purification. 1-Chloro-3-hydroxypropane was purchased from abcr, Texas Red methanethiosulfonate (Texas Red-2-sulfonamidoethyl methanethiosulfonate) was purchased from Toronto Research Chemicals and Texas Red cadaverine was obtained from Invitrogen. 2,2′-Azobis(isobutyronitrile) (AIBN) was recrystallized from diethyl ether and stored at −7 °C. Deuterated solvents were purchased from Deutero GmbH. Dialysis tubings were purchased from CelluSep (MWCO = 1000 g mol−1 or 25000 g mol−1) or from Carl Roth GmbH + Co. KG (Spectra/Por 3 membranes, MWCO = 8000–10000 g mol−1). Thin layer chromatography (TLC) was performed on TLC aluminium sheets (20 × 20 cm, silica gel 60 F254) purchased from Merck and methanol was used as eluent. Anhydrous THF and dioxane were freshly distilled from a sodium/potassium mixture. Azido undecanoyl methacrylate (AzUMA)89 and 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl morpholinium chloride (DMTMM-Cl))90 were prepared modified to literature procedures. (4-Cyanopentanoic acid)dithiobenzoate91 and monopropargyl-functional linear polyglycerol (linPPGm)9 were prepared as described previously. Detailed experimental information and NMR spectra are given in the ESI† (see corresponding Experimental section).
4.2 Copper-catalyzed azide–alkyne cycloaddition of linPPGm and AzUMA
In a typical experiment, linPPGm (1 eq.) was dissolved in methanol (1 ml per 100 mg linPPG) and AzUMA (1.5 eq.) and PMDETA (0.3 eq.) were added. After degassing via three freeze–pump–thaw cycles, CuBr (0.3 eq.) was added under argon. The solution was stirred at room temperature overnight. The reaction was stopped by exposing the catalyst to air and an acidic ion exchange resin (DOWEX 50WX8) was added to remove copper. The colorless solution was neutralized by the addition of basic ion exchange resin (Amberlite IRA67), filtered, and BHT (2.5 mg per 100 mg polymer) was added as a stabilizer. Solvents were removed under reduced pressure at room temperature and the product was stored at −27 °C to prevent premature polymerization. The crude product was precipitated in diethyl ether prior to polymerization to remove residual low molecular weight AzUMA and stabilizer. Yields were between 80% and 100% and characterization data for click-coupled macromonomers can be found in Table S3.† Furthermore, NMR data and SEC traces are given in Fig. S13–S15.†
1H NMR (methanol-d4, 400 MHz): δ (ppm) = 8.03 (s, 1H, triazole), 6.11 and 5.64 (m, 2H, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 4.81 (m, 2H, OCH2CN), 4.43 (t, 2H, NCH2CH2, J = 7.1 Hz), 4.18 (t, 2H, CH2CH2OCO, J = 6.6 Hz), 3.82–3.50 (m, polyether backbone), 3.48 (t, 2H, CH3CH2CH2O, J = 6.7 Hz), 1.98–1.89 (m, 5H, CH3CCH2 and NCH2CH2), 1.75–1.59 (m, 4H, CH3CH2CH2O and CH2CH2OCO), 1.48–1.29 (m, 14H, NCH2CH2(CH2)7CH2CH2OCO), 0.97 (t, 3H, CH3CH2CH2O, J = 7.4 Hz).
C), 4.81 (m, 2H, OCH2CN), 4.43 (t, 2H, NCH2CH2, J = 7.1 Hz), 4.18 (t, 2H, CH2CH2OCO, J = 6.6 Hz), 3.82–3.50 (m, polyether backbone), 3.48 (t, 2H, CH3CH2CH2O, J = 6.7 Hz), 1.98–1.89 (m, 5H, CH3CCH2 and NCH2CH2), 1.75–1.59 (m, 4H, CH3CH2CH2O and CH2CH2OCO), 1.48–1.29 (m, 14H, NCH2CH2(CH2)7CH2CH2OCO), 0.97 (t, 3H, CH3CH2CH2O, J = 7.4 Hz).
13C NMR (methanol-d4, 100 MHz): δ (ppm) = 168.94 (CO), 146.38 (HCCN), 137.92 (CCH2), 125.94 (CCH2), 124.98 (HCCN), 81.56 (CH2CH(CH2OH)), 80.61 (CH2CH(CH2OH), terminal), 74.04 (CH3CH2CH2O), 71.92–70.59 (CH2 polyether backbone), 65.94 (CH2CH2OCO). 64.06 (OCH2CN), 62.63 (CH2OH), 51.38 (NCH2CH2), 31.32 (NCH2CH2), 30.53–30.08, 27.49 and 27.07 (NCH2CH2(CH2)7CH2CH2OCO), 29.69 (CH2CH2OCO), 23.88 (CH3CH2CH2O), 18.43 (CH3CCH2), 10.92 (CH3CH2CH2O).
4.3 RAFT-polymerization of click-coupled macromonomers
In a typical protocol, the macromonomer (30 eq.) was dissolved in water (1 ml per 200 mg macromonomer). (4-Cyanopentanoic acid)dithiobenzoate (CTA, 1 eq.) and AIBN (0.2 eq.) were dissolved in freshly distilled dioxane and added to the macromonomer solution to yield a water/dioxane ratio of 9:1. The solution was degassed via three freeze–pump–thaw cycles and the polymerization was performed for 48–72 h at 65 °C. The reaction was stopped by exposing the solution to air and the solvents were removed under reduced pressure. The residue was dissolved in methanol and precipitated in cold diethyl ether or dialyzed against water (MWCO = 25000).
Yields were at ∼50% and characterization data for graft copolymers P(linPGTzUMA) are given in Table S4 and Fig. S16–S20 in the ESI.† Moreover, further details on the post polymerization endgroup modification towards heterotelechelic graft copolymers can be found in Fig. S21–S24.† Additional information about synthesis of DMTMM-Cl is given in Scheme S3 as well as Fig. S25 and S26.†
C and OH), 4.29 (br, 2H, NCH2CH2), 3.93–3.12 (m, polyether backbone), 1.54–1.45 (m, CH3CH2CH2O), 0.85 (t, 3H, CH3CH2CH2O, J = 7.4 Hz), 1.87–0.75 (NCH2(CH2)9CH2OCO, and polymethacrylate backbone).
13C NMR (DMSO-d6, 100 MHz): δ (ppm) = 144.61 (HCCN), 123.49 (HCCN), 80.14–79.90 (CH2CH(CH2OH)), 79.05–78.84 (CH2CH(CH2OH), terminal), 71.97 (CH3CH2CH2OCH2), 70.63 and 70.15 (PrOCH2CH2O and PrOCH2CH2OCH2), 69.61–69.00 (CH2 polyether backbone), 62.76–62.60 (OCH2CN), 61.10–60.68 (CH2OH), 49.30 (NCH2CH2), 29.89 (NCH2CH2), 29.48–28.39, 26.23–25.45 (NCH2CH2(CH2)7CH2CH2OCO), 22.48 (CH3CH2CH2O), 10.56 (CH3CH2CH2O).
4.4 EPR Spectroscopy
4.5 Negative stain electron microscopy (EM)
The core–shell polymer samples were prepared in pure water to give concentrations of 0.2 wt%. Negatively stained samples were prepared by spreading 5 μL of the dispersion onto a Cu grid coated with a Formvar-film (PLANO, Wetzlar, Germany). After 1 min excess liquid was blotted off with filter paper and 5 μL of 1% aqueous uranyl acetate solution were placed onto the grid and drained off after 1 min. Specimens were air-dried and examined in an EM 900 transmission electron microscope (Carl Zeiss Microscopy GmbH, Oberkochen, Germany). Micrographs were taken with a SSCCD SM-1k-120 camera (TRS, Moorenweis, Germany).4.6 DLS measurements
All DLS data have been obtained by using an ALV-NIBS high performance particle sizer (HPPS) equipped with an ALV-5000/EPP Multiple Tau Digital Correlator (ALV-Laser Vertriebsgesellschaft m. b. H., Langen, Germany). This device facilitates HeNe-LASER irradiation with a typical wavelength of λ = 632.8 nm and 3 mW output power with an automatic attenuator for optimum count rates recorded in a backscattering setup detection angle of 173° relative to the incident monochromatic light. The sample cell temperatures were adjusted in the range of 8–85 °C in steps of 3 to 4 K by a Peltier temperature control unit. All samples have been prepared with a polymer concentration of 0.13–0.20 wt% (4.3–20.7 μM) and have been measured in 1.5 ml PMMA semi-micro cuvettes (BRAND, Wertheim). For sustaining comparability, a 1 mM 16-DSA stock solution in 12.5 mM KOH was used to equip the polymers with the same fatty acid loading as for the EPR measurements in this case well below the critical micelle concentration. The polymer concentrations were adjusted with ultrapure water (Milli-Q) to a final sample volume of 400–600 μl giving slightly alkaline solutions at about pH 8.1. All samples were filtered through Rotilabo cellulose acetate (CA) syringe filters with a pore size of 0.2 μM (CARL ROTH, Karlsruhe, Germany) to minimize adhesion. Data were extracted from the intensity correlation functions by a g2(t)-DLS exponential and a mass weighted regularized fit in the ALV-NIBS software v.3.0 utilizing the CONTIN algorithm.55 While the refractive index was assumed to be constant at nH2O = 1.332 for all temperatures, the water viscosity was corrected for each temperature. Each sample was measured at least 4 times at the same temperature for 60 s and was averaged at least over three individual values. The mean values RH,j,k of the most prominent particle size peaks and their statistical fluctuations are given as the standard deviation as depicted in the error bars in Fig. 4b. The duration of a heating procedure was 8–12 h. The temperature dependent size distributions are given in Fig. S8† as indicated in the main text.4.7 Molecular dynamics (MD) simulations
All molecular models of the coreshell-polymers were constructed using the YASARA-structure software.93 The MD simulations were run in simulation boxes with periodic cell boundaries containing the polymer and an explicit water solvent with 370000–403000 atoms in total. The simulation runtime was 12–13 ns at pH 7.0 in 0.0 M NaCl and T = 298 K applying the AMBER03 forcefield. Due to calculational effort, the model sizes were limited to a molecular weight of 100 kDa (N = 32–37) and are therefore only consulted for generalized phenomenological conclusions.
Acknowledgements
We gratefully acknowledge helpful discussions with Dr. Andreas Kerth and Dr. Christian Schwieger (both MLU Halle-Wittenberg) and Prof. Dr. Rudolf Zentel (JGU Mainz). We are also grateful to Dr. Gerd Hause (Microscopy Unit, Biocenter, MLU Halle-Wittenberg) for recording the cryoTEM image, Christine Rosenauer (MPI for Polymer Research, Mainz) for SEC-MALLS measurements, and to Heike Schimm and Stefanie Weber for continuing technical support. This work was financially supported by the Deutsche Forschungsgemein-schaft (DFG) under grant number HI 1094/5-1 and by the Max Planck Graduate Center with the Johannes Gutenberg-University Mainz (MPGC).Notes and references
- X. S. Wang, S. F. Lascelles, R. A. Jackson and S. P. Armes, Chem. Commun., 1999, 18, 1817 RSC.
- X. S. Wang and S. P. Armes, Macromolecules, 2000, 33, 6640 CrossRef CAS.
- M. Zhang, T. Breiner, H. Mori and A. H. E. Müller, Polymer, 2003, 44, 1449 CrossRef CAS.
- B. H. Tan, H. Hussain, Y. Liu, C. B. He and T. P. Davis, Langmuir, 2009, 26, 2361 CrossRef PubMed.
- Y. Y. Yuan, Q. Du, Y. C. Wang and J. Wang, Macromolecules, 2010, 43, 1739 CrossRef CAS.
- C. Porsch, S. Hansson, N. Nordgren and E. Malmström, Polym. Chem., 2011, 2, 1114 RSC.
- A. Thomas, F. K. Wolf and H. Frey, Macromol. Rapid Commun., 2011, 32, 1910 CrossRef CAS PubMed.
- Y. L. Luo, W. Yu, F. Xu and L. L. Zhang, Polym. Chem., 2012, 50, 2053 CrossRef CAS.
- A. Thomas, K. Niederer, F. Wurm and H. Frey, Polym. Chem., 2014, 5, 899 RSC.
- J. F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459 CrossRef CAS.
- J. F. Lutz, Adv. Mater., 2011, 23, 2237 CrossRef CAS.
- F. Herbst, D. Döhler, P. Michael and W. H. Binder, Macromol. Rapid Commun., 2013, 34, 203 CrossRef CAS PubMed.
- D. Döhler, P. Michael and W. H. Binder, in Self-Healing Polymers: From Principles to Applications, ed. W. H. Binder, Wiley-VCH, Weinheim, Germany, 2013, pp. 5–60 Search PubMed.
- J. Z. Du, L. Y. Tang, W. J. Song, Y. Shi and J. Wang, Biomacromolecules, 2009, 10, 2169 CrossRef CAS PubMed.
- O. G. Schramm, M. A. R. Meier, R. Hoogenboom, H. P. van Erp, J. F. Gohy and U. S. Schubert, Soft Matter, 2009, 5, 1662 RSC.
- J. A. Johnson, Y. Y. Lu, A. O. Burts, Y. H. Lim, M. G. Finn, J. T. Koberstein, N. J. Turro, D. A. Tirrell and R. H. Grubbs, J. Am. Chem. Soc., 2010, 133, 559 CrossRef PubMed.
- Y. Bakkour, V. Darcos, F. Coumes, S. Li and J. Coudane, Polymer, 2013, 54, 1746 CrossRef CAS.
- T. Peters Jr., All about Albumin: Biochemistry, Genetics, and Medical Applications, Academic Press, San Diego, 1995 Search PubMed.
- S. J. Rehfeld, D. J. Eatough and W. Z. Plachy, J. Lipid Res., 1978, 19, 841 CAS.
- R. C. Perkins Jr., N. Abumrad, K. Balasubramanian, L. R. Dalton, A. H. Beth, J. H. Park and C. R. Park, Biochemistry, 1982, 21, 4059 CrossRef CAS.
- T. G. Gantchev and M. B. Shopova, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1990, 1037, 422 CrossRef CAS.
- M. Ge, S. B. Rananavare and J. H. Freed, Biochim. Biophys. Acta, Gen. Subj., 1990, 1036, 228 CrossRef CAS.
- M. J. N. Junk, H. W. Spiess and D. Hinderberger, Angew. Chem., Int. Ed., 2010, 49, 8755 CrossRef CAS PubMed.
- J. Reichenwallner and D. Hinderberger, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 5382 CrossRef CAS PubMed.
- Y. Akdogan, Y. Wu, K. Eisele, M. Schaz, T. Weil and D. Hinderberger, Soft Matter, 2012, 8, 11106 RSC.
- S. L. Kuan, B. Stöckle, J. Reichenwallner, D. Y. Ng, Y. Wu, M. Doroshenko, K. Koynov, D. Hinderberger, K. Müllen and T. Weil, Biomacromolecules, 2013, 14, 367 CrossRef CAS PubMed.
- D. Kurzbach, D. R. Kattnig, B. Zhang, A. D. Schlüter and D. Hinderberger, Chem. Sci., 2012, 3, 2550 RSC.
- D. Kurzbach, W. Hassouneh, J. R. McDaniel, E. A. Jaumann, A. Chilkoti and D. Hinderberger, J. Am. Chem. Soc., 2013, 135, 11299 CrossRef CAS PubMed.
- D. Kurzbach, M. N. Reh and D. Hinderberger, ChemPhysChem, 2011, 12, 3566 CrossRef CAS PubMed.
- D. Kivelson, J. Chem. Phys., 1960, 33, 1094 CrossRef CAS.
- J. H. Freed and G. K. Fraenkel, J. Chem. Phys., 1963, 39, 326 CrossRef CAS.
- J. H. Freed, J. Chem. Phys., 1964, 41, 2077 CrossRef CAS.
- T. J. Stone, T. Buckman, P. L. Nordio and H. M. McConnell, Proc. Natl. Acad. Sci. U. S. A., 1965, 54, 1010 CrossRef CAS.
- J. H. Freed, in Spin Labeling: Theory and Applications, ed. L. J. Berliner, Academic Press, New York, 1976, vol. 1, pp. 53–132 Search PubMed.
- W. L. Hubbell and C. Altenbach, Curr. Opin. Struct. Biol., 1994, 4, 566 CrossRef CAS.
- O. H. Griffith, P. J. Dehlinger and S. P. Van, J. Membr. Biol., 1974, 15, 159 CrossRef CAS PubMed.
- H. J. Steinhoff, A. Savitzky, C. Wegener, M. Pfeiffer, M. Plato and K. Möbius, Biochim. Biophys. Acta, Bioenerg., 2000, 1457, 253 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379 CrossRef CAS.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079 CrossRef CAS.
- O. H. Griffith and A. S. Waggoner, Acc. Chem. Res., 1969, 2, 17 CrossRef CAS.
- D. A. Windrem and W. Z. Plachy, Biochim. Biophys. Acta, Biomembr., 1980, 600, 655 CrossRef CAS.
- D. Hinderberger, in EPR Spectroscopy: Applications in Chemistry and Biology, ed. M. Drescher and G. Jeschke, Springer, Berlin, Heidelberg, 2012, pp. 67–89 Search PubMed.
- A. S. Waggoner, T. J. Kingzett, S. Rottschaefer, O. H. Griffith and A. D. Keith, Chem. Phys. Lipids, 1969, 3, 245 CrossRef CAS PubMed.
- J. Reichenwallner, C. Schwieger and D. Hinderberger, unpublished work.
- M. J. N. Junk, W. Li, A. D. Schlüter, G. Wegner, H. W. Spiess, A. Zhang and D. Hinderberger, Angew. Chem., Int. Ed., 2010, 49, 5683 CrossRef CAS PubMed.
- N. C. Pace and C. Tanford, Biochemistry, 1968, 7, 198 CrossRef CAS PubMed.
- A. V. Finkelstein and O. B. Ptitsyn, Protein physics: a course of lectures, Academic Press, London, San Diego, 2002 Search PubMed.
- M. J. N. Junk, U. Jonas and D. Hinderberger, Small, 2008, 4, 1485 CrossRef CAS PubMed.
- M. J. N. Junk, W. Li, A. D. Schlüter, G. Wegner, H. W. Spiess, A. Zhang and D. Hinderberger, Macromol. Chem. Phys., 2011, 212, 1229 CrossRef CAS.
- D. Kurzbach, M. J. N. Junk and D. Hinderberger, Macromol. Rapid Commun., 2013, 34, 119 CrossRef CAS PubMed.
- R. Dulbecco and M. Vogt, J. Exp. Med., 1954, 99, 167 CrossRef CAS PubMed.
- C. Tanford, J. Mol. Biol., 1972, 67, 59 CrossRef CAS PubMed.
- W. J. Simmonds, Aust. J. Exp. Biol. Med. Sci., 1972, 50, 403 CrossRef CAS PubMed.
- S. W. Provencher, Comput. Phys. Commun., 1982, 27, 229 CrossRef.
- P. R. Majhi and A. Blume, J. Phys. Chem. B, 2002, 106, 10753 CrossRef CAS.
- P. J. Tummino and R. A. Copeland, Biochemistry, 2008, 47, 5481 CrossRef CAS PubMed.
- J. F. W. Keana, in Spin labeling: Theory and applications, ed. L. J. Berliner, Academic Press, New, York, 1979, vol. 2, pp. 115–172 Search PubMed.
- Y. Ma, C. Loyns, P. Price and V. Chechik, Org. Biomol. Chem., 2011, 9, 5573 CAS.
- E. A. Haidasz, D. Meng, R. Amorati, A. Baschieri, K. U. Ingold, L. Valgimigli and L. A. Pratt, J. Am. Chem. Soc., 2016, 138, 5290 CrossRef CAS PubMed.
- R. A. Copeland, Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, Wiley-VCH, New York, 2000 Search PubMed.
- G. Scatchard, Ann. N. Y. Acad. Sci., 1949, 51, 660 CrossRef CAS.
- C. Tanford, Physical Chemistry of Macromolecules, John Wiley & Sons, New York, 1961 Search PubMed.
- T. Hauenschild, J. Reichenwallner, V. Enkelmann and D. Hinderberger, Chem. – Eur. J., 2016, 22, 12825–12838 CrossRef CAS PubMed.
- J. Matsui, Y. Miyoshi, O. Doblhoff-Dier and T. Takeuchi, Anal. Chem., 1995, 67, 4404 CrossRef CAS.
- H. Hussain, K. Y. Mya and C. He, Langmuir, 2008, 24, 13279 CrossRef CAS PubMed.
- A. Gurachevsky, E. Shimanovitch, T. Gurachevskaya and V. Muravsky, Biochem. Biophys. Res. Commun., 2007, 360, 852 CrossRef CAS PubMed.
- J. A. Hamilton, Prog. Lipid Res., 2004, 43, 177 CrossRef CAS PubMed.
- W. Scheider, J. Phys. Chem., 1980, 84, 925 CrossRef CAS.
- F. Kamp and J. A. Hamilton, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2006, 75, 149 CrossRef CAS PubMed.
- J. A. Hamilton, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2007, 77, 355 CrossRef CAS PubMed.
- E. Barta, J. Membr. Biol., 2015, 248, 783 CrossRef CAS PubMed.
- E. Sackmann and H. Träuble, J. Am. Chem. Soc., 1972, 94, 4482 CrossRef CAS PubMed.
- E. Sackmann and H. Träuble, J. Am. Chem. Soc., 1972, 94, 4492 CrossRef CAS PubMed.
- C. J. Scandella, P. Devaux and H. M. McConnell, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 2056 CrossRef CAS.
- H. Träuble and E. Sackmann, Nature, 1973, 245, 210 CrossRef.
- B. N. Brockhouse, Il Nuovo Cimento, 1958, 9, 45 CrossRef CAS.
- B. N. Brockhouse and N. K. Pope, Phys. Rev. Lett., 1959, 3, 259 CrossRef CAS.
- C. T. Chudley and R. J. Elliott, Proc. Phys. Soc., 1961, 77, 353 CrossRef.
- P. A. Egelstaff, J. Chem. Phys., 1970, 53, 2590 CrossRef CAS.
- S. A. Goldman, G. V. Bruno, C. F. Polnaszek and J. H. Freed, J. Chem. Phys., 1972, 56, 716 CrossRef CAS.
- K. A. Valiev and E. N. Ivanov, Phys. Usp., 1973, 16, 1 CrossRef.
- G. V. Bruno and J. H. Freed, J. Phys. Chem., 1974, 78, 935 CrossRef CAS.
- K. A. Earle, D. E. Budil and J. H. Freed, J. Phys. Chem., 1993, 97, 13289 CrossRef CAS.
- K. A. Sharp, A. Nicholls, R. F. Fine and B. Honig, Science, 1991, 252, 106 CAS.
- I. Tunon, E. Silla and J. L. Pascual-Ahuir, Protein Eng., 1992, 5, 715 CrossRef CAS PubMed.
- H. Sahinbas, D. H. W. Grönemeyer, E. Böcher and A. Szasz, J. Ger. Oncol., 2007, 39, 1 Search PubMed.
- R. Haag, Angew. Chem., Int. Ed., 2004, 43, 278 CrossRef CAS PubMed.
- Q. Shen, J. Zhang, S. Zhang, Y. Hao, W. Zhang, W. Zhang, G. Chen, Z. Zhang and X. J. Zhu, Polym. Sci., Part A: Polym. Chem., 2012, 50, 1120 CrossRef CAS.
- M. Kunishima, C. Kawachi, F. Iwasaki, K. Terao and S. Tani, Tetrahedron Lett., 1999, 40, 5327 CrossRef CAS.
- L. Nuhn, M. Hirsch, B. Krieg, K. Koynov, K. Fischer, M. Schmidt, M. Helm and R. Zentel, ACS Nano, 2012, 6, 2198 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42 CrossRef CAS PubMed.
- E. Krieger, T. Darden, S. B. Nabuurs, A. Finkelstein and G. Vriend, Proteins: Struct., Funct., Bioinf., 2004, 57, 678 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on synthesis and physical characterization, as explained in the text. See DOI: 10.1039/c6py01335j |
| This journal is © The Royal Society of Chemistry 2016 |