
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hierarchically porous monolithic silica with varying porosity using bis(trimethoxysilyl)arenes as precursors†
Martin
von der Lehr
,
Rüdiger
Ellinghaus
and
Bernd M.
Smarsly
*
Institute of Physical Chemistry, Justus Liebig University Giessen, Heinrich-Buff-Ring 17, D-35392 Giessen, Germany. E-mail: bernd.smarsly@phys.chemie.uni-giessen.de; Tel: +49 641 9934590
First published on 18th April 2016
Abstract
The ortho-, meta- and para-isomers of bis(trimethoxysilyl)benzene and bis((trimethoxysilyl)phenyl)dimethoxysilane were synthesized from their corresponding diarylbromides. Mixtures of these bis(trimethoxysilyl)arenes and tetramethoxysilane (TMOS) were used as organosilica precursors for the preparation of macro-mesoporous silica monoliths using different amounts of the porogen poly(ethylene glycol) to investigate the effect on the porosity of the final materials. The prepared samples were characterized by nitrogen and dibromomethane sorption measurements, mercury intrusion porosimetry and scanning electron microscopy. The analysis of dibromomethane isotherms indicated a significantly lower polarity of the mesopore surface compared to that of pure SiO2 monoliths, proving the presence of arene units at the mesopore surfaces.
Introduction
Inorganic–organic hybrids have received considerable attention during the last decades as they combine the specific demands of each species in a single material. They are of peculiar interest due to the possibility of adjusting their final physical and chemical characteristics such as porosity, hydrophobicity and functionality depending on the desired application. Such materials can be prepared by sol–gel chemistry routes, known for mild and versatile reaction conditions. In addition, this pathway offers the benefit of straightforward control of the composition of the matrix. For obtaining a hybrid material, in the simplest case a single methyl group is incorporated by using a methyl-substituted alkoxysilane or attaching it to the support.1 Incorporating bifunctionalized/disubstituted organic moieties as an integral component of the inorganic support is an alternative approach. The utilization of molecular building blocks that contain a variable organic fragment attached to two or more trialkoxysilane groups offers a higher degree of linkage per monomer unit and leads to bridged polysilsesquioxanes. In these highly cross-linked three-dimensional networks the organic moiety is covalently attached to trialkoxysilane groups by non-hydrolyzable silicon carbon bonds and can be varied in functionality, length and torsional rigidity by its substitution geometry.2 In contrast to a post-synthetic modification of the silica surface (i.e. by classical grafting3), the utilization of an organically modified precursor in the starting sol–gel mixture offers the advantage of a convenient one-pot strategy.The integration of such compounds into the siliceous matrices of periodic mesoporous organosilicas (PMOs) was intensively described and reviewed by Froeba et al.4 Moreover, PMOs made from silsesquioxanes with bridging phenyl groups were synthesized by Kuschel and Polarz and studied with respect to their catalytic applications.5 Nakanishi et al. reported the integration of alkyltrialkoxysilanes and alkylene-bridged alkoxysilanes as organosilica precursors for the synthesis of silica monoliths.6 Huesing and co-workers utilized different ethylene-glycol-modified derivatives of organoalkoxysilanes such as methyl-, phenyl-, ethylene-, and phenylene-bridged silanes in the presence of a surfactant to achieve monolithic hybrid materials with hierarchically ordered porosity.7 Inorganic–organic hybrid materials assembled by sol–gel polymerization of polyfunctional molecular building blocks were also described by Corriu8,9 and studied in detail by Shea and Loy, i.e. by incorporating bridged hydrocarbons of different lengths as well as arylenes, such as 1,4-phenylene, 1,3,5-phenylene, 4,4′-biphenylene and 4,4′-terphenylene bridging groups.2,10 The polycondensation of the disubstituted silanetriols, 4,4′-bis(trihydroxysilyl)stilbene and 4,4′-bis(trihydroxysilyl)diphenylbuta-1,3-diyne was described by Cerveau and co-workers.11
We recently reported the synthesis of meso-macroporous monolithic silica based on the hydrolysis and condensation of 1,4-bis(trimethoxysilyl)arenes in a modified sol–gel route.12 This previous study showed the proof-of-principle concept of incorporating 1,4-bis(trimethoxysilyl)benzene (para-BTMB) and 1,4-bis((trimethoxysilyl)phenyl)dimethoxysilane (para-BTPMS). Based on the previous study, we demonstrate herein how systematic variations of the porogen used in the synthesis affects the porosity of the final materials, starting from the ortho-, meta- and para-isomers of bis(trimethoxysilyl)arenes. The choice of an arylene bridging group is based on the thermal stability upon annealing being required with respect to template removal. Even if the use of triethoxysilyl analogues is widely more common due to their lower toxicity, the trimethoxy variety was chosen in this study, as it offers a similar hydrolysis behavior to that of TMOS, required for co-condensation.
One major goal and challenge of the present study was the synthesis of the respective precursors, especially the gram-scale synthesis and purification of the ortho-, meta- and para-isomers of BTMB and BTPMS. Since elevated temperatures can result in polymerization reactions, suitable vacuum distillation conditions had to be applied in order to obtain the pure compounds.
A further main objective of this study lies with the assessment of the influence of the precursor on the average pore size, the pore size distribution and pore volume (both meso- and macroporosity) in the monolith materials obtained by the procedure developed by K. Nakanishi.13,14 Such insights are of significant relevance with respect to the crucial role of the combined meso- and macroporosity of this type of monolithic silica, which endows the material with both a high surface area (mesoporosity) and a high pore volume (macroporosity). Evidently, the prepared hybrid materials may have potential applications as chromatographic supports in separation sciences (monolithic HPLC columns), as they provide, compared to pure SiO2 materials, high specific surface areas and a high permeability in combination with a hydrophobic surface resulting from the chemical functionality.15 Moreover, they offer the possibility for further postsynthetic functionalization, i.e. by sulfonating the aromatic moieties which could be useful in acidic catalysis or in solid state proton conductivity applications.16
Since the modification of the interaction between the arylene moieties and species in the liquid and/or gaseous phase lies at the heart of such effects, we performed in-depth physisorption analysis using dibromomethane (DBM), performed at T = 290 K, i.e. at room temperature. Physisorption measurements utilizing DBM provide relevant information on such hybrid materials. First, in contrast to N2 or Ar, DBM is a polar fluid and can be expected to show a different behavior at small p/p0, i.e. at conditions for layer formation, if pure SiO2 materials and the hybrid materials under study are compared. Second, the desorption of DBM fluid should be different for pure SiO2 materials and the hybrid materials, as the contact angle of liquid DBM on the mesopore surface should depend on the hydrophilic/hydrophobic character of the mesopore surface. Hence, we performed an extensive study of DBM sorption in the prepared hybrid materials, in comparison to corresponding monoliths of pure SiO2 as reference.
Experimental details
Chemicals
1,2- and 1,3-dibromobenzene were purchased from abcr chemicals (Karlsruhe, Germany), 1,4-dibromobenzene, tetrachlorosilane, muriatic and acetic acid were purchased from Sigma-Aldrich (Steinheim, Germany), and tetramethyl orthosilicate (TMOS), urea and poly(ethylene glycol) (PEG, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) were obtained from Merck (Darmstadt, Germany). All reagents were used as received without further purification.
000 g mol−1) were obtained from Merck (Darmstadt, Germany). All reagents were used as received without further purification.
Synthesis procedure
The general synthetic procedure is outlined in Scheme 1. According to a literature protocol, chlorotrimethoxysilane was synthesized by the dropwise addition of 88 mL of anhydrous methanol to 80 mL of tetrachlorosilane while a flow of argon was applied and the mixture was stirred for 30 minutes. At a boiling point of 112 °C the crude product was then obtained by distillation.17 To 44 g of chlorotrimethoxysilane and 16 g of magnesium turnings submerged in 300 mL of anhydrous THF, a solution of 32 g of the corresponding dibromobenzene in 400 mL of THF was added with a dropping funnel. Cooling with an ice bath was applied when the exothermic reaction became too intense. The mixture was stirred for 15 hours at room temperature after which the reactants were filtered and most of the solvent removed. Extraction of the residue using dry pentane was carried out under inert conditions using the Schlenk technique. Products were obtained after removal of the solvent and fractional distillation in vacuum. While BTMB was obtained at 121 °C (0.1 mbar) as a clear liquid, the byproduct BTPMS was distilled at 198 °C (0.4 mbar) and was collected as a yellow liquid. In the case of para-BTMP the liquid solidified below 52 °C. Yields were 30% for the compound BTMB and 18% for BTPMS.8 The main reason for such poor yields is the relatively high temperature at which distillation is carried out, forcing an easy polymerization of the silica species and thus adequate vacuum conditions have to be ensured. The purity of the compounds was shown by NMR (see the ESI†).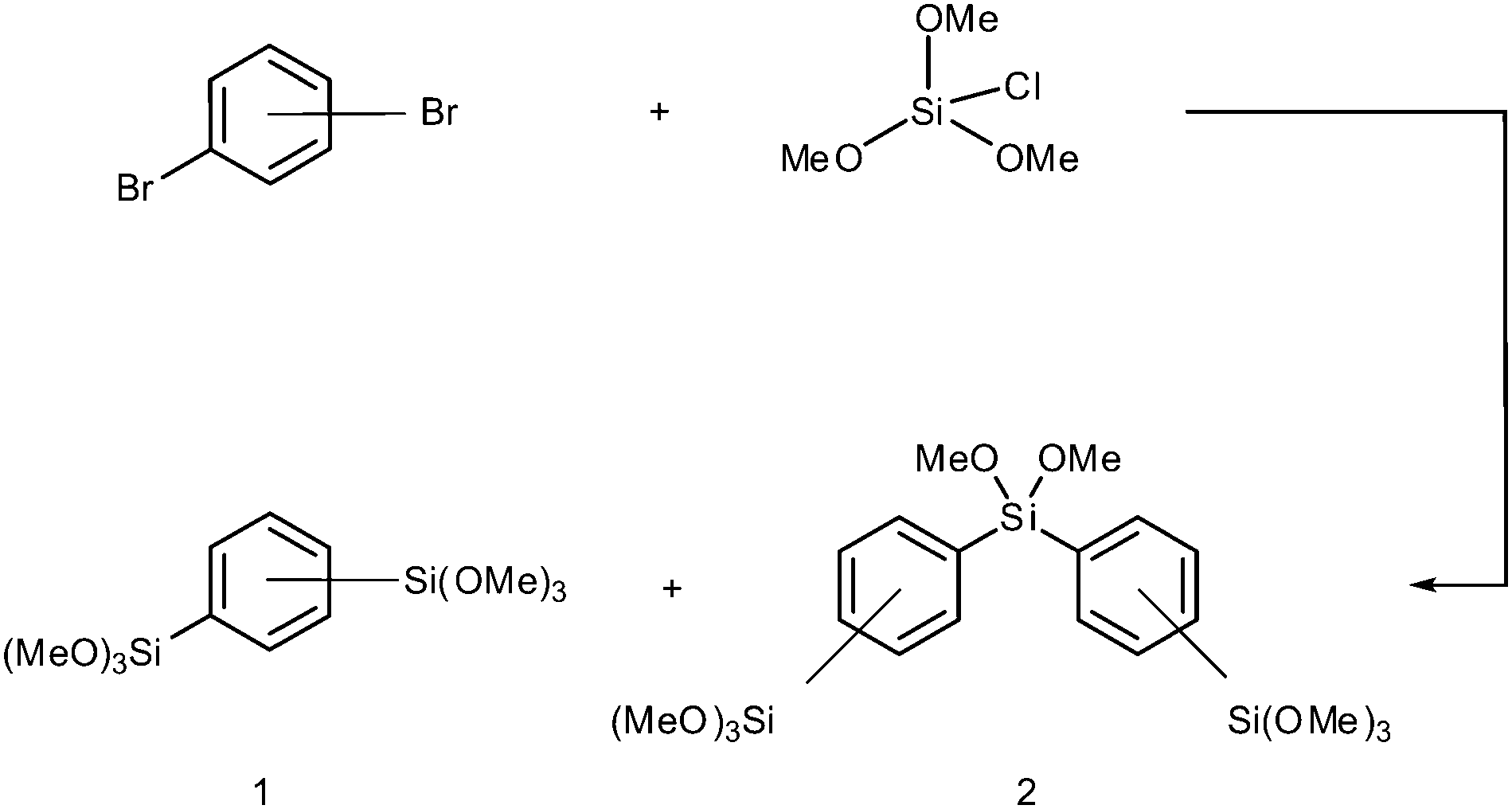 | ||
| Scheme 1 ortho-, meta- and para-substituted bis(trimethoxysilyl)arenes by silylation of dibromobenzene with chlorotrimethoxysilane. (1) Bis(trimethoxysilyl)benzene (BTMB), (2) bis((trimethoxysilyl)phenyl)dimethoxysilane (BTPMS). | ||
Preparation of monolithic hybrid silica rods
The preparation conditions of the monolithic silica rods were similar to those reported elsewhere in the literature.18 Typical conditions are as follows. A mixture (2.5 mL) of TMOS and the corresponding bis(trimethoxysilyl)arene in weight ratios of 95/5 and 90/10, respectively, was added to a solution of acetic acid (0.01 M, 5 mL), urea (0.45 g) and PEG. The amount of PEG was varied in 0.02 g steps in the range of 0.355 g to 0.475 g. As a solid compound, 1,4-bis(trimethoxysilyl)benzene was firstly liquefied at 70 °C and, after the addition of TMOS, stirred at this temperature for 1 hour. After mixing, the solutions were stirred at room temperature for 25 minutes, filtered with a 0.45 μm PTFE filter and charged into a 10 mL plastic vial which was brought into a water bath (40 °C) for approximately 16 hours. The resultant gel was further hydrothermally treated by raising the temperature slowly over a period of 12 hours up to 95 °C and then kept for 15 hours to form mesopores by ammonia generated through the hydrolysis of urea. The obtained alcogels were then washed with methanol for 3 days, replacing the solvent daily. A final calcination was carried out by raising the temperature over 15 hours up to 330 °C, then keeping at this temperature for 10 hours and cooling down afterwards over 15 hours.Measurements/characterization
Physisorption measurements were performed in an automated gas sorption station (AS-6, Quantachrome Corporation, Boynton Beach, USA) utilized for standard characterization measurements of nanostructured matter by nitrogen sorption isotherms. The instrument software supported the standard data reduction algorithms, such as Brunauer–Emmett–Teller (BET), as well as NLDFT kernels for typical pore geometries. The monolithic silica rods were placed in standard glass tubes and stabilized at the measurement temperature with T = 77 K maintained by liquid nitrogen in standard cryostats. Isotherms were measured up to 0.95 of the equilibrium nitrogen pressures p0. Samples were evacuated at ϑ = 120 °C for 12 h before the measurements. For measuring dibromomethane (DBM) sorption, samples were evacuated for 60 h and stabilized at T = 290 K (liquid density 2.477 g cm−3). In this case, a Quadrasorb Evo instrument was used. Mercury intrusion measurements were carried out using Pascal 140/440 porosimeters (Thermo Fisher Scientific, Rodano, Italy) in a pressure range from 0 MPa to 400 MPa. Results were calculated applying the instrument software Sol.I.D. using Washburn's equation with a mercury surface tension of 0.48 N m−1 and a contact angle of intruded mercury of θ = 140°. The morphology of monolithic silica rods was examined with a high resolution scanning electron microscope (HSEM Merlin/Software Smart SEM V05.03, Zeiss, Oberkochen, Germany).Results and discussion
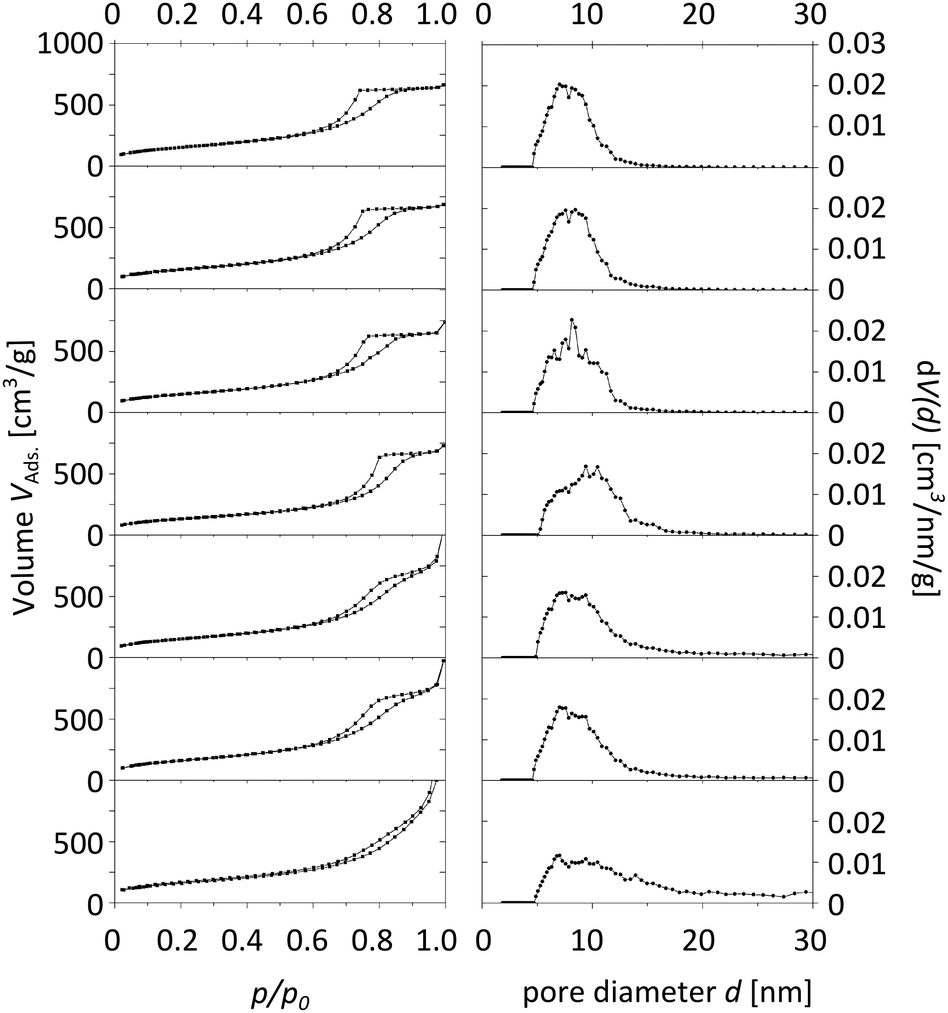
The characterization of the obtained meso-macroporous materials is exemplified for para-BTMB and para-BTPMS test series containing 5% of the organosilica precursor, shown in the following figures. N2 physisorption measurements revealed that an increased PEG amount results in an only moderate change in the shape of the isotherm (Fig. 1 and 2). The mean diameter of the mesopores and the mesopore volume were similar to the values typically observed for such SiO2-based monoliths. Within the given data series, the average mesopore diameters stayed almost constant. Only at significantly higher PEG amounts in the starting solution was a substantial broadening of the mesopore size distributions observed, i.e. the formation of a well-defined porosity was aggravated. Statistical deviations constitute inconsistent pressure tightness of the used plastic vials during the hydrothermal treatment of the wet gels. A comparison of the different isomers' porosity data of both compounds can be seen in Table 1 and Fig. 3. The smallest average mesopore diameters of 7.7 nm are generated using the para-isomers, while the meta-isomers lead to mesopore sizes in the range of 9.5–9.7 nm. The biggest mesopore size of 10.4 nm was achieved using the ortho-isomer. Since the mesoporosity is generated during the hydrothermal treatment by etching SiO2 with ammonia originating from the decomposition of urea, we attribute these differences to two possible mechanisms: slight differences in the surface polarity, caused by the substitution geometry of the incorporated silica precursor, can affect the diffusion of the ammonia-containing liquid into the siliceous matrix. In addition, also a different degree of condensation of the silica's silanol groups based on the ortho-, meta- and para-isomers as organosilica precursors can be discussed as an origin of the different degrees of etching by ammonia. While no corresponding solid-state NMR data were available, we exclude this possible mechanism, since all the prepared materials were subjected to a final treatment at 330 °C for 10 hours, which can be expected to result in a comparable condensation. In conclusion, the successful synthesis of meso- and macroporous monoliths was only possible by using a weight ratio of 95/5 and 90/10 of both TMOS/BTMB and TMOS/BTPMS, respectively. | ||
| Fig. 1 Physisorption isotherms (left) and pore size distributions (right) of TMOS/BTMB hybrids with 5% of organosilica precursor and increasing amounts of PEG (from top to bottom). The mesopore diameter distributions were calculated using NLDFT adsorption branch analysis assuming a cylindrical pore shape. | ||
 | ||
| Fig. 2 Physisorption isotherms (left) and pore size distributions (right) of TMOS/BTPMS hybrids with 5% organosilica precursor and increasing amounts of PEG (from top to bottom). | ||
| PEG content [g] | BET surf. area (SBET) [m2 g−1] | Mesopore volume [cm3 g−1] | Mesopore diam. [nm] | Macropore volume [cm3 g−1] | Macropore diam. [μm] | |
|---|---|---|---|---|---|---|
| p-BTMB (5%) | 0.355 | 560 | 0.95 | 7.5 | 2.12 | 2.9 |
| 0.375 | 580 | 1.06 | 7.5 | 2.16 | 1.0 | |
| 0.395 | 570 | 1.04 | 7.5 | 2.22 | 1.0 | |
| 0.415 | 530 | 1.01 | 8.1 | 2.29 | 1.1 | |
| 0.435 | 530 | 0.97 | 7.5 | 2.37 | 0.3 | |
| 0.455 | 520 | 1.53 | 9.4 | 2.33 | 0.3 | |
| 0.475 | 590 | 2.52 | 7.0 | 2.09 | 0.1 | |
| p-BTPMS (5%) | 0.355 | 550 | 0.97 | 7.0 | 2.07 | 1.5 |
| 0.375 | 550 | 1.02 | 8.4 | 2.19 | 1.4 | |
| 0.395 | 530 | 1.04 | 8.1 | 2.34 | 1.4 | |
| 0.415 | 470 | 1.06 | 9.4 | 2.31 | 1.1 | |
| 0.435 | 550 | 1.40 | 7.5 | 2.33 | 0.4 | |
| 0.455 | 570 | 1.31 | 7.0 | 2.39 | 0.5 | |
| 0.475 | 570 | 2.43 | 7.0 | 2.10 | 0.7 | |
| m-BTMB (5%) | 0.355 | 510 | 0.93 | 9.4 | — | — |
| 0.375 | 470 | 0.90 | 9.4 | — | — | |
| 0.395 | 500 | 0.94 | 9.4 | — | — | |
| 0.415 | 470 | 0.96 | 9.4 | — | — | |
| 0.435 | 470 | 0.94 | 9.4 | — | — | |
| 0.455 | 500 | 1.03 | 9.4 | — | — | |
| 0.475 | 470 | 1.26 | 11.6 | — | — | |
| m-BTPMS (5%) | 0.355 | 520 | 1.13 | 10.4 | — | — |
| 0.375 | 560 | 1.23 | 9.0 | — | — | |
| 0.395 | 590 | 1.19 | 9.4 | — | — | |
| 0.415 | 560 | 1.26 | 9.4 | — | — | |
| 0.435 | 300 | 0.81 | 9.4 | — | — | |
| 0.455 | 590 | 1.55 | 9.4 | — | — | |
| 0.475 | 560 | 2.09 | 9.4 | — | — | |
| o-BTMB (5%) | 0.355 | 450 | 0.96 | 10.4 | — | — |
| 0.375 | 440 | 0.97 | 10.4 | — | — | |
| 0.395 | 430 | 1.01 | 10.4 | — | — | |
| 0.415 | 460 | 1.14 | 9.4 | — | — | |
| 0.435 | 440 | 1.11 | 10.4 | — | — | |
| 0.455 | 460 | 1.41 | 10.4 | — | — | |
| 0.475 | 440 | 2.47 | 11.6 | — | — | |
| o-BTPMS (5%) | 0.355 | 470 | 0.93 | 9.4 | — | — |
| 0.375 | 410 | 0.97 | 10.4 | — | — | |
| 0.395 | 440 | 1.02 | 10.4 | — | — | |
| 0.415 | 400 | 1.01 | 11.6 | — | — | |
| 0.435 | 440 | 1.14 | 10.4 | — | — | |
| 0.455 | 410 | 1.16 | 10.4 | — | — | |
| 0.475 | 420 | 1.67 | 10.4 | — | — | |
| p-BTMB (10%) | 0.355 | 560 | 0.95 | 7.5 | — | — |
| 0.375 | 580 | 1.06 | 7.5 | — | — | |
| 0.395 | 570 | 1.04 | 7.5 | — | — | |
| 0.415 | 530 | 1.01 | 8.1 | — | — | |
| 0.435 | 530 | 0.97 | 7.5 | — | — | |
| 0.455 | 520 | 1.53 | 9.4 | — | — | |
| 0.475 | 590 | 2.52 | 7.0 | — | — | |
| p-BTPMS (10%) | 0.355 | 550 | 0.97 | 7.0 | — | — |
| 0.375 | 550 | 1.02 | 8.4 | — | — | |
| 0.395 | 530 | 1.04 | 8.1 | — | — | |
| 0.415 | 470 | 1.06 | 9.4 | — | — | |
| 0.435 | 550 | 1.40 | 7.5 | — | — | |
| 0.455 | 570 | 1.31 | 7.0 | — | — | |
| 0.475 | 570 | 2.43 | 7.0 | — | — | |
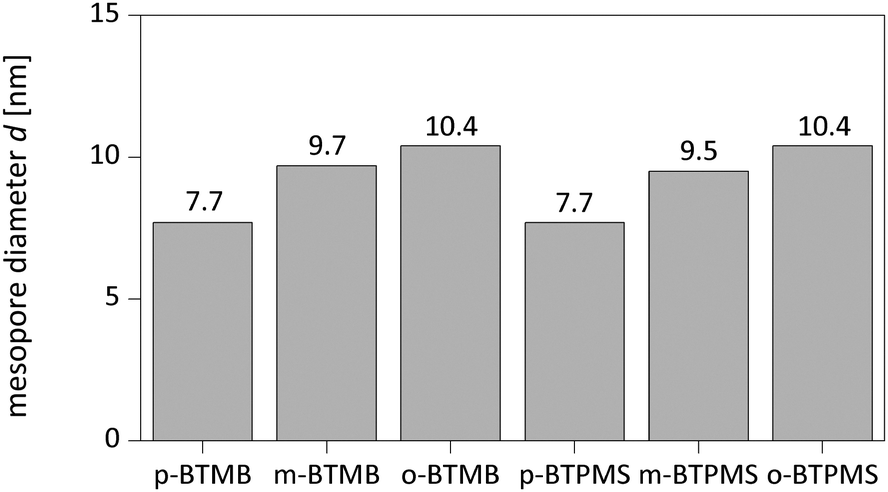 | ||
| Fig. 3 Mesopore diameters of silica hybrids with different isomers and 5% precursor. | ||
Mesopore size distributions using different evaluation methods are given in Fig. 4 and the corresponding values are presented in Table 2. The NLDFT model for cylindrical pores utilizing kernels for the adsorption branch of the isotherm was chosen in this study, as it is known to give reasonable results for the pore size distribution for silica materials of this kind.19 A main advantage of density functional theory (DFT) methods is the determination of an accurate and valid pore size distribution across the complete mesopore (and micropore) size range. In contrast to classical calculation methods like the BJH method, which typically underestimates the mesopore size systematically, the calculation via adsorption branch kernels takes the delayed condensation of a metastable fluid correctly into account.20
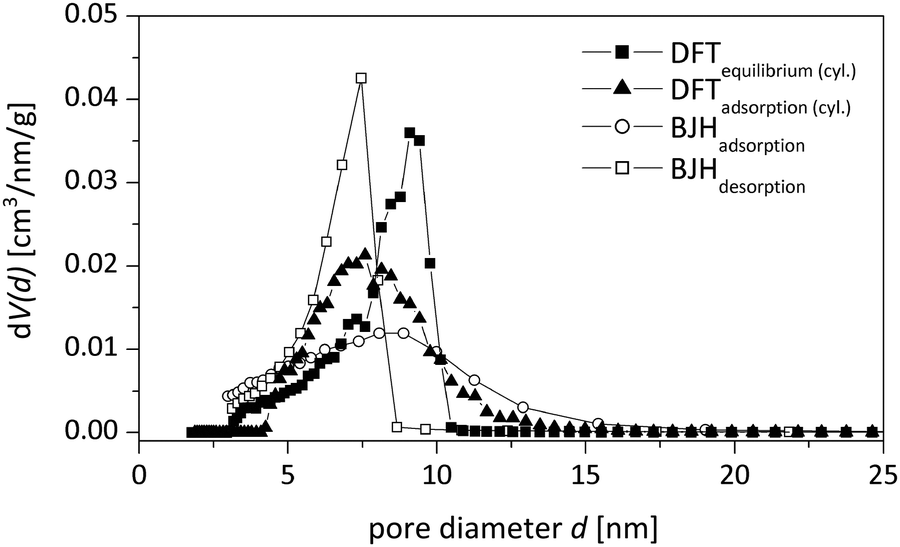 | ||
| Fig. 4 Mesopore size distributions for the sample p-BTMB (5%, 0.355 g PEG) obtained by different evaluation methods: NLDFT equilibrium model for cylindrical pore shapes (■), NLDFT adsorption branch model for cylindrical pore shapes (▲), BJH adsorption branch model (○) and BJH desorption branch model (□). | ||
| NLDFT (des) | NLDFT (ads, cyl) | NLDFT (ads, cyl/sph) | BJH (ads) | BJH (des) | |
|---|---|---|---|---|---|
| Mesopore volume [cm3 g−1] | 0.94 | 0.95 | 0.92 | 0.93 | 0.97 |
| Average mesopore diameter [nm] | 9.0 | 7.5 | 12.9 | 8.8 | 7.4 |
As shown in Fig. 5 and 6 in a direct comparison of the N2 physisorption analysis, the mesoporosity of the monoliths prepared using para-BTMB and para-BTPMS is almost identical to the mesoporosity observed in pure SiO2 monoliths, prepared under standard conditions.18 The pore size distributions and also the mesopore volumes are thus almost indistinguishable, suggesting that our synthetic procedure is sufficiently optimized to produce such monoliths in a highly reproducible fashion. While the analysis of N2 physisorption experiments (performed at T = 77 K), combined with the aforementioned evaluation procedure, provides accurate and meaningful structural parameters describing the mesoporosity, other relevant information on the mesopore network cannot be obtained. In particular, the modified polarity of the silica surface (resulting from the incorporated aromatic moiety) cannot be determined from N2 physisorption measurements, not even in a qualitative way. A further aspect is that sorption experiments performed near room temperature are helpful to judge the presence of possible swelling effects under those conditions, being relevant for applications in chromatography, e.g. by using polar/non-polar solvents. Such insights into the polarity of the surface of the prepared hybrid materials were addressed in the present study by performing additional physisorption experiments utilizing dibromomethane (DBM) as a polar adsorbate at T = 290 K.
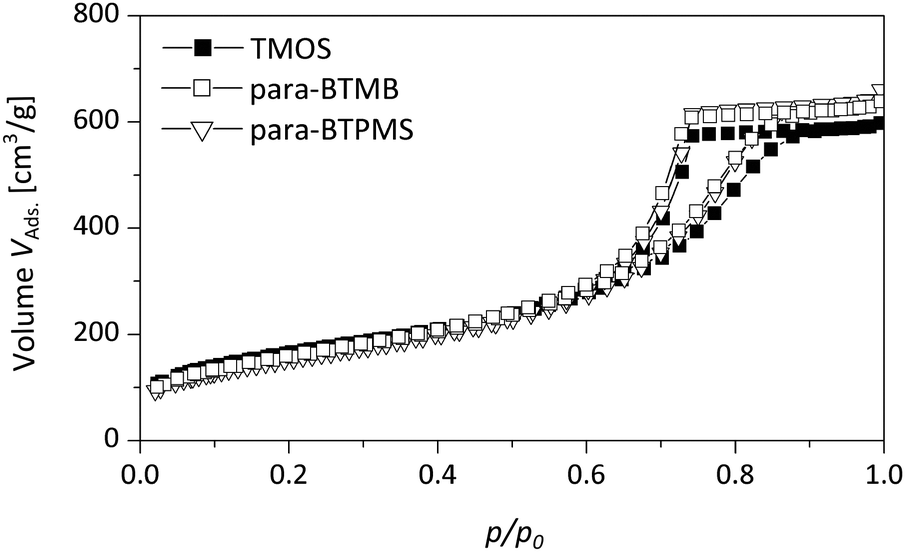 | ||
| Fig. 5 Nitrogen physisorption isotherms of a pure TMOS based reference monolith compared to the hybrid samples containing the para-isomer of BTMB/BTPMS (5%, 0.355 g PEG). | ||
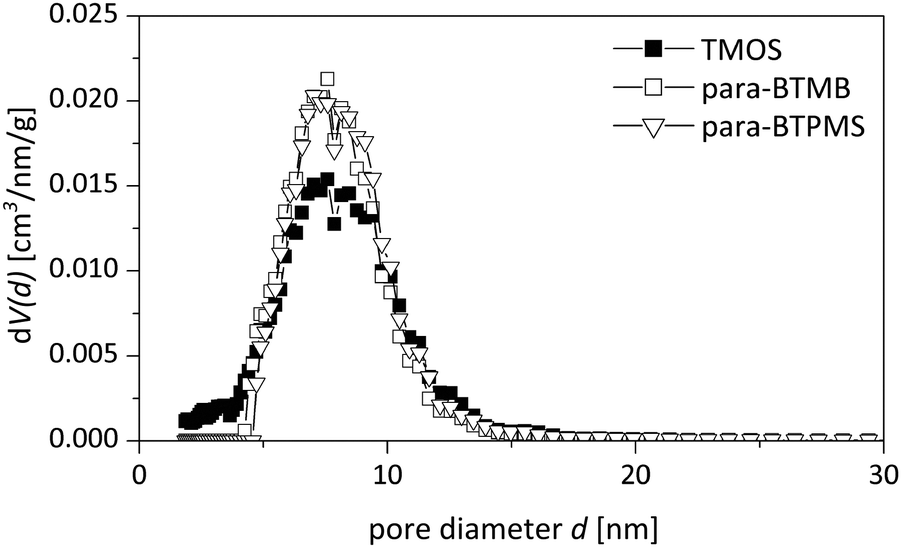 | ||
| Fig. 6 Pore size distribution from nitrogen physisorption (NLDFT adsorption branch analysis with cylindrical pore shapes) of a pure TMOS based reference monolith compared to the hybrid samples containing the para isomer of BTMB/BTPMS (5%, 0.355 g PEG). | ||
Fig. 7 presents DBM isotherms comparing a pure SiO2 monolith (prepared using TMOS) and monoliths generated using para-BTMB and para-BTPMS ((5%, 0.355 g PEG) see Table 1). The latter can be classified as type V hysteresis loops according to IUPAC recommendations, while N2 isotherms exhibit type IV hysteresis behavior (see above). This difference is in accordance with the fact that DBM generally does not exhibit the formation of layers on siliceous surfaces. In general, it should be noted that the DBM isotherms are of high quality, showing a well-defined plateau at large pressures and the absence of non-closure behavior upon desorption.21
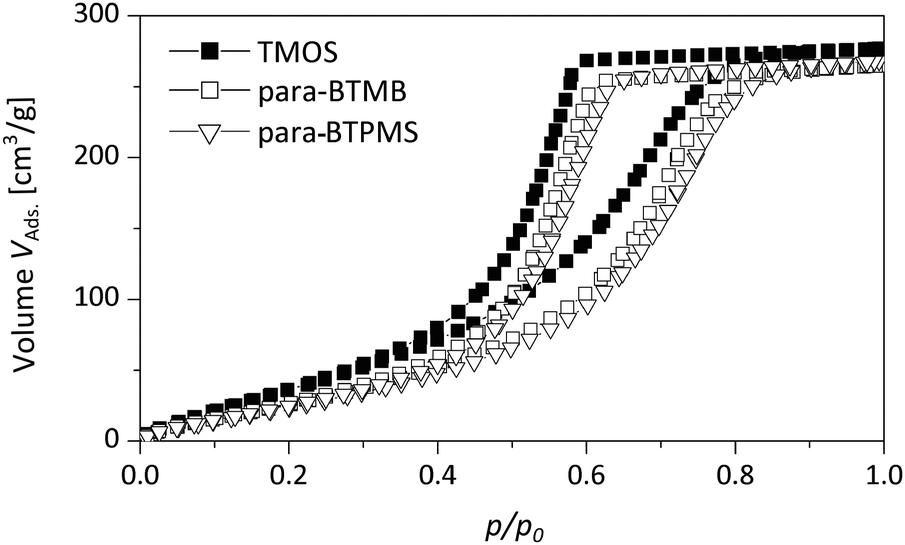 | ||
| Fig. 7 Dibromomethane sorption isotherms of a pure TMOS based reference monolith compared to the hybrid samples containing the para-isomer of BTMB/BTPMS (5%, 0.355 g PEG). | ||
Furthermore, these isotherms feature interesting characteristics with respect to the sorption and interaction behavior as a function of composition. First, the DBM uptake of the hybrid materials is lower, in terms of the adsorbed volume, than the DBM uptake obtained for the monolith made up of pure SiO2, even for small p/p0. This finding suggests a reduced interaction of DBM with the mesopore surfaces within the hybrid materials compared to pure SiO2, possessing a certain content of aromatic moieties, which is plausible taking the polar nature of DBM into account. Second, both condensation and desorption branches are located at larger p/p0, if compared to pure SiO2 (see Fig. 7). This behavior is in line with the different contact angle of DBM on a pure SiO2 surface and a surface additionally containing non-polar moieties, resulting in different values for the partial pressure of pore emptying according to the Kelvin equation.
Since the DBM isotherms feature a well-defined closure upon desorption, the presence of swelling or other effects related to kinetically hindered desorption can be excluded. These results are in contrast to DBM sorption investigations performed recently by our group.21 In these studies, porous SiO2 spheres were post-functionalized by alkyl chains, and the isotherm showed pronounced non-closure effects along the desorption branch. While the origin of this peculiar difference in the DBM sorption properties has not been studied in detail, we speculate that the mesopore network of the materials described in ref. 21 exhibits a hindrance towards emptying for DBM under the applied conditions. In conclusion, these comparative sorption experiments using DBM reveal a reduced interaction of DBM with the organically modified surface, which is reasonable taking into account the unpolar nature of the precursors used. Furthermore, the sorption experiments using DBM allow for an estimation of the contact angle, following the procedure described in literature.21 This approach is based on the Kelvin equation
| ln(p/p0) = −2γVLcosθ/RTrm | (1) |
Furthermore, the DBM sorption properties of hybrid monoliths prepared from different isomers were studied (Fig. 8 and Fig. S1 in the ESI†). All of the hybrid materials feature well-defined isotherms, confirming the well-defined mesopore structure and a high degree of cross-linking (absence of swelling). For the hybrids containing the para-isomers the lower closure point of the hysteresis is observed at ca. p/p0 = 0.3 (BTMB) and p/p0 = 0.2 (BTPMS). The closure point for samples with the built-in meta-isomer is shifted to higher relative pressures at ca. p/p0 = 0.35 (BTMB) and p/p0 = 0.3 (BTPMS). For samples functionalized with the ortho-isomers the closure point is located at ca. p/p0 = 0.45 (BTMB) and p/p0 = 0.4 (BTPMS). These difference can be attributed to the differences in the pore-size distributions (see nitrogen physisorption analysis), and pore-blocking of cavitation effects can be excluded for DBM sorption performed at T = 290 K.
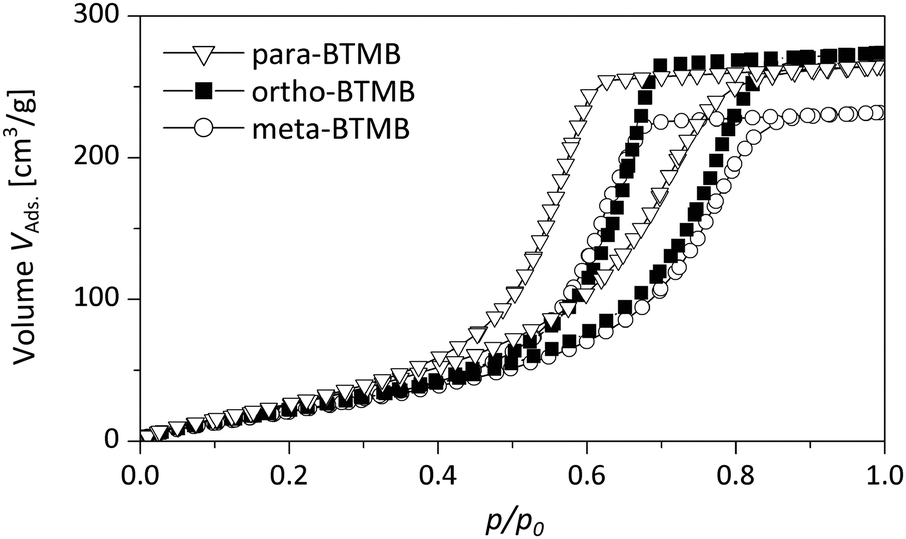 | ||
| Fig. 8 Dibromomethane sorption isotherms of silica hybrids containing the different isomers of BTMB (5%, 0.355 g PEG). | ||
The systematic variation of the PEG ratio used in the starting mixture exhibited a systematic effect on the macroporosity as indicated by Hg intrusion porosimetry and SEM observations (Fig. 9 and 10): with an increasing amount of PEG acting as the porogen, the average macropore diameter decreases until the typical macropore morphology finally collapses at the highest PEG content used. These trends are in accordance with the results known for pure silica monoliths.21
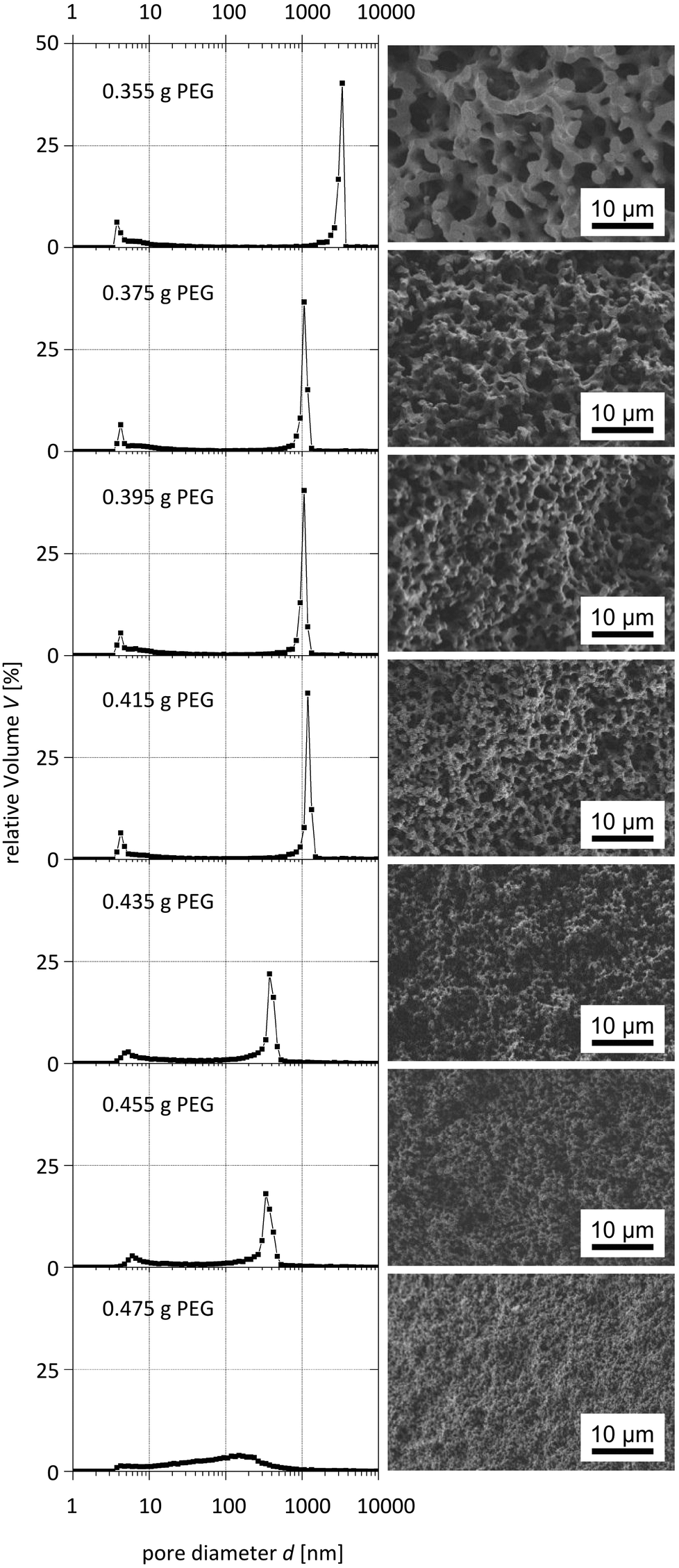 | ||
| Fig. 9 Pore size distributions obtained from mercury intrusion porosimetry (left) and corresponding SEM images (right) of para-TMOS/BTMB (5%) hybrids. | ||
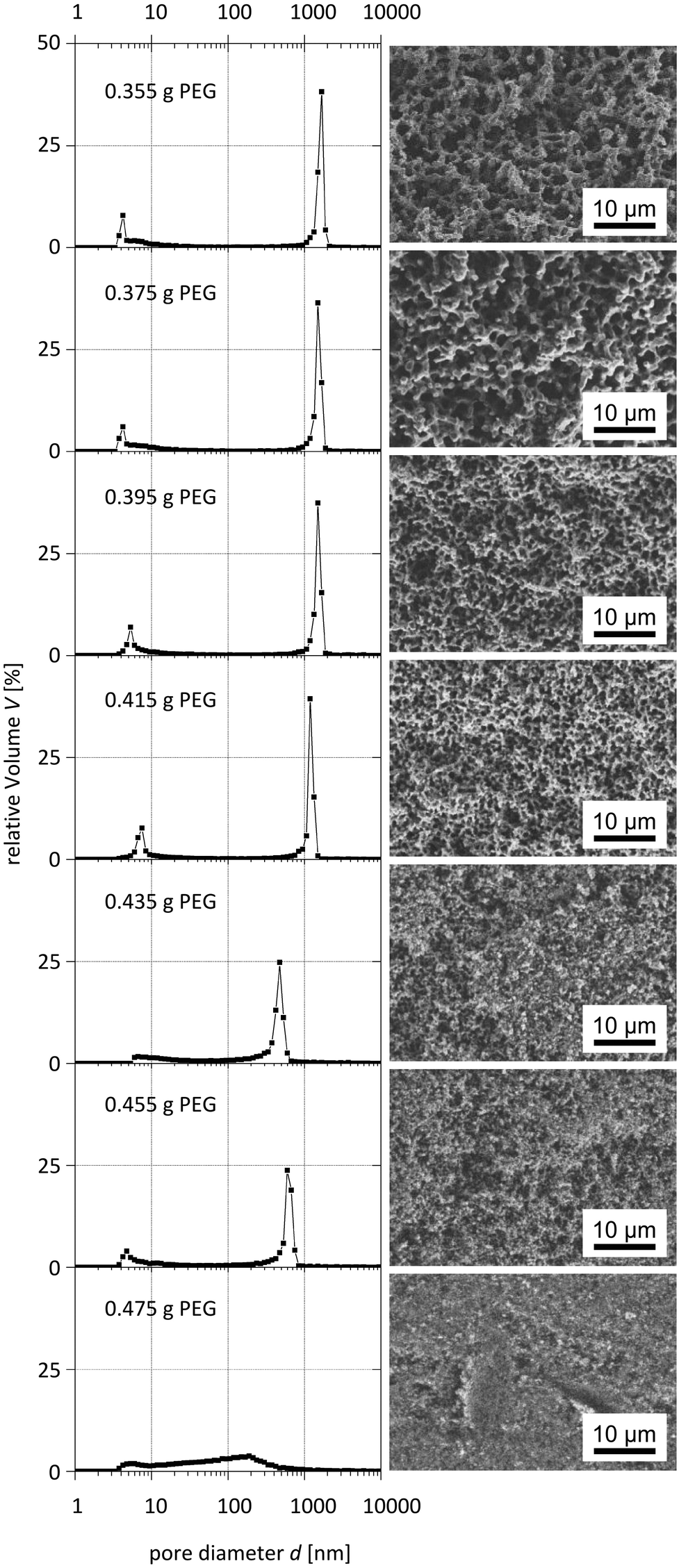 | ||
| Fig. 10 Pore size distributions obtained from mercury intrusion porosimetry (left) and corresponding SEM images (right) of para-TMOS/BTPMS (5%) hybrids. | ||
Interestingly, one can also observe a dependence of the mesoporosity on the amount of porogen, which suggests that a molecular templating mechanism of PEG chains occurs. This interpretation is supported by the observation of increasing mesopore volumes when using the highest PEG amount and leads to the assumption that two kinds of mesopore species are present. This interpretation is supported by the observation of increasing mesopore volumes when using the highest PEG amount and leads to the assumption that two kinds of mesopore species are present. Whilst one is the result of chemical etching of the silica surface in the hydrothermal treatment, the other species is created via PEG templating on a molecular level.
Furthermore, the reduced macropore volume observed at the highest concentrations of PEG used (Fig. 11) indicates that above a certain amount of PEG the spinodal decomposition mechanism is affected, i.e. an increasing fraction of PEG does not participate in this intricate separation process, but is incorporated into the siliceous matrix and results in mesopores after calcination. Our experiments also showed that the synthesis of silica monoliths with a homogeneous bimodal porosity can only be realized in narrow ranges of the BTMB and BTPMS content.
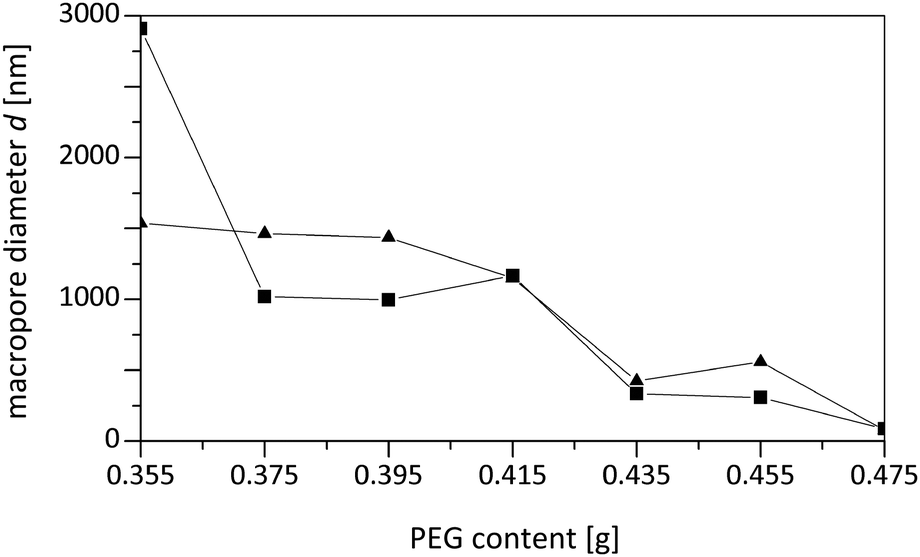 | ||
| Fig. 11 Average macropore diameter of meso-macroporous TMOS/BTMB- and TMOS/BTPMS-based hybrids (para-isomers with 5% precursor content) as a function of the varied amount of porogen in the starting feed solution. | ||
Conclusions
In the present study the ortho-, meta- and para-isomers of bis(trimethoxysilyl)benzene (BTMB) and bis((trimethoxysilyl)phenyl)dimethoxysilane (BTPMS) were synthesized and used as bifunctionalized organosilica precursors, in mixtures with tetramethyl orthosilicate (TMOS) as SiO2 precursors. The obtained compounds were utilized for the preparation of monolithic silica hybrids in an adapted sol–gel-route resulting in the formation of a macroporous skeleton by phase separation induced by poly(ethylene glycol), according to the synthesis developed by K. Nakanishi.14The amount of polymer was varied spanning a quite wide range to adjust defined macropore morphology comparable to pure silica monoliths. The porosity was characterized by N2 and dibromomethane sorption, Hg-intrusion and SEM. Similar trends regarding the macro- and mesoporosity as known from TMOS-based monoliths were observed over a relatively large range of PEG concentrations. Hybrids with well-defined and bicontinuous macroporosity and pronounced mesoporosity were obtained up to weight ratios of 95/5 and 90/10 of TMOS/BTMB and TMOS/BTPMS, respectively (Fig. 12). While such amounts of the bis(trimethoxysilyl)arenes appear to be small, even such moderate contents of organic moieties are sufficient to substantially change the materials' properties, as revealed by physisorption experiments using dibromomethane. Using a previously described approach, the hybrid materials possess a contact angle of the mesopore surface of ca. 30° with respect to dibromomethane, which significantly exceeds the contact angle derived for corresponding pure SiO2 monoliths by the same procedure. Thereby, it is proven that the arene units are accessible from the mesopore space. We have recently demonstrated that using a weight ratio of 5/95 of phenyltrimethoxysilane (PTMS) and tetramethoxysilane (TMOS) as precursors yielded monolithic silica hybrid capillaries, which exhibited a different interaction towards hydrocarbons compared to capillaries containing pure silica possessing an analogous porosity.15 This substantial impact of small contents of aromatic moieties on the interaction can be attributed to the homogeneous distribution of the organic moieties in combination with the high porosity and high surface area, which enhance this interaction. Thus, the results of this study will be utilized for the analysis of hydrophobicity and separation characteristics as well as tests of column performances in HPLC and the preparation of capillary columns.
 | ||
| Fig. 12 SEM images of TMOS/BTMB and TMOS/BTPMS hybrids with different isomers, each containing 5% of organosilica precursor and 0.355 g of PEG. | ||
Acknowledgements
We would like to thank Merck KGaA for financial support, in particular Dr. Karin Cabrera. Moreover, we thank Christoph Weidmann for performing the physisorption measurements and Dr. Heike Hausmann for NMR analysis. This project was supported by the Laboratory of Materials Research (LaMa) of JLU.References
- (a) T. Hara, S. Makino, Y. Watanabe, T. Ikegami, K. Cabrera, B. Smarsly and N. Tanaka, J. Chromatogr. A, 2010, 1217, 89–98 CrossRef CAS PubMed; (b) S. Bruns, T. Hara, B. M. Smarsly and U. Tallarek, J. Chromatogr. A, 2011, 1218, 5187–5194 CrossRef CAS PubMed.
- K. J. Shea and D. A. Loy, Chem. Mater., 2001, 13, 3306–3319 CrossRef CAS.
- A. El Kadib, R. Chimenton, A. Sachse, F. Fajula, A. Galarneau and B. Coq, Angew. Chem., Int. Ed., 2009, 48, 4969–4972 CrossRef CAS PubMed.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., 2006, 118, 3290–3328 CrossRef.
- (a) A. Kuschel and S. Polarz, Adv. Funct. Mater., 2008, 18, 1272–1280 CrossRef CAS; (b) A. Kuschel, M. Drescher, T. Kuschel and S. Polarz, Chem. Mater., 2010, 22, 1472–1482 CrossRef CAS; (c) J. Gehring, D. Schleheck, M. Luka and S. Polarz, Adv. Funct. Mater., 2014, 24, 1140–1150 CrossRef CAS.
- K. Kanamori and K. Nakanishi, Chem. Soc. Rev., 2011, 40, 754 RSC.
- (a) N. Hüsing, D. Brandhuber and P. Kaiser, J. Sol–Gel Sci. Technol., 2006, 40, 131–139 CrossRef; (b) S. Hartmann, D. Brandhuber and N. Hüsing, Acc. Chem. Res., 2007, 40, 885–894 CrossRef CAS PubMed; (c) M. Weinberger, T. Fröschl, S. Puchegger, H. Peterlik and N. Hüsing, Silicon, 2009, 1, 19–28 CrossRef CAS; (d) D. Brandhuber, H. Peterlik and N. Huesing, Small, 2006, 2, 503–506 CrossRef CAS PubMed.
- R. J. P. Corriu, J. J. E. Moreau, P. Thepot and M. W. C. Man, Chem. Mater., 1992, 4, 1217–1224 CrossRef CAS.
- (a) A. Mehdi, C. Reye and R. Corriu, Chem. Soc. Rev., 2011, 40, 563 RSC; (b) R. Mouawia, A. Mehdi, C. Rey and R. Corriu, New J. Chem., 2006, 30, 1077 RSC.
- (a) K. J. Shea, D. A. Loy and O. Webster, J. Am. Chem. Soc., 1992, 114, 6700–6710 CrossRef CAS; (b) D. A. Loy and K. J. Shea, Chem. Rev., 1995, 95, 1431–1442 CrossRef CAS.
- G. Cerveau, S. Chappellet and R. J. P. Corriu, J. Mater. Chem., 2003, 13, 2885 RSC.
- M. von der Lehr, C. Weidmann, S. Mascotto and B. M. Smarsly, Chem. Ing. Tech., 2013, 85, 1700–1706 CrossRef CAS.
- K. Nakanishi and K. Kanamori, J. Mater. Chem., 2005, 15, 3776 RSC.
- K. Nakanishi, Bull. Chem. Soc. Jpn., 2006, 79, 673–691 CrossRef CAS.
- R. Meinusch, K. Hormann, R. Hakim, U. Tallarek and B. M. Smarsly, RSC Adv., 2015, 5, 20283–20294 RSC.
- M. Sharifi, C. Köhler, P. Tölle, T. Frauenheim and M. Wark, Small, 2011, 7, 1086–1097 CrossRef CAS PubMed.
- J. Suzuki, A. Shimojima, Y. Fujimoto and K. Kuroda, Chem. – Eur. J., 2008, 14, 973–980 CrossRef CAS PubMed.
- T. Hara, S. Mascotto, C. Weidmann and B. M. Smarsly, J. Chromatogr. A, 2011, 1218, 3624–3635 CrossRef CAS PubMed.
- M. Thommes, B. Smarsly, M. Groenewolt, P. I. Ravikovitch and A. V. Neimark, Langmuir, 2006, 22, 756–764 CrossRef CAS PubMed.
- M. Thommes, Chem. Ing. Tech., 2010, 82, 1059–1073 CrossRef CAS.
- (a) D. Stoeckel, C. Kübel, M. O. Loeh, B. M. Smarsly and U. Tallarek, Langmuir, 2015, 31, 7391–7400 CrossRef CAS PubMed; (b) D. Stoeckel, C. Kübel, K. Hormann, A. Höltzel, B. M. Smarsly and U. Tallarek, Langmuir, 2014, 30, 9022–9027 CrossRef CAS PubMed; (c) D. Stoeckel, D. Wallacher, G. A. Zickler, M. Thommes and B. M. Smarsly, Langmuir, 2015, 31, 6332–6342 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nj00758a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |