
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reduction of dichloro(diaza-phospha)stibanes – isolation of a donor-stabilized distibenium dication†‡
Alexander
Hinz
a,
Julia
Rothe
b,
Axel
Schulz
*ab and
Alexander
Villinger
a
aInstitut für Chemie, Universität Rostock, Albert-Einstein-Str. 3a, 18059 Rostock, Germany. E-mail: axel.schulz@uni-rostock.de
bAbteilung Materialdesign, Leibniz-Institut für Katalyse e.V. an der Universität Rostock, Albert-Einstein-Str. 29a, 18059 Rostock, Germany
First published on 3rd August 2015
Abstract
A reaction of antimonytrichloride SbCl3 with potassium bis(terphenylimino)phosphide K[(TerN)2P] smoothly afforded a novel class of mixed diazadipnictanes, namely dichloro(diaza-phospha)stibane [Ter2N2P(III)Sb(III)Cl2], which is considered to exist as open chain-like and cyclic isomers in an equilibrium. [Ter2N2PSbCl2] is a versatile starting material for reduction and halide abstraction experiments. Halide abstraction led to the formation of a cyclic diazastibaphosphenium cation [P(μ-NTer)2SbCl]+. Upon reduction of [Ter2N2PSbCl2], the transient existence of the novel mixed biradicaloid [P(μ-NTer)2Sb] was proven by a trapping experiment with an alkyne, while reduction in the absence of trapping agents afforded the eight-membered heterocycle [Sb2-{μ-(TerN)2P}2]. This constitutional isomer of a dimerized biradicaloid features a bonding situation that indicates the presence of a donor-stabilized [Sb2]2+ ion.
Introduction
As early as 1894 Michaelis and Schroeter discovered the first dichloro-diphosphadiazane (Fig. 1, species A with R = Ph) upon reaction of aniline hydrochloride with phosphorus(III) chloride.1 Nowadays, these species are well known with respect to their oxidation, substitution, halide abstraction and coordination behaviour.2 Only in the early 1980s, Paine et al. investigated the reduction of such cyclic species,3–5 which led to the isolation of α-(RN)4P4 (C, R = tBu), with a P4N4 core isostructural to α-P4S4 and S4N4.6 Later, Wright et al. were able to convert the α- into the β-isomer photochemically (D).7 However, upon reduction of a dichloro-diphosphadiazane, in the first step a biradicaloid (species B) is formed. If the steric bulk of the substituent is sufficient to prevent dimerization, these biradicaloids can be isolated, e.g. 1,3-diphospha-2,4-diazane-1,3-diyl [P(μ-NTer)]2 (Ter = 2,6-dimesityl-phenyl).8 Singlet biradicaloids fascinated chemists in theory and synthesis equally in the past few decades since they are highly reactive species and putative intermediates in the processes of bond breaking and bond formation.9–11 The term biradicaloids used throughout this paper refers to a subset of biradicals with two radical centers interacting significantly. Pioneering work in this field was carried out by Niecke et al. who discovered 1,3-dichloro-2,4-diphosphacyclobutane-1,3-diyl [ClC(μ-PMes*)]2 (Fig. 2 species E, Mes* = 2,4,6-tri-tertbutyl-phenyl) in 1995.12 The reactivity of this particular biradicaloid was investigated with respect to photochemical isomerization, reduction and oxidation as well as acid–base reactivity afterwards.13–17 Yoshifuji et al. reported on a different synthetic approach to a 1,3-diphosphacyclobutane-2,4-diyl (F) bearing the bulky substituent not on the phosphorus but on the carbon atom.18–23 In 2002, Bertrand et al. succeeded in the isolation of another biradicaloid, [iPr2P(μ-BtBu)]2 (G), which featured intriguing bond-stretch isomerism in dependence of the steric congestion upon changing the substituents.24,25 Furthermore, radical-type as well as ionic-type characteristic reactions were observed.26,27 In the groups of Power and Schnepf, several biradicaloid main group cluster compounds were investigated.28–30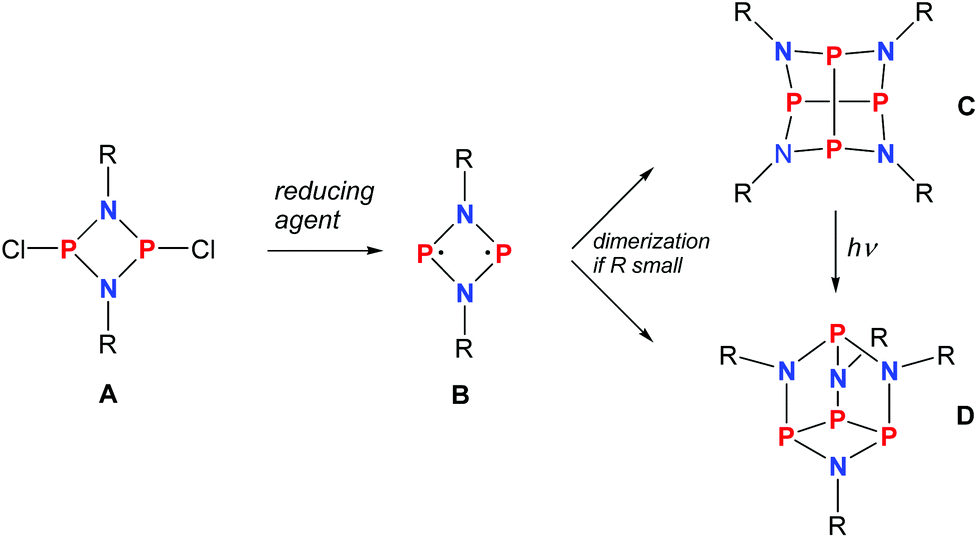 | ||
| Fig. 1 Reduction of cyclodichlorodiphosphadiazane A yielding biradicloid B or its dimers C and D depending on the steric strain. | ||
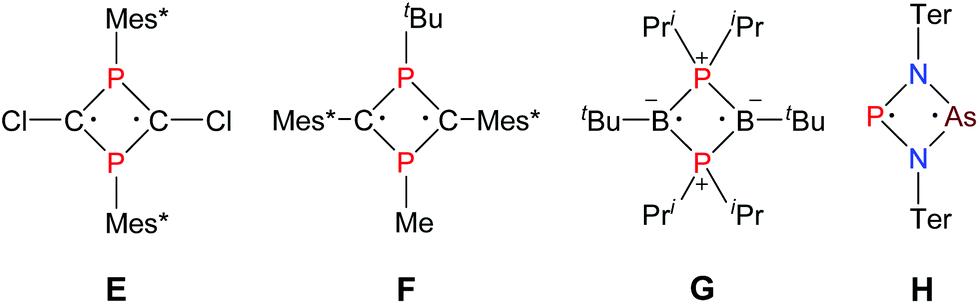 | ||
| Fig. 2 Selected known biradicaloids. | ||
Currently, several examples of cyclobutane-1,3-diyl derivatives are known (Fig. 2),31–37 however all known singlet biradicaloids feature equivalent radical centres. The first biradicaloid with different radical centres, 1-arsa-3-phospha-2,4-diazane-1,3-diyl [P(μ-NTer)2As] (H), was recently obtained by reduction of a cyclic dichloro-arsa-phospha-diazane and features remarkable regioselectivity in the activation of small molecules.38 To evoke even larger differences between the radical centres, we sought to generate 1-stiba-3-phospha-2,4-diazane-1,3-diyl [P(μ-NTer)Sb], which is in the focus of this contribution.
Results and discussion
Synthesis of NPN-substituted dichlorostibanes
The reaction of potassium bis(terphenylimino)phosphide (K[(TerN)2P], 1, δ(31P) 322 ppm, Fig. 3)38 with antimony(III) chloride in toluene at ambient temperature led to the formation of a mixture of two constitutional isomers: (i) open-chain dichlorostibane 2a (Fig. 3) and trans-2b as indicated by temperature variable 31P NMR studies (Fig. 4).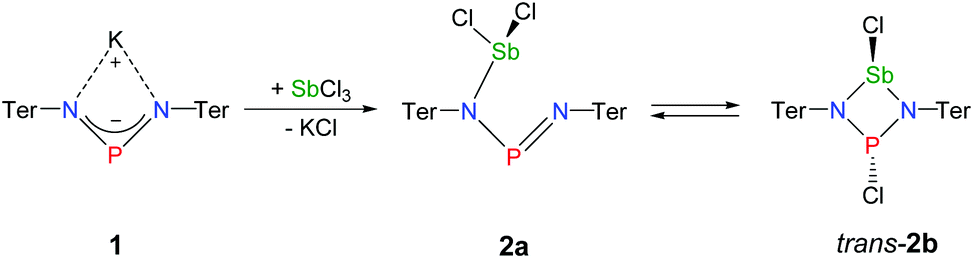 | ||
| Fig. 3 Preparation of NPN-substituted dichlorostibane 2a and its equilibrium with trans-2b as indicated by 31P NMR studies. | ||
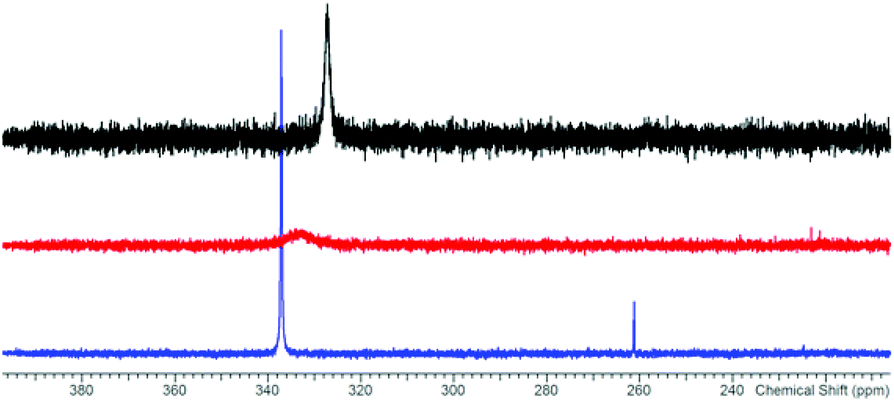 | ||
| Fig. 4 31P NMR spectra of 2 in [D8]-toluene at −80 (blue), +20 (red), and +100 °C (black). | ||
The redissolved compound 2 exhibits a very broad 31P NMR resonance (Fig. 4, δ(31P, 300 K, 500 MHz) = 332 ppm, ν1/2 = 900 Hz) under ambient conditions indicating intramolecular dynamics. To shed some light into this dynamics, density functional theory at the pbe1pbe level (for details see the ESI‡) was applied to study the potential energy surface (PES) of 2M (Fig. 5, M = model with the terphenyl group substituted by phenyl).39,40 In the gas phase, initially three isomers of 2M were considered: 2a, as well as the cyclic 1,3-dichloro-1-stiba-3-phospha-2,4-diazanes cis- and trans-[ClP(μ-NPh)2SbCl] 2b. These three isomers were found to be very similar in energy (relative to 2a: cis-2b +2.6, trans-2b +0.1 kJ mol−1). In addition, a higher-lying cyclic isomer 2c (30.3 kJ mol−1) bearing a SbCl2 group was located at the PES when studying the intrinsic reaction for the different isomerization processes as depicted in Fig. 5. Isomer 2c is a typical intermediate lying in a very shallow energy valley, therefore cannot be observed in the 31P NMR spectra. At low temperatures, two singlets were observed in the 31P NMR spectra of a sample of redissolved crystalline 2a (193 K, 337.1 and 261.2 ppm), which were assigned to 2a and trans-2b (computed δ(31P): cis-2b 242, trans-2b 262, 2a 333 ppm). At −80 °C, a ratio of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was found for both species, so the coalesced signal is expected to occur at 331.2 ppm (observed at 100 °C: 327.2 ppm). We do want to stress that the assignment of trans-2b to the NMR resonance at 261.2 ppm is solely based on comparison with computational data. The 31P NMR data with a coalescence temperature of 313 K indicate a Gibbs activation energy for the rearrangement reaction 2a → trans-2b of 49.5 kJ mol−1. From a van't Hoff plot, the difference in the energy of both isomers was estimated to be 6.2 kJ mol−1 (cf. ΔE(0 K) = 0.1 kJ mol−1 for the gas phase). Interestingly, single crystal X-ray studies revealed the exclusive presence of thermodynamically more stable open-chain species 2a in the solid state (yield: 72%, Fig. 6 left). The thermodynamical preference of the open chain species 2a over 2b and 2c is a clear difference to the lighter homologues. For example, the reaction of 1 with arsenic(III) chloride led to the formation of the cyclic arsa-phospha-diazane [ClP(μ-NTer)2AsCl].38
:1 was found for both species, so the coalesced signal is expected to occur at 331.2 ppm (observed at 100 °C: 327.2 ppm). We do want to stress that the assignment of trans-2b to the NMR resonance at 261.2 ppm is solely based on comparison with computational data. The 31P NMR data with a coalescence temperature of 313 K indicate a Gibbs activation energy for the rearrangement reaction 2a → trans-2b of 49.5 kJ mol−1. From a van't Hoff plot, the difference in the energy of both isomers was estimated to be 6.2 kJ mol−1 (cf. ΔE(0 K) = 0.1 kJ mol−1 for the gas phase). Interestingly, single crystal X-ray studies revealed the exclusive presence of thermodynamically more stable open-chain species 2a in the solid state (yield: 72%, Fig. 6 left). The thermodynamical preference of the open chain species 2a over 2b and 2c is a clear difference to the lighter homologues. For example, the reaction of 1 with arsenic(III) chloride led to the formation of the cyclic arsa-phospha-diazane [ClP(μ-NTer)2AsCl].38
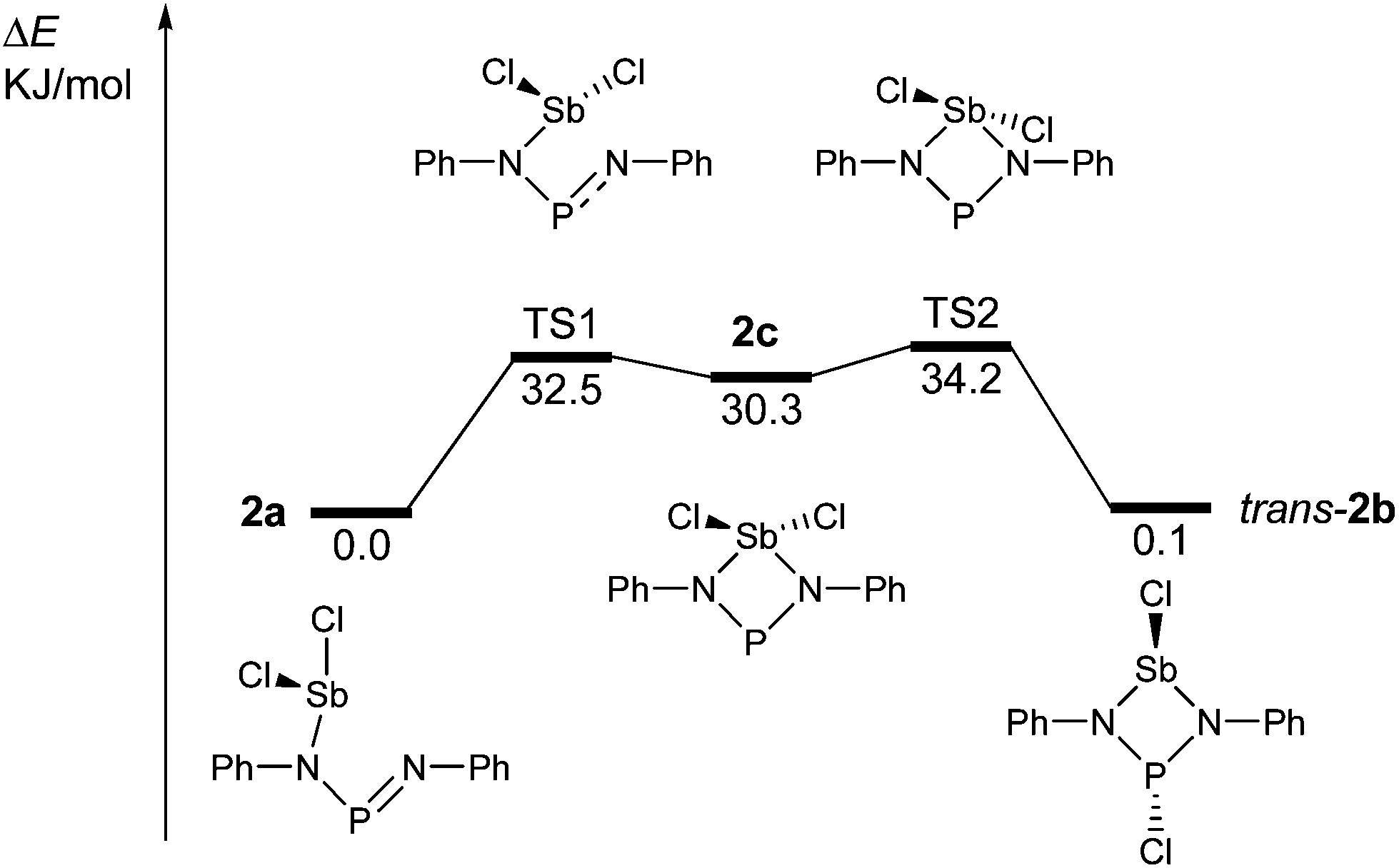 | ||
| Fig. 5 Potential energy surface of 2M (M = model, terphenyl group substituted by phenyl; the isomerisation process, leading to cis-2b, was omitted for clarity, see the ESI;‡ TS = transition state). | ||
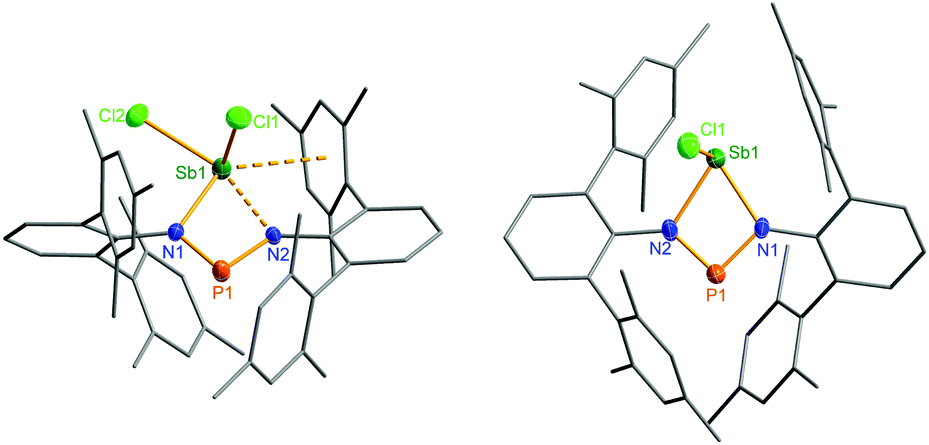 | ||
| Fig. 6 Molecular structure of 2a (left) and 3 (right). Thermal ellipsoids are drawn at 50% probability (173 K). Selected bond lengths [Å] and angles [°]: 2: Sb1–N1 2.133(4), Sb1–N2 2.392(4), Sb1–Cl1 2.358(2), Sb1–Cl2 2.451(2), P1–N1 1.623(4), P1–N2 1.598(4), N2–P1–N1 96.0(2). 3: Sb1–N1 2.136(3), Sb1–N2 2.171(3), Sb1–Cl1 2.342(1), P1–N2 1.625(3), P1–N1 1.643(4), N2–P1–N1 91.7(2). | ||
The structural motif observed for 2a resembles the recently studied NCN-substituted LSbCl2-compounds (e.g. L = amidinate donors) of Ragogna et al.,41,42 Jones et al.,43 Dehnicke et al.,44 and S. Schulz et al.45 which are described as base stabilized dihalopnictanes. In accord with this concept and the fact, that only the open-chain species 2a was observed in the solid state, a short N–Sb bond length (N1–Sb1 2.133(4) Å, Fig. 6), clearly corresponding to a single bond (∑rcov(N–Sb) 2.11 Å), and a significantly longer Sb–N distance, indicating only a secondary interaction (N3–Sb1 2.392(4) Å), are observed. Furthermore, the in-plane Sb–Cl (Sb1–Cl2 2.451(2) Å) bond is elongated compared to the out-of-plane Sb–Cl bond (Sb1–Cl1 2.358(2) Å), as it is found for all these base stabilized pnictogen halides. NBO analyses (NBO = natural bond orbital)46,47 indicate the donation of electron density from the lone pair (LP) located at the imino-N2 atom into the in-plane antibonding ∑*(Sb–Cl2) orbital.40 In addition, Menshutkin type (dispersion) interactions between one mesityl group of the terphenyl substituent with the Sb atom is observed, a common feature of such complexes.
Halide abstraction in 2
Ring closure in 2a was easily achieved by addition of a Lewis acid such as GaCl3via halide abstraction (Fig. 7), affording the cyclic red cationic species (3, δ(31P) 374 ppm, Fig. 6 right) and the GaCl4− ion. The formal charge in 3 is located on the dicoordinated P atom, hence it will subsequently be denoted as the phosphenium cation, even though the NBO charges indicate stronger charge localization on Sb than on P (Sb +1.61, P +1.36e). The large positive charge at the Sb center mainly arises from strongly polarized Sb–N σ-bonds. Interestingly, upon halide abstraction the phosphenium cation is formed rather than the stibenium cation. Also there is no exchange of the Cl− ion between the P and Sb atoms of the four-membered ring in solution, which can be observed on the NMR time scale, in accord with computation indicating that the P+ centred species is considerably favoured over the Sb+ centred ion by 59.3 kJ mol−1. The hypothetical stibenium cation [ClP(μ-NTer)Sb]+, that could result from Cl shifting from Sb to P, would have been expected to exhibit green colour48,49 (see computations in the ESI‡). The P+ center in [ClSb(μ-NTer)P]+ can be better stabilized by delocalization of the nitrogen lone pairs (LP) into the empty p atomic orbitals (AO) of the P+ ion than the Sb+ center in hypothetical [ClP(μ-NTer)Sb]+ due to the relative orbital mismatch for the donation of an N lone pair into a 3p (P) or 5p (Sb) acceptor orbital in accordance with NBO analysis data. This formal p-LP(N) → p-AO(P) hyperconjugation accounts for a significant π-bond character along the N–P–N unit (see below). Thus both P–N distances are rather short and display a double bond character (P1–N2 1.625(3), P1–N1 1.643(4) Å, cf. ∑rcov(N–P) 1.82 Å),38,49 while the Sb–N distances correspond well with single bonds (Sb1–N1 2.136(3), Sb1–N2 2.171(3), ∑rcov(N–Sb) 2.11 Å).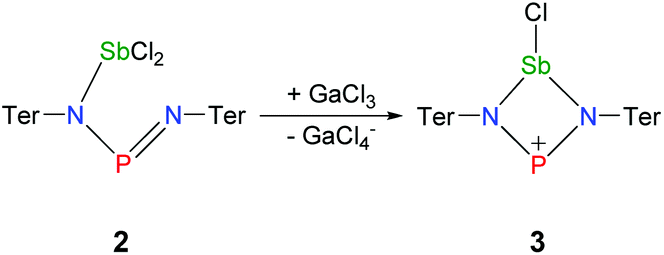 | ||
| Fig. 7 Ring closure in 2 by halide abstraction affording 3. | ||
Reduction of 2
Reduction of the yellow dichlorostibane 2 was studied using different reducing agents such as Mg in thf (Fig. 8 and 9) and KC8 (Fig. 10 and 11) in benzene at ambient temperature. Both reactions are rather slow and need ambient temperature. Treatment of 2 with magnesium turnings in thf led to a species, which was first characterized by a singlet 31P NMR resonance at 351 ppm (4, Fig. 9) indicating a di-coordinated phosphorus atom. Interestingly, only a minuscule change of the yellow colour was observed. Single crystal X-ray structure elucidation unequivocally revealed the presence of a magnesium salt of the bis(terphenylimino)-phosphide anion (4). Remarkably, there is quite a significant difference in the 31P NMR shifts when the cation is changed from potassium as in 1 (K[(TerN)2P], δ(31P) = 322–324 ppm for different solvates) to magnesium in 4 (MgCl[(TerN)2P]·thf, δ(31P) = 351 ppm) or lithium (Li[(TerN)2P]: 350 ppm, cf. ESI 4.1.‡). This effect can be rationalized by consideration of the hardness (in the sense of the hard-and-soft-acid and base concept) and different cation radii (Li+: 0.73, Mg2+: 0.71 and K+: 1.51 Å for the coordination number four). Li+ and Mg(Cl)+ feature a small but very similar ion radius and thus a stronger downfield shift. The K+ ion is considerably larger, hence the cation–anion separation is larger and the charge transfer is of lesser extent as confirmed by NBO analyses (NBO charge K 0.90675; Li 0.82680; second order perturbation theory analysis, maximal contribution: N–K 3, N–Li 13 kcal mol−1).40 From these data it can be concluded, that strongly interacting ion pairs (with a larger charge transfer) display a 31P NMR shift for [(TerN)2P]− around 350 ppm, while for weakly interacting ion pairs this value is shifted to higher field (320–300 ppm).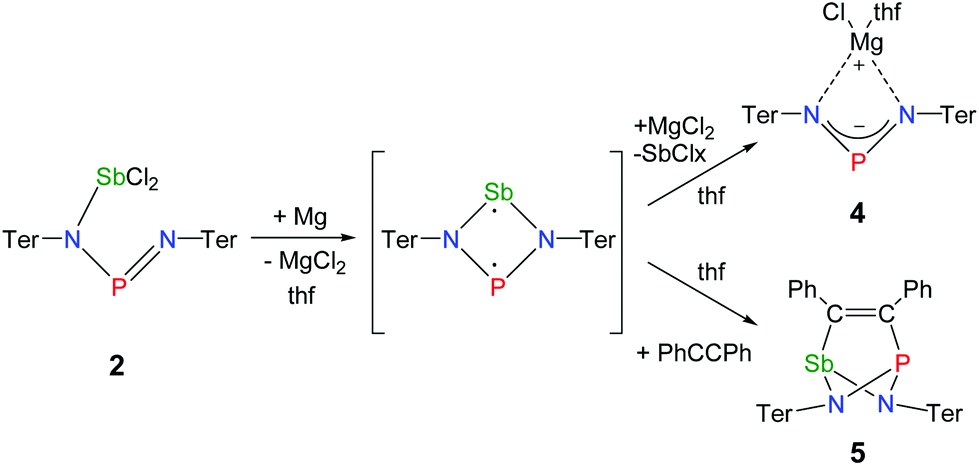 | ||
| Fig. 8 Reduction of 2 with magnesium and trapping of the transient biradicaloid. | ||
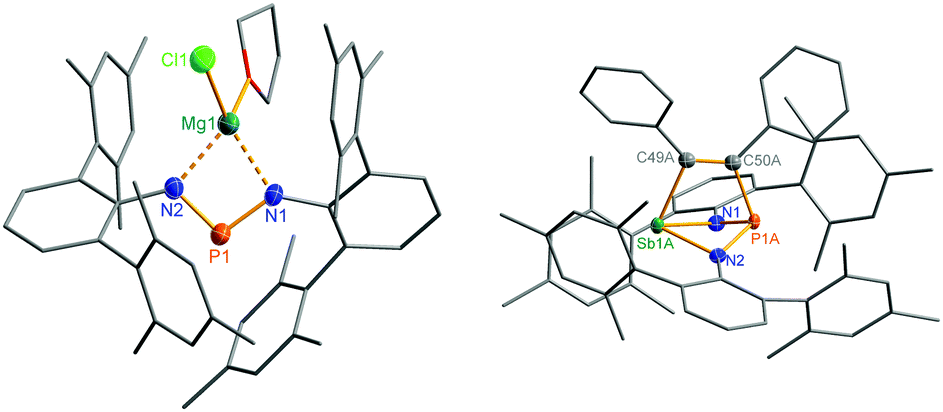 | ||
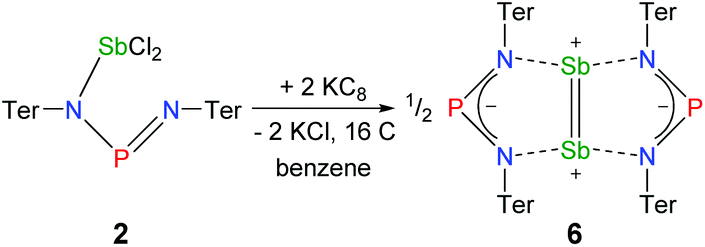
| Fig. 9 Molecular structures of 4 and 5. Thermal ellipsoids are drawn at 50% probability (173 K). Selected bond lengths [Å] and angles [°]: 4: P1–N2 1.607(2), P1–N1 1.615(2), Cl1–Mg1 2.269(2), Mg1–N1 2.102(2), Mg1–N2 2.088(2), N2–P1–N1 98.3(2). 5: Sb1A–C49A 2.228(8), Sb1A–P1A 2.804(2), P1A–C50A 1.947(8), C49A–C50A 1.337(6), C50A–C49A–Sb1A 107.7(4), C49A–C50A–P1A 113.3(4). | ||
 | ||
| Fig. 10 Reduction of 2 in a non-polar solvent affords 6. | ||
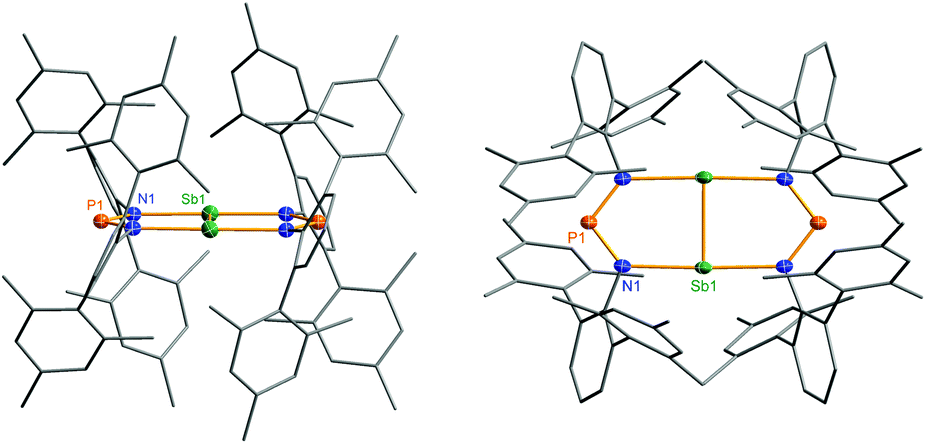 | ||
| Fig. 11 Molecular structure of 6 (left view: along the Sb–Sb bond, right: topview). Thermal ellipsoids are drawn at 50% probability (173 K). Selected bond lengths [Å] and angles [°]: 6: Sb1–Sb1′ 2.6438(4), Sb1–N1 2.372(2), P1–N1 1.613(2), N1–Sb1–N1′′ 177.81(9), N1–Sb1–Sb1′ 88.91(4), N1–P1–N1′′′ 104.6(2). | ||
To prove the intermediate formation of a biradicaloid species [P(μ-NTer)2Sb] as postulated in Fig. 8, trapping experiments were carried out. Indeed, trapping of the intermediately formed biradicaloid [P(μ-NTer)2Sb] was possible by addition of a suitable alkyne, e.g. diphenylacetylene, affording the [2.1.1]bicyclic species 5 (Fig. 9 right) in good yield (40%) as a yellow crystalline substance. The 31P and 13C NMR data as well as the single crystal X-ray data (Fig. 9 right) confirmed the structure of 5 (δ(31P) = 219.0, δ(13C) = 183.95, 181.84 ppm) in accord with the experimental data observed for [(TerNPCPh)2] (δ(31P) = 246.2, δ(13C) = 171.09 ppm).50 Interestingly, here the resonances for the bridging C atoms of the former alkyne were observed as doublets due to coupling with only one P nucleus (2JC49–P = 6.6, 1JC50–P = 54 Hz, cf. doublet of doublets, JCP = 50, 8.5 Hz in [(TerNPCPh)2]). The bicyclic trapping product 5 features a strained and distorted [2.1.1]heterobicycle as the central structural motif. The Sb–P distance is considerably longer than expected for a single bond (Sb1–P1 2.804(2), ∑rcov(N–Sb) 2.51 Å, Fig. 9). Both the Sb–C and the P–C bonds are elongated with respect to the sum of covalent radii (Sb1–C49 2.228(8), ∑rcov(Sb–C) 2.15; P1–C50 1.947(8), ∑rcov(P–C) 1.86 Å). In contrast, the C–C distance clearly corresponds to a double bond (C49–C50 1.337(6), ∑rcov(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) 1.34 Å) manifesting the addition of the triple bond of diphenylacetylene in 5.
C) 1.34 Å) manifesting the addition of the triple bond of diphenylacetylene in 5.
The solvent clearly influences the reduction process, since the obtained product 4 contains coordinated solvent molecules. To avoid the formation of decomposition product 4, the reduction was carried out with KC8 in benzene, which is a less coordinating solvent (Fig. 10). After addition of two equivalents of KC8 to a solution of 2 in benzene the colour changed to dark green after five minutes often indicating the formation of a distibene intermediate.51–58 Finally, within the course of three hours the color of the reaction mixture turned orange. After filtration and concentration orange block-shaped crystals could be isolated in moderate yield (22%). Surprisingly, instead of the expected biradicaloid [P(μ-NTer)2Sb], single crystal X-ray analysis revealed the formation of an eight-membered heterocycle of the type [Sb2-{μ-(TerN)2P}2] (6, δ(31P) 325.7 ppm, Fig. 11) with a strong Sb–Sb bond. Presumably, the green color of the reaction mixture at the beginning arises from the intermediate formation of blue-colored biradicoid [P(μ-NTer)2Sb] (cf. [P(μ-NTer)2As] is violet) which isomerizes to give orange [Sb2-{μ-(TerN)2P}2] 6, in accord with TD-DFT computation of the biradicaloid (TD = time dependent, see the ESI‡).40 In contrast to biradicoid [E(μ-NTer)2E] (E = P, As), which does not dimerize, biradicaloid [P(μ-NTer)2Sb] dimerizes along the Sb atoms to give 6 rather than forming a E4N4 cage compound as illustrated in Fig. 1 if smaller substituents are utilized. The result is that compound 6 is devoid of biradicaloid character. Notably, for the phenyl substituted model compound 6M, all possible cage compounds are thermodynamically more favoured compared to the eight-membered heterocyclic isomer (ca. 35–79 kJ mol−1, see the ESI‡). This situation changes dramatically when the terphenyl substituted experimentally observed compound 6 is considered. In this case, the cage compounds do not represent a minimum on the PES anymore. Obviously, the folding about the Sb–Sb axis, which would lead to the formation of α-cage compound 6, is hindered for steric reasons. Bulkier substituents may cause stabilization of the monomeric stiba-phospha-diazanediyl and prevent dimerization. It is worth mentioning that in an attempt to obtain the putative intermediate [(Ter2N2PSbCl)2] by reduction with one equivalent of KC8, only the unreacted starting material and 6 were isolated.55
Structure and bonding of 6
Heterocycle 6 crystallizes in the orthorhombic space group Pnnn with only one quarter of the compound in the asymmetric unit (Fig. 11). The most prominent structural feature is the highly symmetric, planar eight-membered N4P2Sb2 heterocycle featuring a very short Sb–Sb bond length of 2.6438(4) Å (cf. Tbt2Sb2 2.642(1), Ter2Sb2 2.6558(5), [Ar*N(SiiPr3)]2Sb2 2.7104(5), Ar* = 2,6-bis(diphenylmethyl)-4-isopropyl-phenyl)52,55–60 displaying a considerable degree of π-bonding (cf. ∑rcov(Sb–Sb) = 2.8 vs. ∑rcov(SbSb) = 2.66 Å).59 Hence, the eight-membered heterocycle can also be considered as two condensed five-membered rings. The Sb–N bond lengths in 6 amount to 2.372(2) Å, which is considerably larger than the sum of covalent radii (∑rcov(Sb–N) = 2.11 Å).59 Moreover, the short P–N distances of 1.613(2) Å are in the typical range of PN double bonds (∑rcov(PN) = 1.62 Å).59 Therefore, it can be deduced that the symmetrically bridging NPN ligand retains its anionic nature in the bridging mode, as indicated by these phosphorus–nitrogen bond lengths, which is similar to analogous structures with μ-N,N′ bridging complexes (cf. averaged P–N values 1.601(2) in 1, 1.611 Å in 4). The molecular structure of heterocycle 6 is reminiscent to the (NCN)2P2 scaffold of [P2{μ-(MesN)2CNMes}2].42,61 However, unlike the planar N4P2Sb2 bicyclic fragment in 6, the (NCN)2P2 scaffold has a puckered arrangement of the two five-membered P2N2C rings, where the two planes are 100.4° to each other (Fig. 4).42,61 Additionally, the molecular geometry about the phosphorus atoms is trigonal pyramidal and the P–P distances are in the range of typical P–P single bonds. Recently, Jones et al. reported on base-stabilized diarsenes [As2-{μ-(ArN)2-CR}2] (Ar = 2,6-diisopropyl-phenyl, R = N(cHex)2, N(iPr)2, tBu)43 displaying also a planar structure of the central structural motif along with a AsAs double bond similar to the situation found in 6 (Fig. 10). It is interesting to note that the isovalence-electronic triazenide species, [N(μ-NTer)2Sb],62 exists as a monomeric compound and does not dimerize to form the distibenium bicyclic species.
According to DFT calculations,40 compound 6 possesses a closed shell singlet ground state, hence it does not exhibit the typical high reactivity of biradicaloids, e.g. it does not react with small molecules as alkynes, phosphaalkynes, isonitriles or diphenyldiazene. In the 31P NMR spectrum, a singlet resonance at 325.7 ppm is observed for 6, which is very similar to the value found for the “free” solvated [(TerN)2P]− ion as in the potassium salt 1 (322–324 ppm, vide supra), also indicating the ionic nature of the interaction between the [(TerN)2P]− and [Sb2]2+ moieties. To gain further insight into the bonding situation of 6, MO (MO = molecular orbital), NBO and ELF (electron localization function)63–65 computations were carried out. Analysis of the frontier orbitals supports the existence of a highly localized SbSb double bond, as illustrated by the HOMO−1 (Fig. 12, HOMO−1 π bond, HOMO−2 σ bond, HOMO = highest occupied molecular orbital). The LUMO (lowest unoccupied molecular orbital) represents the antibonding counterpart, thus a π bond order of one can be considered. While the HOMO is NPN ligand centered, HOMO−12 features large coefficients for the lone pairs at the Sb atoms, which possess considerable s character.
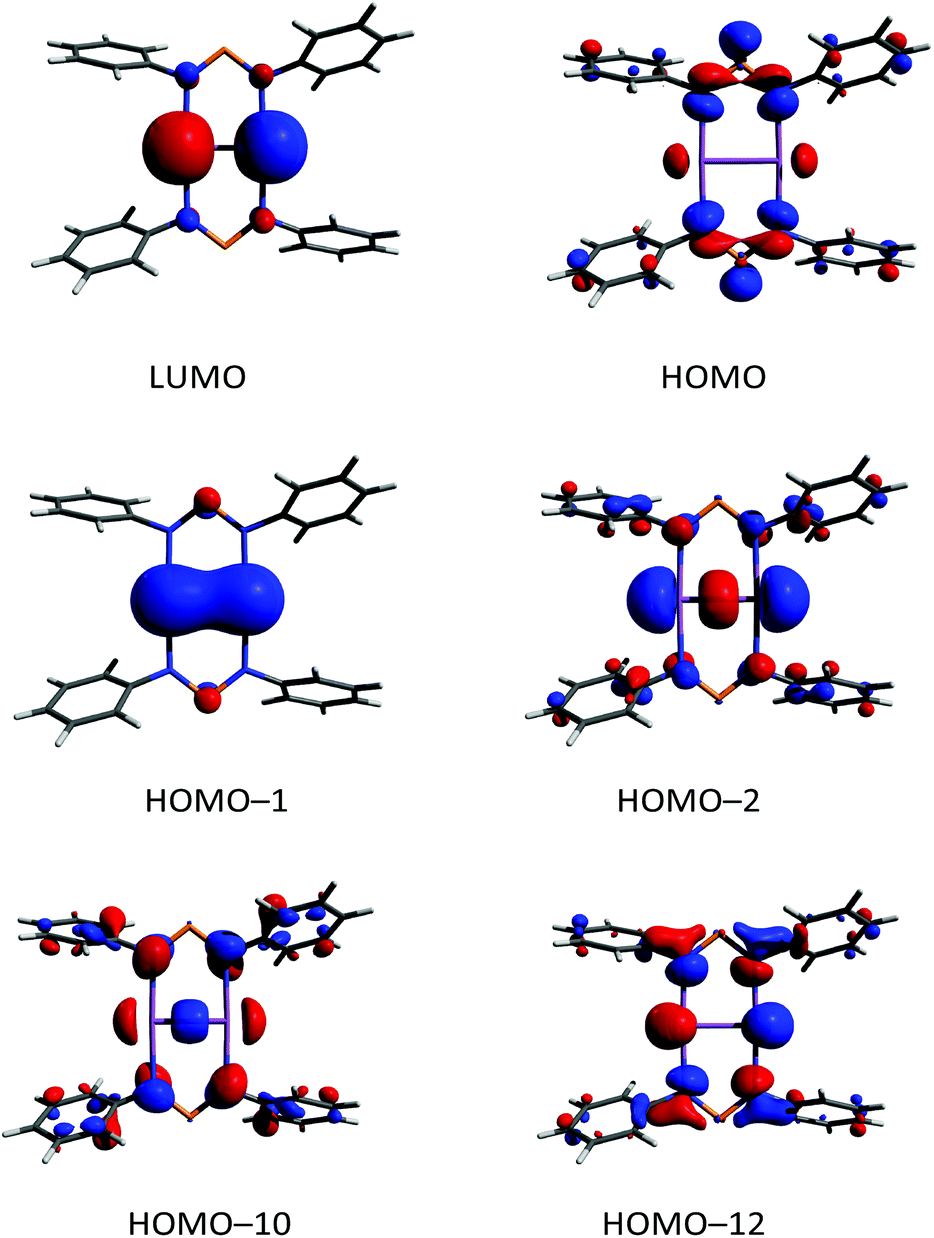 | ||
| Fig. 12 Selected Kohn–Sham orbitals of 6. | ||
In accord with this MO picture, NBO analyses find as the best Lewis representation a [Sb2]2+ ion bearing a double bond that interacts with two [(TerN)2P]− ions (Fig. 10). For the SbSb double bond, there are two contributions, a σ and a π bond, both being mainly formed by interactions of p atomic orbitals located at both Sb centres. Additionally, a lone pair predominantly carrying s-character is located on both Sb atoms. However, there are a strong donor–acceptor interactions (LP(N) → p-AO(Sb) hyperconjugation) between the anionic NPN ligands and the [Sb2]2+ cation, accompanied by a considerable charge transfer. The computed NLMO/NPA bond (NLMO = natural molecular orbital, NPA natural population analysis) indices amount to 0.99 for the Sb–Sb σ-bond and 0.94 for the π-bond, while only a value of 0.17 was found for each Sb–N bond. The total valency for each antimony was estimated to be 2.29 in accord with the picture of a base stabilized [Sb2]2+ ion. The NBO data are in good agreement with the values reported by Jones et al. for the base-stabilized diarsene [As2-{μ-(ArN)2-CR}2].43 However, the electronic stabilization invoked by the amidinate or guanidate ligand was not sufficient to enable the preparation of the corresponding distibene. Base stabilized antimony cations were the focus of recent research.66 For instance, stibinostibonium ions have been published by Breunig et al. ([Me3Sb–SbMe2]2+)67 and our group ([(Me3Sb)2SbMe]2+)68,69 as well as a series of different interpnictogen Sb cations by Burford et al. (e.g. [Sb4(PMe3)4]4+).66,70–73
The ELF of 6 also indicates the presence of a double bond as depicted in Fig. 13.63–65,74–76 In 6, the SbSb double bond domain is characterized by a dumb-bell shaped region of localized electrons (ELF = 0.75) in accord with the description of a classical double bond in a planar environment (e.g. in ethylene C2H4) and in contrast to the situation of non-classical double bonds (slipped double bonds) usually found between heavy main group elements.63,74 However, as pointed out by Grützmacher and Fässler dipnictogens of the type REER (E = N–Bi) fall within the category of classical double bonds displaying a strong resemblance to the situation in ethylene.74
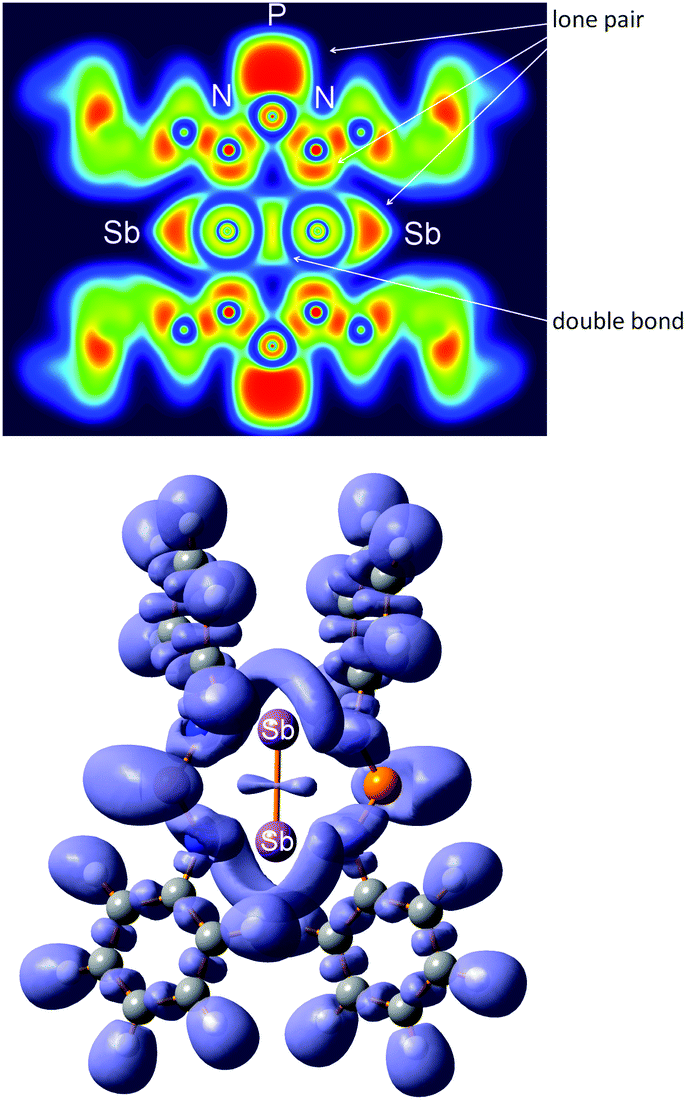 | ||
| Fig. 13 Top: two-dimensional (2D) cross section through the molecule in 6. Bottom: 3D representation of the ELF at 0.75. | ||
Finally, we want to address the question why dimerization occurred via Sb–Sb linkage and ring opening in the biradicaloid intermediate [P(μ-NTer)2Sb] rather than cage formation (vide supra). For this reason, we computed the biradical character β (β = c22/(c12 + c22) utilizing the method reported by Miliordios et al.77 The coefficients c1 and c2 were obtained from the complete active space self-consistent field (CASSCF(2,2)) level of theory. According to these computations, the biradical character of [P(μ-NTer)2Sb] amounts to only 8%, which is rather small for dipnictadiazanediyls (cf. 25–33% for [E(μ-NTer)]2, E = P, As, cf. ESI 4.4.‡).62 Nevertheless, biradicaloid reactivity was experimentally observed for [P(μ-NTer)2Sb] (“monomeric” 6). In contrast, isovalence-electronic [N(μ-NTer)2E] (E = P, As, Sb) do not activate small molecules, e.g. alkynes, to form [2.1.1]bicycles, but possess similar β values compared to [P(μ-NTer)2Sb] ([N(μ-NTer)2E], β: E = P 15%, As 10%, Sb 6%, Bi 0%). Thus the success of the trapping reaction of [P(μ-NTer)2Sb] with alkynes to form [2.1.1]bicycle 5 was unexpected (see Fig. 9), as well as the formal dimerization to 6 if no trapping reagent was available. We had expected a kinetically stabilized [P(μ-NTer)2Sb] biradical as it was the case for [E(μ-NTer)2E] (E = P, As) or [N(μ-NTer)2E] (E = P–Sb). However, compared to the lighter congeners [P(μ-NTer)2Sb] features rather long N–Sb bonds and acute N–Sb–N angles due to predominantly ionic bonding thus providing less steric protection around the antimony and allowing dimerization. Interestingly, for 6 the computed biradical character amounts to zero and therefore, a conventional closed-shell species is anticipated. This confirms the observation, that no activation of small molecules bearing multiple bonds was achieved, even though steric congestion contributes to the inertness of 6 as well.
Conclusions
In conclusion, the initially targeted biradicaloid [P(μ-NTer)2Sb] remains elusive, its existence as transient species, however, could be corroborated by trapping with diphenylacetylene forming a [2.1.1]bicycle. In the absence of a trapping reagent, [P(μ-NTer)2Sb] dimerizes via Sb–Sb double bond formation affording a novel planar eight-membered N4P2Sb2 heterocycle (6). Due to the highly polar Sb–N bond situation, the formal dimer 6 might also be regarded as a donor stabilized [SbSb]2+ ion in accord with MO, NBO and ELF considerations.
Experimental section
All manipulations were carried out under oxygen- and moisture-free conditions under argon using standard Schlenk or drybox techniques. The preparation of 1 was reported earlier.38 Full experimental and computational data are available in the ESI.‡Synthesis of 2
188 mg of K[(TerN)2P] (0.259 mmol) were dissolved in 5 ml of toluene. To the yellow solution, a solution of 60 mg SbCl3 (0.263 mml) in 3 ml toluene was added. The solution became turbid after a few minutes and was stirred for 3 hours to ensure completion of the reaction. The solution was filtered over a sinter padded with kieselguhr (Celite, G4) and the residue was washed with further 3 ml of toluene. The filtrate was concentrated to incipient crystallization (1 ml) and left undisturbed overnight at 4 °C, resulting in the deposition of yellow crystals. The mother liquor was removed via syringe and the crystals were dried in vacuo, yielding 163 mg (0.186 mmol, 72%) of the product.Mp: 215 °C (dec.). EA for C48H50N2PSbCl2 found (calc.): C 65.03 (65.62), H 5.78 (5.74), N 3.26 (3.19). 1H NMR (298 K, C6D6, 250.1 MHz): 2.08 (s, 24 H, o-CH3), 2.24 (s, 12 H, p-CH3), 6.81 (s, 8 H, m-CHMes), 6.83–6.91 (m, 6 H, CH). 31P NMR (298 K, C6D6, 121.5 MHz): 331.5 (br s).
Synthesis of 3
To a solution of [Ter2N2PSbCl2] (191 mg, 0.217 mmol) in dichloromethane (4 ml), a solution of GaCl3 (39 mg, 0.221 mmol) is added dropwise at −80 °C. The initially yellow solution immediately turns red and is stirred for further 15 minutes at the same temperature before being warmed to 20 °C. The solution is then concentrated until crystallization commences (0.5 ml) and left undisturbed overnight, which leads to the formation of red needle-shaped crystals (168 mg, 0.159 mmol, 73%). Crystals suitable for X-ray structure elucidation were obtained by repeated recrystallization from dichloromethane between 25 °C and 4 °C.Mp: 258 °C (dec.). EA for C48H50N2PSbGaCl5 found (calc.): C 54.53 (54.66), H 5.48 (4.78), N 2.93 (2.66). 1H NMR (298 K, CD2Cl2, 250.1 MHz): 1.90 (s, 24 H, o-CH3), 2.44 (s, 12 H, p-CH3), 7.03 (d, 3JHH = 7.7 Hz, 4 H, m-CH), 7.06 (s, 8 H, m-CHMes), 7.28 (t, 3JHH = 7.7 Hz, 2 H, p-CH). 31P NMR (298 K, CD2Cl2, 121.5 MHz): 374.0 (s).
Synthesis of 4
[Ter2N2PSbCl2] (200 mg, 0.228 mmol) and magnesium turnings (80 mg) were combined in a flask. To this mixture, 10 ml THF was added and the suspension was stirred overnight at ambient temperature with a glass stirring bar. Out of the initially yellow solution a black precipitate was formed. Volatiles were removed in vacuo and the residue was extracted with 10 ml benzene and washed with an additional 3 ml of benzene. The combined filtrate was concentrated to incipient crystallization (approx. 2 ml) and left undisturbed overnight, resulting in the deposition of light yellow crystals. The supernatant was removed via syringe and the crystals were dried in vacuo (130 mg, 0.159 mmol, 70%).Mp: 110 °C (dec.). EA for C52H58N2PMgClO found (calc.): C 75.92 (76.37), H 7.36 (7.15), N 3.14 (3.43). 1H NMR (298 K, C6D6, 250.1 MHz): 1.21 (m, 4 H, OCH2CH2), 1.98 (s, 12 H, o-CH3), 2.03 (s, 12 H, o-CH3), 2.23 (s, 12 H, p-CH3), 3.21 (m, 4 H, OCH2CH2), 6.80–6.89 (m, 6 H, m-/p-CH), 6.87 (s, 8 H, m-CHMes). 31P NMR (298 K, C6D6, 121.5 MHz): 351.8 (s).
Synthesis of 5
[Ter2N2PSbCl2] (180 mg, 0.205 mmol), diphenylacetylene (53 mg) and magnesium turnings (80 mg) were combined in a flask. To the mixture, 10 ml thf was added. After stirring with a glass-covered stirring bar, the initially yellow solution turned orange and a black precipitate formed. Volatiles were removed in vacuo and the residue was extracted with 5 ml benzene and then washed with another portion of 3 ml of benzene. The extract was then concentrated to incipient crystallization and left undisturbed overnight, affording orange crystals. The mother liquor was removed via syringe and the crystals were dried in vacuo (82 mg, 0.083 mmol, 40%).Mp. 231 °C (dec.). EA found (calc.): C 68.93 (69.39), H 6.00 (5.64), N 2.86 (2.61). 1H NMR (298 K, C6D6, 250.1 MHz): 2.05 (s, 12 H, o-CH3), 2.09 (s, 12 H, o-CH3), 2.20 (s, 12 H, p-CH3), 6.54 (s, 8 H, CHMes), 6.68 (s, 6 H, m-/p-CH), 6.82–7.23 (m, 10 H, CHPh). 31P NMR (298 K, C6D6, 121.5 MHz): 219.0 (s).
Synthesis of 6
[Ter2N2PSbCl2] (215 mg, 0.245 mmol) was dissolved in 5 ml of benzene while stirring with a glass stirring bar. To the solution, KC8 (70 mg, 0.518 mmol) was added. The solution adopted a dark green colour after 5 minutes. To ensure completion of the reaction, the suspension was stirred for 3 hours at ambient temperature. The suspension was filtered over a sinter padded with kieselguhr (Celite) and the residue was washed with another portion of 2 ml of benzene. The combined filtrate was concentrated to incipient crystallization (approx. 1 ml) and left undisturbed overnight. Orange block-shaped crystals were obtained. The supernatant was removed via syringe and the crystals were dried in vacuo (43 mg, 0.027 mmol, 22%).Mp: 110 °C (dec.). EA found (calc.): C 70.80 (70.98), H 6.98 (6.76), N 3.48 (3.45). 1H NMR (298 K, C6D6, 250.1 MHz): 2.03 (s, 48 H, o-CH3), 2.25 (s, 24 H, p-CH3), 6.72 (s, 16 H, m-CHMes), 6.80–6.93 (m, 12 H, m-/p-CH). 31P NMR (298 K, C6D6, 121.5 MHz): 326.0 (s).
Acknowledgements
The authors thank the DFG (SCHU 1170/11-1) for financial support. M.Sc. Jonas Bresien is gratefully acknowledged for setting up and maintaining Gaussian and NBO software on the cluster computer. Dr Dirk Michalik is acknowledged for recording temperature-variable NMR spectra. A.H. thanks the GDCh for financial support.Notes and references
- A. Michaelis and G. Schroeter, Chem. Ber., 1894, 27, 490–497 CrossRef PubMed.
- M. S. Balakrishna, D. J. Eisler and T. Chivers, Chem. Soc. Rev., 2007, 36, 650–664 RSC.
- D. A. DuBois, E. N. Duesler and R. T. Paine, Organometallics, 1983, 2, 1903–1905 CrossRef CAS.
- D. A. DuBois, E. N. Duesler and R. T. Paine, Organometallics, 1984, 3, 1913–1915 CrossRef CAS.
- D. A. DuBois, E. N. Duesler and R. T. Paine, Inorg. Chem., 1985, 24, 3–5 CrossRef CAS.
- D. A. DuBois, E. N. Duesler and R. T. Paine, J. Chem. Soc., Chem. Commun., 1984, 488–489 RSC.
- H. Bladt, S. G. Calera, J. Goodman, R. J. Less, V. Naseri, A. Steiner and D. S. Wright, Chem. Commun., 2009, 6637–6639 RSC.
- T. Beweries, R. Kuzora, U. Rosenthal, A. Schulz and A. Villinger, Angew. Chem., 2011, 123, 9136–9140 ( Angew. Chem., Int. Ed. , 2011 , 50 , 8974–8978 ) CrossRef PubMed.
- M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed.
- F. Breher, Coord. Chem. Rev., 2007, 251, 1007–1043 CrossRef CAS PubMed.
- G. He, O. Shynkaruk, M. W. Lui and E. Rivard, Chem. Rev., 2014, 114, 7815–7880 CrossRef CAS PubMed.
- E. Niecke, A. Fuchs, F. Baumeister, M. Nieger and W. W. Schoeller, Angew. Chem., 1995, 107, 640–642 ( Angew. Chem., Int. Ed. , 1995 , 34 , 555–557 ) CrossRef PubMed.
- M. Sebastian, A. Hoskin, M. Nieger, L. Nyulaszi and E. Niecke, Angew. Chem., 2005, 117, 1429–1432 ( Angew. Chem., Int. Ed. , 1995 , 44 , 1405–1408 ) CrossRef PubMed.
- O. Schmidt, A. Fuchs, D. Gudat, M. Nieger, W. Hoffbauer, E. Niecke and W. W. Schoeller, Angew. Chem., 1998, 110, 995–998 ( Angew. Chem., Int. Ed. , 1998 , 37 , 949–952 ) CrossRef.
- E. Niecke, A. Fuchs, M. Nieger, O. Schmidt and W. W. Schoeller, Angew. Chem., 1999, 111, 3216–3219 ( Angew. Chem., Int. Ed. , 1999 , 38 , 3028–3031 ) CrossRef.
- M. Blättner, M. Nieger, A. Ruban, W. W. Schoeller and E. Niecke, Angew. Chem., 2000, 112, 2876–2879 ( Angew. Chem., Int. Ed. , 2000 , 39 , 2768–2771 ) CrossRef.
- M. Sebastian, M. Nieger, D. Szieberth, L. Nyulászi and E. Niecke, Angew. Chem., 2004, 116, 647–651 ( Angew. Chem., Int. Ed. , 2004 , 43 , 637–641 ) CrossRef PubMed.
- S. Ito, M. Kikuchi, H. Sugiyama and M. Yoshifuji, J. Organomet. Chem., 2007, 692, 2761–2767 CrossRef CAS PubMed.
- S. Ito, J. Miura, N. Morita, M. Yoshifuji and A. J. Arduengo, C. R. Chim., 2010, 13, 1180–1184 CrossRef CAS PubMed.
- H. Sugiyama, S. Ito and M. Yoshifuji, Chem. – Eur. J., 2004, 10, 2700–2706 CrossRef CAS PubMed.
- M. Yoshifuji, A. J. Arduengo III, T. A. Konovalova, L. D. Kispert, M. Kikuchi and S. Ito, Chem. Lett., 2006, 35, 1136–1137 CrossRef CAS.
- S. Ito, M. Kikuchi, J. Miura, N. Morita and M. Yoshifuji, J. Phys. Org. Chem., 2012, 25, 733–737 CrossRef CAS PubMed.
- H. Sugiyama, S. Ito and M. Yoshifuji, Angew. Chem., 2003, 115, 3932–3934 ( Angew. Chem., Int. Ed. , 2003 , 42 , 3802–3804 ) CrossRef PubMed.
- V. Gandon, J.-B. Bourg, F. S. Tham, W. W. Schoeller and G. Bertrand, Angew. Chem., 2007, 120, 161–165 ( Angew. Chem., Int. Ed. , 2008 , 47 , 155–159 ) CrossRef PubMed.
- J.-B. Bourg, A. Rodriguez, D. Scheschkewitz, H. Gornitzka, D. Bourissou and G. Bertrand, Angew. Chem., 2007, 120, 5843–5847 ( Angew. Chem., Int. Ed. , 2007 , 46 , 5741–5745 ) CrossRef PubMed.
- H. Amii, L. Vranicar, H. Gornitzka, D. Bourissou and G. Bertrand, J. Am. Chem. Soc., 2004, 126, 1344–1345 CrossRef CAS PubMed.
- G. Fuks, N. Saffon, L. Maron, G. Bertrand and D. Bourissou, J. Am. Chem. Soc., 2009, 131, 13681–13689 CrossRef CAS PubMed.
- A. F. Richards, M. Brynda, M. M. Olmstead and P. P. Power, Organometallics, 2004, 23, 2841–2844 CrossRef CAS.
- C. Schenk, A. Kracke, K. Fink, A. Kubas, W. Klopper, M. Neumaier, H. Schnöckel and A. Schnepf, J. Am. Chem. Soc., 2011, 133, 2518–2524 CrossRef CAS PubMed.
- C. Schrenk, A. Kubas, K. Fink and A. Schnepf, Angew. Chem., 2011, 123, 7411–7415 ( Angew. Chem., Int. Ed. , 2011 , 50 , 7273–7277 ) CrossRef PubMed.
- X. Wang, Y. Peng, M. M. Olmstead, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2009, 131, 14164–14165 CrossRef CAS PubMed.
- P. Henke, T. Pankewitz, W. Klopper, F. Breher and H. Schnöckel, Angew. Chem., 2009, 121, 8285–8290 ( Angew. Chem., Int. Ed. , 2009 , 48 , 8141–8145 ) CrossRef PubMed.
- H. Cox, P. B. Hitchcock, M. F. Lappert and L. J.-M. Pierssens, Angew. Chem., 2004, 116, 4600–4604 ( Angew. Chem., Int. Ed. , 2004 , 43 , 4500–4504 ) CrossRef PubMed.
- K. Takeuchi, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2011, 133, 12478–12481 CrossRef CAS PubMed.
- S. H. Zhang, H. W. Xi, K. H. Lim, Q. Meng, M. B. Huang and C. W. So, Chem. – Eur. J., 2012, 18, 4258–4263 CrossRef CAS PubMed.
- H. Sugiyama, S. Ito and M. Yoshifuji, Chem. – Eur. J., 2004, 10, 2700–2706 CrossRef CAS PubMed.
- P. P. Power, Chem. Rev., 2003, 103, 789–810 CrossRef CAS PubMed.
- A. Hinz, A. Schulz and A. Villinger, Angew. Chem., 2014, 127, 678–682 ( Angew. Chem., Int. Ed. , 2014 , 54 , 668–672 ) CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- Computations were carried out at the pbe1pbe level of theory utilizing a 6-31G(d,p) basis for H, C, N, P atoms and a relativistic pseudopotential was used for Sb: ECP46MDF 4 46.
- A. L. Brazeau, A. S. Nikouline and P. J. Ragogna, Chem. Commun., 2011, 47, 4817–4819 RSC.
- A. L. Brazeau, M. M. Hänninen, H. M. Tuononen, N. D. Jones and P. J. Ragogna, J. Am. Chem. Soc., 2012, 134, 5398–5414 CrossRef CAS PubMed.
- S. P. Green, C. Jones, G. Jin and A. Stasch, Inorg. Chem., 2007, 46, 8–10 CrossRef CAS PubMed.
- C. Ergezinger, F. Weller and K. Dehnicke, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1988, 43, 1119–1124 CAS.
- B. Lyhs, S. Schulz, U. Westphal, D. Bläser, R. Boese and M. Bolte, Eur. J. Inorg. Chem., 2009, 2009, 2247–2253 CrossRef PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- M. Lehmann, A. Schulz and A. Villinger, Angew. Chem., 2012, 124, 8211–8215 ( Angew. Chem., Int. Ed. , 2012 , 51 , 8087–8091 ) CrossRef PubMed.
- D. Michalik, A. Schulz, A. Villinger and N. Weding, Angew. Chem., 2008, 120, 6565–6568 ( Angew. Chem., Int. Ed. , 2008 , 47 , 6465–6468 ) CrossRef PubMed.
- A. Hinz, R. Kuzora, U. Rosenthal, A. Schulz and A. Villinger, Chem. – Eur. J., 2014, 20, 14659–14673 CrossRef CAS PubMed.
- T. Sasamori, Y. Arai, N. Takeda, R. Okazaki, Y. Furukawa, M. Kimura, S. Nagase and N. Tokitoh, Bull. Chem. Soc. Jpn., 2002, 75, 661–675 CrossRef CAS.
- N. Tokitoh, Y. Arai, T. Sasamori, R. Okazaki, S. Nagase, H. Uekusa and Y. Ohashi, J. Am. Chem. Soc., 1998, 120, 433–434 CrossRef CAS.
- T. Sasamori and N. Tokitoh, Dalton Trans., 2008, 1395–1408 RSC.
- T. Sasamori, N. Takeda and N. Tokitoh, J. Phys. Org. Chem., 2003, 16, 450–462 CrossRef CAS PubMed.
- B. Twamley, C. D. Sofield, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 1999, 121, 3357–3367 CrossRef CAS.
- M. Sakagami, T. Sasamori, H. Sakai, Y. Furukawa and N. Tokitoh, Bull. Chem. Soc. Jpn., 2013, 86, 1132–1143 CrossRef CAS.
- M. Sakagami, T. Sasamori, H. Sakai, Y. Furukawa and N. Tokitoh, Chem. – Asian J., 2013, 8, 690–693 CrossRef CAS PubMed.
- T. Sasamori, E. Mieda, N. Nagahora, K. Sato, D. Shiomi, T. Takui, Y. Hosoi, Y. Furukawa, N. Takagi, S. Nagase and N. Tokitoh, J. Am. Chem. Soc., 2006, 128, 12582–12588 CrossRef CAS PubMed.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- D. Dange, A. Davey, J. A. B. Abdalla, S. Aldridge and C. Jones, Chem. Commun., 2015, 51, 7128–7131 RSC.
- H. W. Roesky, H. Zamankhan, W. S. Sheldrick, A. H. Cowley and S. K. Mehrotra, Inorg. Chem., 1981, 20, 2910–2915 CrossRef CAS.
- A. Hinz, A. Schulz and A. Villinger, J. Am. Chem. Soc., 2015, 137, 3975–3980 CrossRef CAS PubMed.
- T. E. Fässler and A. Savin, Chemie unserer Zeit, 1997, 31, 110–120 CrossRef PubMed.
- A. Savin, O. Jepsen, J. Flad, O. K. Andersen, H. Preuss and H. G. von Schnering, Angew. Chem., 1992, 104, 186–188 ( Angew. Chem., Int. Ed. , 1992 , 31 , 187–188 ) CrossRef CAS PubMed.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397 CrossRef CAS PubMed.
- A. P. M. Robertson, P. A. Gray and N. Burford, Angew. Chem., 2014, 127, 6162–6182 ( Angew. Chem., Int. Ed. , 2014 , 53 , 6050–6069 ) CrossRef PubMed.
- H. Althaus, H. J. Breunig and E. Lork, Chem. Commun., 1999, 1971–1972 RSC.
- C. Hering, M. Lehmann, A. Schulz and A. Villinger, Inorg. Chem., 2012, 51, 8212–8224 CrossRef CAS PubMed.
- C. Hering, J. Rothe, A. Schulz and A. Villinger, Inorg. Chem., 2013, 52, 7781–7790 CrossRef CAS PubMed.
- S. S. Chitnis, Y. Y. Carpenter, N. Burford, R. McDonald and M. J. Ferguson, Angew. Chem., 2013, 125, 4963–4966 ( Angew. Chem., Int. Ed. , 2013 , 52 , 4863–4866 ) CrossRef PubMed.
- S. S. Chitnis, A. P. M. Robertson, N. Burford, J. J. Weigand and R. Fischer, Chem. Sci., 2015, 6, 2559–2574 RSC.
- E. Conrad, N. Burford, U. Werner-Zwanziger, R. McDonald and M. J. Ferguson, Chem. Commun., 2010, 46, 2465–2467 RSC.
- A. P. M. Robertson, N. Burford, R. McDonald and M. J. Ferguson, Angew. Chem., 2014, 126, 3548–3551 ( Angew. Chem., Int. Ed. , 2014 , 53 , 3480–3483 ) CrossRef PubMed.
- H. Grützmacher and T. Fässler, Chem. – Eur. J., 2000, 6, 2317–2325 CrossRef.
- A. Savin, R. Nesper, S. Wengert and T. E. Fässler, Angew. Chem., 1997, 125, 1892–1918 ( Angew. Chem., Int. Ed. , 1997 , 36 , 1808–1832 ) CrossRef PubMed.
- H.-J. Flad, F. Schautz, Y. Wang, M. Dolg and A. Savin, Eur. Phys. J. D, 1999, 6, 243–254 CrossRef CAS.
- E. Miliordos, K. Ruedenberg and S. S. Xantheas, Angew. Chem., 2013, 125, 5848–5851 ( Angew. Chem., Int. Ed. , 2013 , 52 , 5736–5739 ) CrossRef PubMed.
Footnotes |
| † This contribution is dedicated to Prof. Dr Ekkehart Hahn on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Additional experimental details, full characterization of all compounds and computational details. CCDC 1404259–1404264. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02711j |
| This journal is © The Royal Society of Chemistry 2016 |