
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon-based cores with polyglycerol shells – the importance of core flexibility for encapsulation of hydrophobic guests†
Maike C.
Lukowiak
a,
Benjamin
Ziem
a,
Katharina
Achazi
a,
Gesine
Gunkel-Grabole
a,
Chris S.
Popeney
a,
Bala N. S.
Thota
a,
Christoph
Böttcher
a,
Anke
Krueger
b,
Zhibin
Guan
c and
Rainer
Haag
*a
aInstitut für Chemie und Biochemie, Freie Universität Berlin, Takustr. 3, 14195 Berlin, Germany. E-mail: haag@chemie.fu-berlin.de
bInstitut für Organische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
cDepartment of Chemistry, University of California, 5042D Frederick Reines Hall, Irvine, California 92697-2025, USA
First published on 8th December 2014
Abstract
Two core–shell nanoparticles with polyglycerol shells and sp3 carbon cores with different flexibilities (soft dendritic polyethylene and hard nanodiamond) were synthesized, their encapsulation capacities were compared, and their ability to transport into tumor cells was investigated. The nanocarrier with a soft core was superior to the hard one.
The poor solubility of hydrophobic compounds in aqueous media strongly limits their application, especially in the case of newly developed chemotherapeutic drugs.1–3 Since polymeric nanocarriers may present a solution to this problem, polymer therapeutics have become very interesting for researchers.4–6 Amphiphilic polymers can assemble into supramolecular micelles or vesicles in aqueous solution, which enables their application as nanocarriers.7–10 In particular, unimolecular amphiphilic core–shell nanotransporters that resemble covalently bound micelles have been shown to be beneficial because of their higher stability under dilution conditions.11–15 Guests can be encapsulated within unimolecular core–shell micelles either in the core, in the shell, or at the interface of core and shell. The encapsulation mechanism, however, is not necessarily unimolecular but can be also based on aggregates of unimolecular micelles with the encapsulated guest.16–20 While the release of the guest remains the focus of many research projects,21–25 there is still poor knowledge of the factors that influence the encapsulation of guest molecules in nanocarriers.26,27 The earliest studies in this area were mostly conducted on the encapsulation of hydrophilic compounds in organic solvents, however, this knowledge is not necessarily transferable to the encapsulation of hydrophobic compounds in aqueous solutions.28–31 Hence, an investigation of structure–property relationships for transport capacities is very important for improving the design of the next carrier generations. This manuscript describes the synthesis of two core–shell nanoparticles that both have a polyglycerol (PG) shell and sp3 carbon cores, but with different core flexibilities. Further, their encapsulation capacity for hydrophobic compounds in aqueous solution is investigated and compared. The soft core was dendritic polyethylene (PE)32 and the hard core was nanodiamond (ND) (Fig. 1). The rigid, impenetrable nanodiamond core does not permit the penetration of the guest molecules into the sp3 diamond lattice structure. Nevertheless, effective guest encapsulation will be possible also with impenetrable cores if interfacial mechanisms predominate the encapsulation process. Thus, a comparison of these otherwise similar systems will provide insight into the fundamental processes involved in guest encapsulation in core–shell nanocarriers.
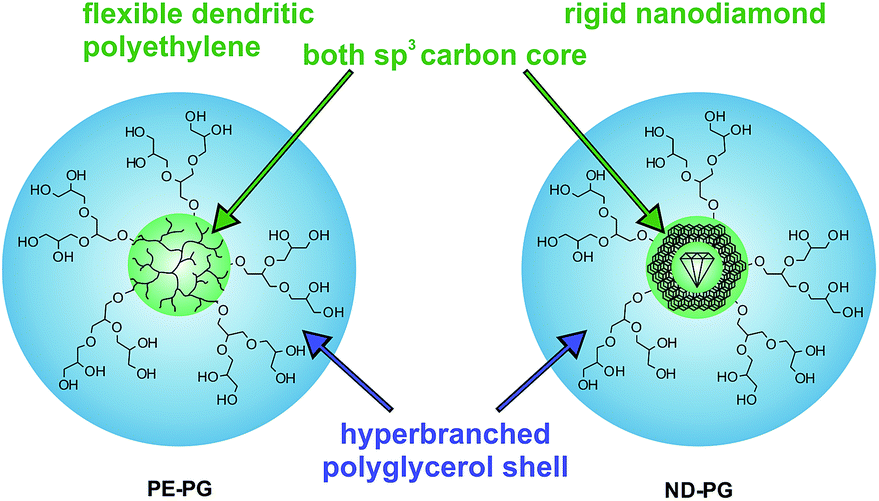 | ||
| Fig. 1 Schematic representation of core–shell nanoparticles PE-PG with a dendritic polyethylene core and ND-PG with a nanodiamond core both with hyperbranched polyglycerol shells. | ||
The synthesis of PE-PG core–shell copolymer was performed similarly to a previously reported method by tandem coordination, and ring-opening hyperbranched polymerization (Scheme S1†).14 A chain walking polymerization (CWP)32 of ethylene at low pressure (0.1 atm) with siloxy-functionalized comonomer and deprotection of the siloxy groups resulted in dendritic hydroxyl-functionalized polyethylene PE-OH. Unlike a previous approach, the copolymerization was performed with a methyl-substituted Brookhart's Pd(II) α-diimine CWP catalyst instead of the isopropyl-substituted version.14,33 This led to much lower molecular weight PE-OH with number-averaged molecular weight Mn of 26 kDa and polydispersity (Mw/Mn) of 1.7 as determined by gel permeation chromatography (GPC) coupled with a multi-angle laser light scattering (MALLS) detector in THF. The incorporation ratio of the hydroxyl-functionalized comonomer was 4 mol% based on 1H NMR, which corresponded to 30 calculated OH-groups per PE-OH copolymer. PE-PG core–shell copolymer was yielded after ring-opening polymerization of glycidol from the macroinitiator PE-OH and was purified by extensive dialysis to remove any unbound PG. PE-PG was water-soluble and had a much bigger number-averaged molecular weight Mn of 268 kDa, as was determined by GPC-MALLS in water (Fig. S1†). Based on 1H NMR (Fig. S2†), the molar ratio of glycerol (PG shell) to olefin (PE core) was approximately 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which corresponded to an average of 175 polymerized glycerol units per hydroxy group. The synthesis of ND-PG (Scheme S2†) was also performed in two steps by first oxidizing ND to NDox34 and subsequently grafting-from glycidol to build up the hyperbranched PG shell based on the reported procedures.35,36 The reaction products were characterized by IR, elemental analysis (EA), transmission electron microscopy (TEM), and NMR. Details about synthesis and characterization can be found in the ESI.† Both core–shell nanoparticles contained the same weight ratio of around 90% of PG as determined by thermogravimetric analysis (TGA) (Fig. 2a). This large amount of PG was necessary for good water solubility and dispersibility of the nanodiamond system. The sizes of the nanoparticles were investigated with dynamic light scattering (DLS) and cryo-TEM. Both systems showed tendencies to aggregate as seen in DLS in phosphate buffered saline (PBS) solution with average intensity-based particle diameters of 119 ± 3 nm for PE-PG and 159 ± 6 nm for ND-PG, respectively (Fig. 2b). Aggregates in the same size range could be observed in cryo-TEM (Fig. 2d), however, cryo-TEM revealed corresponding individual particle sizes in the range of 3–10 nm for PE-PG and 3–9 nm for ND-PG (Fig. 2c and d).
:1, which corresponded to an average of 175 polymerized glycerol units per hydroxy group. The synthesis of ND-PG (Scheme S2†) was also performed in two steps by first oxidizing ND to NDox34 and subsequently grafting-from glycidol to build up the hyperbranched PG shell based on the reported procedures.35,36 The reaction products were characterized by IR, elemental analysis (EA), transmission electron microscopy (TEM), and NMR. Details about synthesis and characterization can be found in the ESI.† Both core–shell nanoparticles contained the same weight ratio of around 90% of PG as determined by thermogravimetric analysis (TGA) (Fig. 2a). This large amount of PG was necessary for good water solubility and dispersibility of the nanodiamond system. The sizes of the nanoparticles were investigated with dynamic light scattering (DLS) and cryo-TEM. Both systems showed tendencies to aggregate as seen in DLS in phosphate buffered saline (PBS) solution with average intensity-based particle diameters of 119 ± 3 nm for PE-PG and 159 ± 6 nm for ND-PG, respectively (Fig. 2b). Aggregates in the same size range could be observed in cryo-TEM (Fig. 2d), however, cryo-TEM revealed corresponding individual particle sizes in the range of 3–10 nm for PE-PG and 3–9 nm for ND-PG (Fig. 2c and d).
 | ||
| Fig. 2 (a) Normalized TGA profiles of PE-PG (blue) and ND-PG (red). (b) Particle diameter distribution by intensity from DLS of PE-PG (blue) and ND-PG (red). (c) Cryo-TEM image of a vitrified aqueous solution of PE-PG. Scale bar: 20 nm. (d) Cryo-TEM image of a vitrified aqueous solution of ND-PG. Scale bar: 20 nm. | ||
With both core–shell nanoparticles PE-PG and ND-PG in hand, the next step was to examine their capability as nanocarriers for hydrophobic compounds in aqueous solution. Pyrene (PY) and Nile red (NR), which are both commonly used hydrophobic fluorescent dyes, were used as model compounds in this study. The encapsulation experiment of PY was performed by solid uptake method and vigorous stirring for 24 hours. After filtration, a part of the solution was lyophilized and redissolved in methanol to enable the calculation of the transport capacity based on the known molar extinction coefficient following Lambert–Beer law. For PE-PG a transport capacity of 0.40 ± 0.06 mg PY per g PE-PG was determined from the absorbance spectra (Fig. S10,† blue curve). In contrast, an uptake of pyrene was not detected for ND-PG in the absorbance spectra (Fig. S10,† red curve). Fluorescence spectra of pyrene (Fig. 3a) from the ND-PG solution also showed only a little intensity which was comparable with the intensity of the pyrene blank experiment solution. Thus, no transport capacity of PY for ND-PG higher than its natural water solubility was found. Based on the well-known sensitivity of PY fluorescence spectra, in particular on the intensity ratio (I3/I1) of the third emission band (385 nm) to the first emission band (374 nm), information about the dyes' local environment could be accessed.37 A I3/I1 ratio of 0.89 ± 0.01 was calculated for PY/PE-PG at a carrier concentration of 1 mg mL−1. In comparison, the reported I3/I1 ratio of PY in non-polar solvents like chloroform was 0.78, in toluene 0.90, or in n-hexane 1.65, and in polar solvents like water 0.63, in ethylene glycol 0.61, or in methanol 0.75.37 The latter two compositionally resemble an environment very similar to the PG shell. From these examples we can conclude that PY/PE-PG experienced a relatively more hydrophobic environment due to the dendritic PE core. In DLS measurements there was no appreciable difference in the size distribution of the aggregates for neither of the core–shell nanoparticle solutions after PY uptake experiment (Fig. 3b).
 | ||
| Fig. 3 (a) Fluorescence spectra of PY of PE-PG (blue), ND-PG (red) at concentrations of 1 mg mL−1, and PY blank (black). (b) Particle diameter distribution by intensity from DLS of unloaded PE-PG (blue continuous curve) and ND-PG (red continuous curve) and after encapsulation experiment with PY (dashed curves) at concentrations of 1 mg mL−1. | ||
Encapsulation experiments with the dye NR were performed following similar experimental details (ESI†). While the NR transport capacity for PE-PG was 0.58 ± 0.22 mg NR per g PE-PG, it was considerably smaller for ND-PG with only 0.07 ± 0.02 mg NR per g ND-PG (Fig. S11†). In the DLS measurement of the core–shell nanoparticle solutions with NR, the size for ND-PG/NR showed no appreciable change, while the size distribution for PE-PG/NR increased from 119 ± 3 nm before encapsulation to 146 ± 5 nm (Fig. S12†). For better comparison all transport capacities can be found in Table 1.
Furthermore, the cell uptake of the NR loaded nanoparticles was investigated. A549 lung tumor cells were incubated with loaded and unloaded nanocarriers as control for 4 hours. Fixed cells were visualized by confocal fluorescence microscopy. Further negative controls were conducted with just PBS, cell culture medium, and the blank experiment solution of NR without the presence of carrier. NR was used as a positive control and was initially dissolved in DMSO and diluted to the required concentrations with medium. Since PE-PG/NR had a higher transport capacity, a concentration dependent study could be performed for this nanotransporter. As expected due to the higher loading, the PE-PG core–shell copolymer showed much stronger NR fluorescence than ND-PG at the same nanocarrier concentration (Fig. 4a and b), but it was also possible to use PE-PG at higher dilutions. The NR fluorescence of A549 cells was quantified by flow cytometry, also after 4 h of incubation with NR loaded nanocarriers (Fig. 4c). The same trend was observed as in qualitative confocal fluorescence microscopy. In summary, the loading capacity and thus efficiency to transport the guest molecules into tumor cells of PE-PG with flexible core was better than for ND-PG with rigid core. Due to the extremely low loading capacities of ND-PG, its applicability as nanocarrier is very limited.
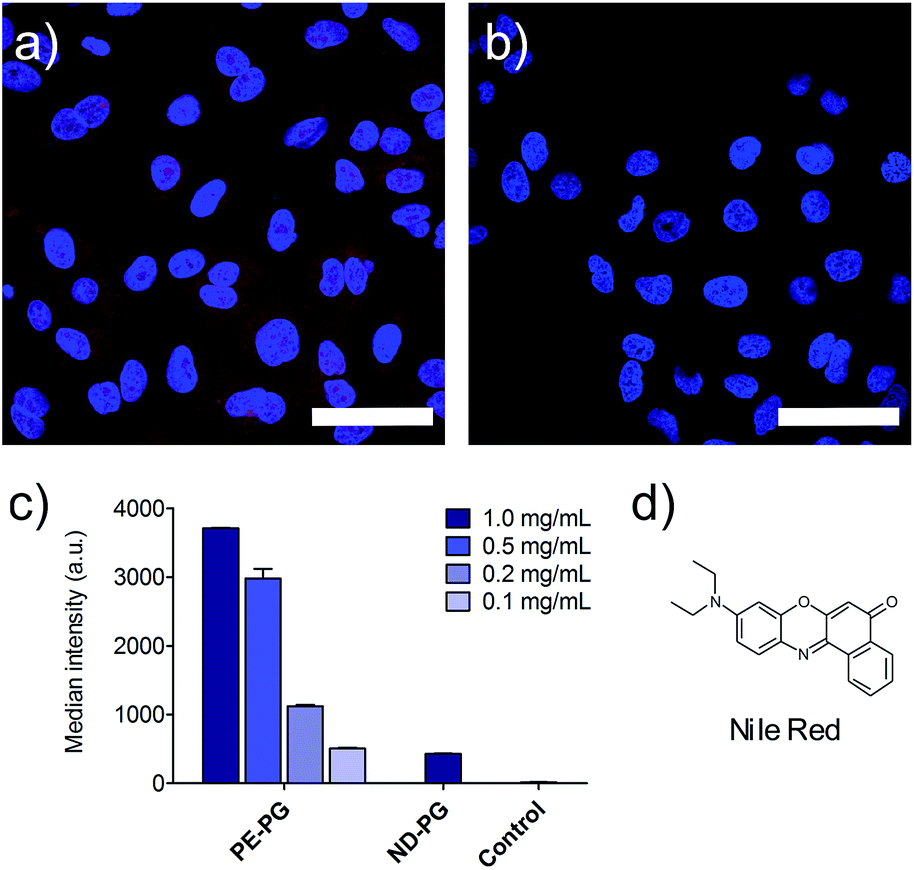 | ||
| Fig. 4 (a) Confocal fluorescence microscopy image of A549 cells after 4 h incubation with PE-PG (1 mg mL−1) with NR (2 μM). Scale bar: 50 μm. (b) Confocal fluorescence microscopy image of A549 cells after 4 h incubation with ND-PG (1 mg mL−1) with NR (0.2 μM). Scale bar: 50 μm. (c) Median fluorescence intensity from three flow cytometry measurements of A549 cells after 4 h incubation with PE-PG and ND-PG both loaded with NR and of the non-treated control. Concentrations of the nanocarriers are given in the legend. (d) Structure of NR. | ||
Conclusions
In this study the transport capacity of core–shell nanocarriers with different core flexibilities was investigated. Specifically, two core–shell nanoparticles that both contained a carbon core and the same polyglycerol shell were synthesized and characterized. The first had a soft dendritic polyethylene core and the second a hard nanodiamond core. The encapsulation capacities in aqueous solution were studied for two hydrophobic model compounds, pyrene and Nile red. Only the nanocarrier with the flexible core was able to encapsulate the dye pyrene. This nanocarrier was also superior to the one with a rigid nanodiamond core in the uptake of the dye Nile red. Nonetheless, both nanocarriers were able to transport their cargo (NR) into tumor cells. The low transport capacity of ND-PG demonstrates that interfacial mechanisms are not predominant in this system. Instead, it is suggested that the flexible core plays a major role in the encapsulation process of hydrophobic guest molecules. In conclusion, this study shows that the flexibility of the core of nanocarriers has dramatic effects on their encapsulation/transport properties and this knowledge will be helpful for the design of better nanocarriers in the future.Acknowledgements
This work was supported by the U.S. National Science Foundation (CHE-0723497) and the Deutsche Forschungsgemeinschaft (DFG HA 2549/10-1) under joint grant and the SFB 1112. The authors would like to thank Christina Kühl and Dr Florian Paulus for technical assistance. Dr Pamela Winchester is acknowledged for proofreading the manuscript. M.L. thanks Mohsen Adeli for helpful discussions.Notes and references
- V. P. Torchilin, J. Controlled Release, 2001, 73, 137–172 CrossRef CAS.
- U. Gupta, H. B. Agashe, A. Asthana and N. K. Jain, Biomacromolecules, 2006, 7, 649–658 CrossRef CAS PubMed.
- P. Kesharwani, K. Jain and N. K. Jain, Prog. Polym. Sci., 2014, 39, 268–307 CrossRef CAS PubMed.
- R. Haag and F. Kratz, Angew. Chem., Int. Ed., 2006, 45, 1198–1215 CrossRef CAS PubMed.
- R. Duncan, Curr. Opin. Biotechnol., 2011, 22, 492–501 CrossRef CAS PubMed.
- R. Duncan and M. J. Vicent, Adv. Drug Delivery Rev., 2013, 65, 60–70 CrossRef CAS PubMed.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- G. S. Kwon and T. Okano, Adv. Drug Delivery Rev., 1996, 21, 107–116 CrossRef CAS.
- R. P. Brinkhuis, F. P. J. T. Rutjes and J. C. M. van Hest, Polym. Chem., 2011, 2, 1449–1462 RSC.
- G. Gunkel-Grabole, S. Sigg, M. Lomora, S. Lörcher, C. G. Palivan and W. P. Meier, Biomater. Sci., 2015, 3, 25–40 RSC.
- M. Liu, K. Kono and J. M. Fréchet, J. Controlled Release, 2000, 65, 121–131 CrossRef CAS.
- G. R. Newkome, C. N. Moorefield, G. R. Baker, M. J. Saunders and S. H. Grossman, Angew. Chem., Int. Ed., 1991, 30, 1178–1180 CrossRef.
- C. J. Hawker, K. Wooley and J. Fréchet, J. Chem. Soc., Perkin Trans. 1, 1993, 1287–1297 RSC.
- C. S. Popeney, M. C. Lukowiak, C. Böttcher, B. Schade, P. Welker, D. Mangoldt, G. Gunkel, Z. Guan and R. Haag, ACS Macro Lett., 2012, 1, 564–567 CrossRef CAS.
- H. Liu, S. Farrell and K. Uhrich, J. Controlled Release, 2000, 68, 167–174 CrossRef CAS.
- M. R. Radowski, A. Shukla, H. von Berlepsch, C. Böttcher, G. Pickaert, H. Rehage and R. Haag, Angew. Chem., Int. Ed., 2007, 46, 1265–1269 CrossRef CAS PubMed.
- I. N. Kurniasih, H. Liang, J. P. Rabe and R. Haag, Macromol. Rapid Commun., 2010, 31, 1516–1520 CrossRef CAS PubMed.
- E. Fleige, B. Ziem, M. Grabolle, R. Haag and U. Resch-Genger, Macromolecules, 2012, 45, 9452–9459 CrossRef CAS.
- E. Fleige, R. Tyagi and R. Haag, Nanocarriers, 2013, 1, 1–9 Search PubMed.
- F. Paulus, D. Steinhilber, P. Welker, D. Mangoldt, K. Licha, H. Depner, S. Sigrist and R. Haag, Polym. Chem., 2014, 5, 5020–5028 RSC.
- E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866–884 CrossRef CAS PubMed.
- M. Calderón, M. Quadir, M. Strumia and R. Haag, Biochimie, 2010, 92, 1242–1251 CrossRef PubMed.
- A. V Ambade, E. N. Savariar and S. Thayumanavan, Mol. Pharm., 2005, 2, 264–272 CrossRef PubMed.
- C. Oerlemans, W. Bult, M. Bos, G. Storm, J. F. W. Nijsen and W. E. Hennink, Pharm. Res., 2010, 27, 2569–2589 CrossRef CAS PubMed.
- C. Alvarez-Lorenzo and A. Concheiro, Chem. Commun., 2014, 50, 7743–7765 RSC.
- A. Kowalczuk, R. Trzcinska, B. Trzebicka, A. H. E. Müller, A. Dworak and C. B. Tsvetanov, Prog. Polym. Sci., 2014, 39, 43–86 CrossRef CAS PubMed.
- M. Irfan and M. Seiler, Ind. Eng. Chem. Res., 2010, 49, 1169–1196 CrossRef CAS.
- J. F. Jansen, E. M. de Brabander-van den Berg and E. W. Meijer, Science, 1994, 266, 1226–1229 CAS.
- D. Wan, H. Pu, M. Jin, H. Pan and Z. Chang, React. Funct. Polym., 2010, 70, 916–922 CrossRef CAS PubMed.
- D. Wan, H. Pu and M. Jin, Macromolecules, 2010, 43, 3809–3816 CrossRef CAS.
- D. Wan, Y. Lai, M. Jin and H. Pu, Macromol. Chem. Phys., 2011, 212, 1910–1917 CAS.
- Z. Guan, P. M. Cotts, E. F. McCord and S. J. McLain, Science, 1999, 283, 2059–2062 CrossRef CAS.
- G. Chen, X. S. Ma and Z. Guan, J. Am. Chem. Soc., 2003, 125, 6697–6704 CrossRef CAS PubMed.
- V. F. Loktev, V. I. Makal'skii, I. V. Stoyanova, A. V. Kalinkin and V. A. Likholobov, Carbon, 1991, 29, 817–819 CrossRef CAS.
- L. Zhao, T. Takimoto, M. Ito, N. Kitagawa, T. Kimura and N. Komatsu, Angew. Chem., Int. Ed., 2011, 50, 1388–1392 CrossRef CAS PubMed.
- L. Zhao, T. Chano, S. Morikawa, Y. Saito, A. Shiino, S. Shimizu, T. Maeda, T. Irie, S. Aonuma, H. Okabe, T. Kimura, T. Inubushi and N. Komatsu, Adv. Funct. Mater., 2012, 22, 5107–5117 CrossRef CAS.
- K. Kalyanasundaram and J. Thomas, J. Am. Chem. Soc., 1977, 99, 2039–2044 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and analytic data. See DOI: 10.1039/c4tb01858c |
| This journal is © The Royal Society of Chemistry 2015 |