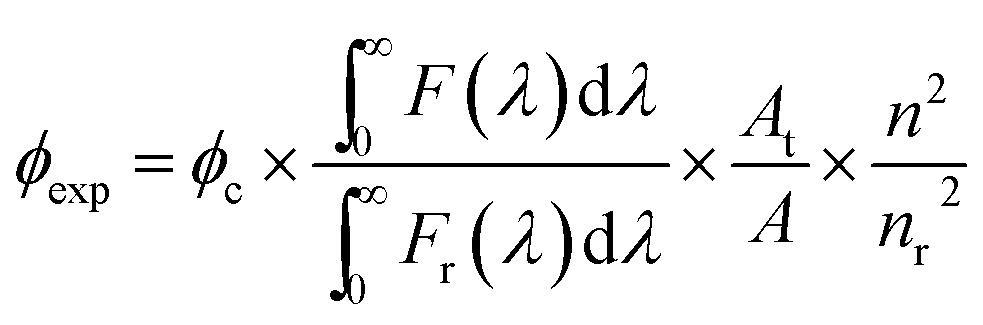DOI:
10.1039/C4RA06061J
(Paper)
RSC Adv., 2014,
4, 37973-37978
Synthesis of a borylated boron–dibenzopyrromethene dye enabling the visual detection of H2O2 vapor†
Received
21st June 2014
, Accepted 13th August 2014
First published on 13th August 2014
Abstract
Given our interest in the development of reaction-based chemosensors, we developed a novel boron–dibenzopyrromethene dye with a pinacolboryl group (1), blue in color (λmax = 621 nm, ε = 8.74 × 104 M−1 cm−1) with red emission (λmax = 643 nm, λex = 550 nm) in THF. H2O2-mediated oxidation of the pinacolboryl group was found to induce a significant fluorescence decrease at 642 nm (λex = 550 nm) in EtOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) at 25 °C, enabling us to detect trace levels of H2O2 visually. The time-dependent response was investigated to evaluate the pseudo-first-order rate constant of 1.69 min−1 under basic conditions, meaning that its fluorescence was decreased by 80% in 2 min. Such a remarkable response capability motivated us to test the suitability of 1 for the detection of H2O2 vapor. For these experiments, a 1-coated TLC plate was fabricated by a spin-coating method and then placed in sealed bottles with H2O2 vapor. It was found that the increasing vapor concentration of H2O2 could be visually monitored by a change in red emission (ΔR value). Based on this, we estimated the detection limit of this method to be 8.43 ppb. We also found that the 1-coated TLC plate could selectively detect H2O2 vapor over common solvents tested.
:1 v/v) at 25 °C, enabling us to detect trace levels of H2O2 visually. The time-dependent response was investigated to evaluate the pseudo-first-order rate constant of 1.69 min−1 under basic conditions, meaning that its fluorescence was decreased by 80% in 2 min. Such a remarkable response capability motivated us to test the suitability of 1 for the detection of H2O2 vapor. For these experiments, a 1-coated TLC plate was fabricated by a spin-coating method and then placed in sealed bottles with H2O2 vapor. It was found that the increasing vapor concentration of H2O2 could be visually monitored by a change in red emission (ΔR value). Based on this, we estimated the detection limit of this method to be 8.43 ppb. We also found that the 1-coated TLC plate could selectively detect H2O2 vapor over common solvents tested.
1. Introduction
There has been growing interest in the development of boronic acid or boronate-based chemosensors because of their versatile reactivity toward analytes.1 For instance, these boron-based functional groups can bind to cis-diols (e.g. saccharides),2 catechols,3 fluoride,4 cyanide,5 and reactive oxygen species such as superoxide, hypochlorous acid or nitrating species.6 A H2O2-triggered response, based on the oxidative conversion of boronates to phenols, is one of the distinctive features of those systems that have been widely used as noninvasive detection tools in biological systems.7 In addition, trace detection of peroxide-based explosives is of significant current importance for security purposes.8 This is because, compared to aromatic explosives such as 2,4,6-trinitrotoluene (TNT), it is difficult to detect explosives that do not contain a nitro group, such as triacetone triperoxide (TATP) and hexamethylene triperoxide diamine (HMTD).9 Therefore, boronate-appended dyes would be highly valuable for use as indicators to monitor H2O2 produced by UV or the acid-catalyzed decomposition of TATP or HMTD.10 Given the demand for real-time detection,11 we decided to develop a visual H2O2-detection system with outstanding optical properties.
4,4-Difuoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) dyes composed of boron–dipyrrin complexes have emerged as promising candidates because of their excellent optical properties, which include high molecular extinction coefficients of absorption, high fluorescence quantum yield, and excellent photostability.12 As such, they have been applied in a wide range of research areas, including chemosensors,13 biological labels,14 bio-imaging,15 organic light-emitting diodes,16 photodynamic therapy,17 light-harvesting arrays,18 and solar-cell devices.19 For their use as reaction-based chemosensors, several BODIPY derivatives have been proposed.20 However, to the best of our knowledge, BODIPY and its congeners capable of detecting vapor peroxides involving H2O2 have not been reported as yet. Therefore, the direct introduction of a boronate reactive group into the chromophore is a novel approach. We have currently focused on modification of the structure of BODIPY by extending π-conjugation to provide new types of boron–dibenzopyrromethene dyes.21 Synthetic acceptability of the dye skeleton led us to prepare a pinacolborylated probe (1). In this study, the synthesis and characteristics are described from the standpoint of preparing an H2O2-detection system; the addition of H2O2 in an EtOH/H2O (1:1 v/v) solution of 1 induced a bathochromic shift in the absorption band, accompanied by a remarkable decrease in its fluorescence intensity through deboronation. Its fast response was observed when tetrabutylammonium hydroxide (TBAOH)10d,22 was added to the solution under the conditions. A test strip composed of a TLC plate coated with 1 was applied for the visual detection of H2O2 vapor.
2. Experimental section
2.1 General
NMR spectra were taken by a Bruker Avance 500 (1H: 500 MHz, 13C: 125 MHz) spectrometer. In 1H and 13C NMR measurements, chemical shifts (δ) are reported downfield from the internal standard Me4Si. Fast atom bombardment (FAB) mass spectra were obtained on a JEOL JMS-700 spectrometer where m-nitrobenzylalcohol was used as a matrix. The absorption and fluorescence spectra were measured using a Shimadzu UV-3600 and a JASCO FP-6300 spectrophotometers, respectively. Elemental analyses were performed on an Exeter Analytical, Inc. CE-440F Elemental Analyzer. Photographic images were recorded using a NIKON D3200 digital single-lens reflex camera.
2.2 Materials
Unless otherwise indicated, reagents used for the synthesis were commercially available and were used as supplied. 1-(2-(2-Methoxybenzoyl)phenyl)ethanone 2 (ref. 23) was prepared through two steps from 2-hydroxyacetophenone. Synthesis of 1-(4-bromo-2-(2-methoxybenzoyl)phenyl)ethanone 3 was conducted according to our previous paper.21b
2.3 Synthesis
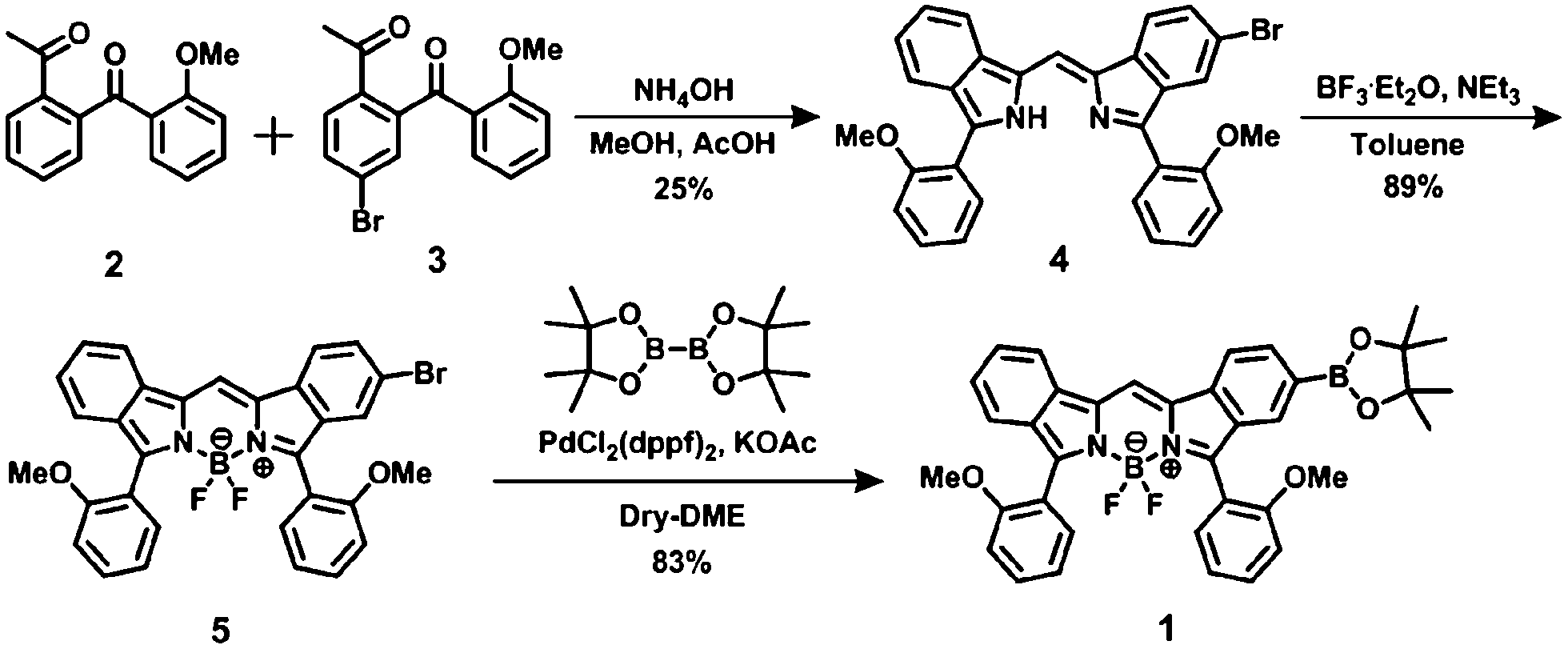
5-Bromo-1-((3-(2-methoxyphenyl)-2H-isoindol-1-yl)-methylene)-3-(2-methoxyphenyl)-1H-isoindole (4). Compounds 2 (1.190 g, 4.680 mmol) and 3 (1.559 g, 4.680 mmol) were dissolved in MeOH (47 mL) and AcOH (27 mL). To the solution was added NH4OH (21 mL) under an icy condition. The resulting mixture was stirred for overnight at 45 °C. After treating with NaHCO3 aqueous solution (150 mL), the precipitation was then collected. It was chromatographed on silica gel (Wacogel C-300) using hexane/benzene (3:2 v/v) as an eluent to give 314.7 mg of 4 as a deep-blue solid in 25% yield. 1H NMR (500 MHz, DMSO-d6) δ (ppm) 3.75 (s, 6H), 7.18 (t, 2H, J = 7.45 Hz), 7.25 (d, 1H, J = 6.25 Hz), 7.26 (d, 1H, J = 6.30 Hz), 7.30 (td, 1H, J = 7.53 and 0.93 Hz), 7.43 (td, 1H, J = 7.60 and 0.60 Hz), 7.47 (dd, 1H, J = 8.00 and 1.58 Hz), 7.49–7.52 (m, 2H), 7.83 (d, 1H, J = 8.20 Hz), 7.90 (dd, 1H, J = 7.55 and 1.60 Hz), 7.92 (dd, 1H, J = 7.60 and 1.60 Hz), 7.96 (d, 1H, J = 1.35 Hz), 8.14 (s, 1H), 8.16 (t, 2H, J = 8.80 Hz); FAB MS: m/z 534 [M]+, 536 [M + 2]+.
Difluoro[5-bromo-1-[[3-(2-methoxyphenyl)-2H-isoindole-1-yl]methylene]-3-(2-methoxyphenyl)-1H-isoindolato-N1,N2]-boron (5). Et3N (0.8 mL, 5.771 mmol) was adeed to a solution of 4 (1.179 g, 2.201 mmol) in dry toluene (83 mL), followed by the addition of BF3·Et2O (3 mL, 23.99 mmol) at 80 °C. The mixture was stirred overnight at 100 °C. The solution was poured into water (40 mL) and extracted with AcOEt (120 mL), the organic layer being washed with H2O (300 mL) and dried with Na2SO4. After removal of the solvent in vacuo, the residue was chromatographed on silica gel (Wacogel C-300) using a gradient of hexane (50% → 0% v/v) in benzene as an eluent to give 5 (1.147 g) in 89% yield. Dye 5 is present as a mixture of conformation isomers at room temperature. As such, integration values for the 1H NMR data were based on that (3H) of OMe signal of the anisole unit. 1H NMR (500 MHz, DMSO-d6) δ (ppm) 3.67 (s, 3H), 3.68 (s, 3H), 3.72 (s, 3H), 3.73 (s, 3H), 7.00 (t, 1H, J = 6.63 Hz), 7.02 (t, 1H, J = 6.82 Hz), 7.07 (t, 1H, J = 7.12 Hz), 7.08 (t, 1H, J = 6.93 Hz), 7.17 (d, 1H, J = 8.20 Hz), 7.18 (d, 1H, J = 8.70 Hz), 7.20 (d, 1H, J = 8.60 Hz), 7.22 (d, 1H, J = 7.95 Hz), 7.31 (t, 2H, J = 7.80 Hz), 7.33 (d, 2H, J = 8.45 Hz), 7.37 (d, 2H, J = 7.80 Hz), 7.39 (s, 2H), 7.44 (d, 1H, J = 8.00 Hz), 7.45 (d, 1H, J = 8.00 Hz), 7.49 (t, 2H, J = 7.93 Hz), 7.50 (t, 2H, J = 7.60 Hz), 7.60 (t, 2H, J = 8.10 Hz), 7.63 (d, 2H, J = 9.25 Hz), 8.12 (d, 2H, J = 8.70 Hz), 8.16 (d, 2H, J = 8.05 Hz), 8.71 (s, 2H); FAB MS: m/z 582 [M]+, 584 [M + 2]+.
Difluoro[5-pinacolboryl-1-[[3-(2-methoxyphenyl)-2H-isoindole-1-yl]methylene]-3-(2-methoxyphenyl)-1H-isoindolato-N1,N2]boron (1). Compound 5 (1.120 g, 1.923 mmol) and KOAc (1.953 g, 19.90 mmol) were dissolved in dry 1,2-dimethoxyethane (DME) (33.5 mL). The solution was degassed by three freeze–pump–thaw cycles. After adding bis(pinacolate)diboron (1.330 g, 5.237 mmol) and a degassed solution of PdCl2(dppf)2 (0.3800 g, 0.4569 mmol) in dry DME (33.5 mL) to the solution, the resulting solution was stirred at 80 °C for 10 h under a N2 atmosphere, poured into water (10 mL), and then extracted with AcOEt. The organic layer was washed with water (200 mL) and dried with Na2SO4. After the removal of solvent in vacuo, the material was chromaographed on silica gel (Wacogel C-300) using a gradient of hexane (50% → 66.7%) in CH2Cl2 as an eluent. In this way, 1.005 g of 1 was obtained in 83% yield. Because 1 is present as a mixture of conformation isomers at room temperature (Fig. S3 in ESI†), integration values for the 1H NMR data were based on that (3H) of OMe signal of the anisole unit. 1H NMR (500 MHz, DMSO-d6) δ (ppm) 1.28 (s, 24H), 3.64 (s, 3H), 3.67 (s, 3H), 3.70 (s, 3H), 3.70 (s, 3H), 7.02 (td, 1H, J = 7.45 and 0.77 Hz), 7.03 (td, 1H, J = 7.43 and 0.80 Hz), 7.06 (td, 1H, J = 7.53 and 0.70 Hz), 7.09 (td, 1H, J = 7.45 and 0.72 Hz), 7.18 (d, 1H, J = 8.90 Hz), 7.19 (d, 1H, J = 8.05 Hz), 7.20 (d, 1H, J = 9.55 Hz), 7.23 (d, 1H, J = 8.30 Hz), 7.26–7.32 (m, 4H), 7.35 (dt, 1H, J = 7.50 and 1.80 Hz), 7.37–7.41 (m, 2H), 7.44 (d, 1H, J = 7.60 Hz), 7.49 (td, 2H, J = 7.93 and 1.57 Hz), 7.51 (td, 2H, J = 7.93 and 1.55 Hz), 7.55–7.59 (m, 2H), 7.59 (d, 2H, J = 0.70 Hz), 7.74 (d, 2H, J = 8.20 Hz), 8.14 (d, 2H, J = 8.20 Hz), 8.16 (d, 2H, J = 8.25 Hz), 8.71 (s, 1H), 8.71 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ (ppm) 24.55, 55.43, 55.46, 55.50, 55.57, 83.69, 111.4, 111.5, 111.6, 117.3, 117.4, 118.9, 119.1, 119.1, 119.3, 119.5, 119.8, 119.9, 120.0, 123.2, 125.3, 126.2, 129.5, 129.5, 130.3, 130.3, 131.1, 131.2, 131.3, 133.4, 133.7, 157.3, 157.4, 157.5; FAB MS: m/z 630 [M]+; elemental analysis: calcd for C37H34B2F2N2O4: C, 70.51; H, 5.44; N, 4.44, found: C, 70.28; H, 5.50; N, 4.44%.
2.4 Determination of fluorescence quantum yield
The fluorescence quantum yield (ϕexp) of 1 was calculated from the equation below.24
where F(λ) and Fr(λ) describe the measured fluorescence intensities of the dye and the reference, respectively, and A and AR describe the corresponding absorbances at the excitation wavelength. Because Rhodamine B was used as the reference, the excitation wavelength was fixed at 535 nm. The refractive indexes are n = 1.41 for THF (solvent) and nr = 1.36 for EtOH.
2.5 Vapor detection
An EtOH solution (7 μL) containing 1 (25 μM) and TBAOH (250 μM) was spin-coated on a TLC plate (2 (length) × 1.5 (width) cm, RP-modified silica plate, Merck) at a rate of 1500 rpm for 20 s and dried for 5 min. Various concentrations of H2O2 were obtained by diluting the commercial 35 wt% H2O2 solution with pure water, and these were then put in a bottle containing the sensing plate and then sealed for 24 h to reach saturated vapor pressure. The vapor concentration was determined by using the following equation; yH2O2 = (YH2O2 × XH2O2 × PsH2O2)/P, where y, Y, X, Ps, and P are defined as the molar fraction in the vapor phase, the activity coefficient, the molar fraction in the liquid phase, the vapor pressure of H2O2 (1.801 mmHg), and atmospheric pressure (1 atm), respectively.25 Because a binary system of H2O/H2O2 was employed as the solution, the activity coefficient of H2O2 was calculated from Margules' equation; YH2O2 = exp[XH2O2 (−1.2661 + 0.2932 × XH2O)].26 In this way, the vapor concentration of H2O2 could be evaluated. In addition, saturated vapor concentrations of the commonly used solvents were evaluated according to the literature.27
3. Results and discussion
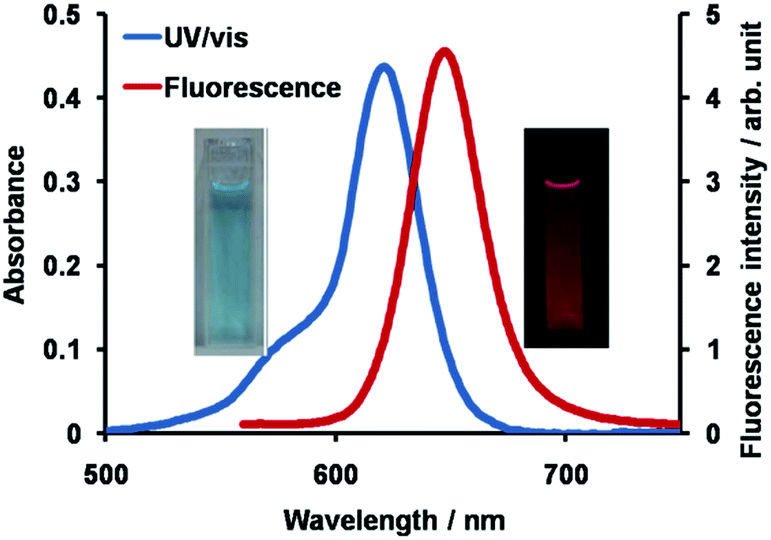
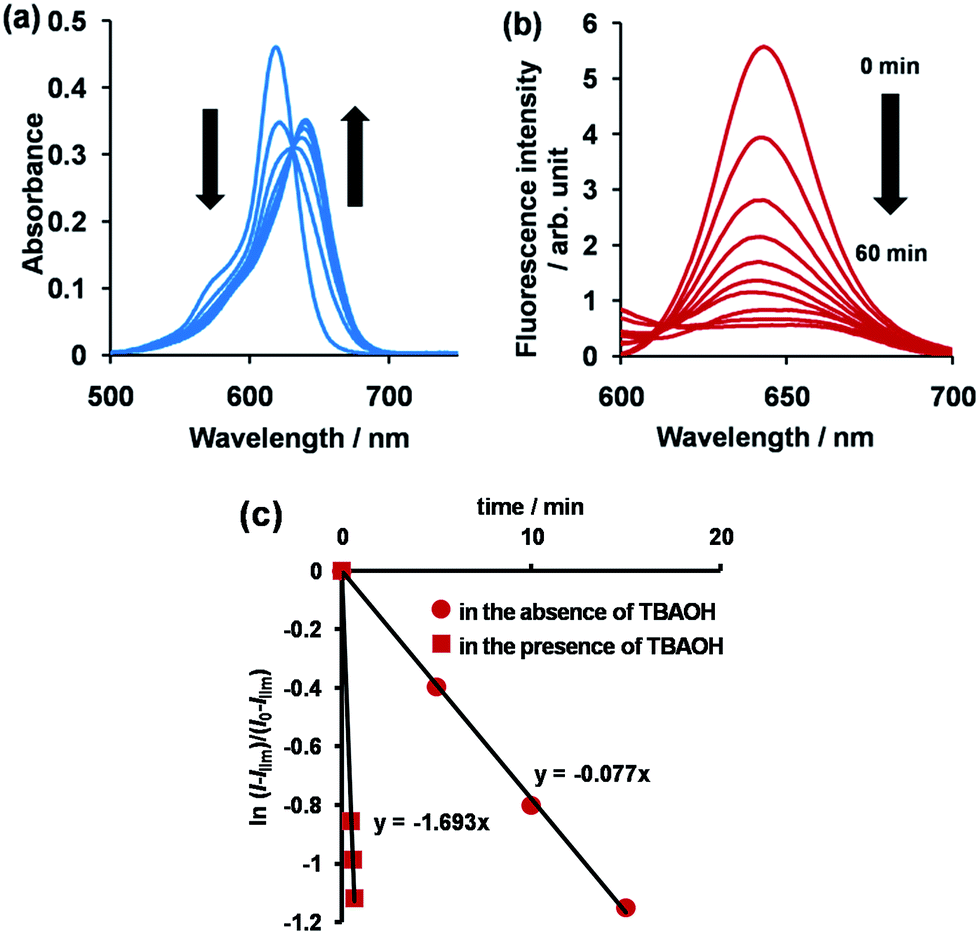
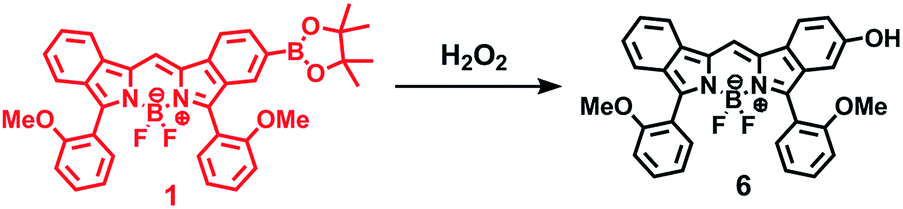
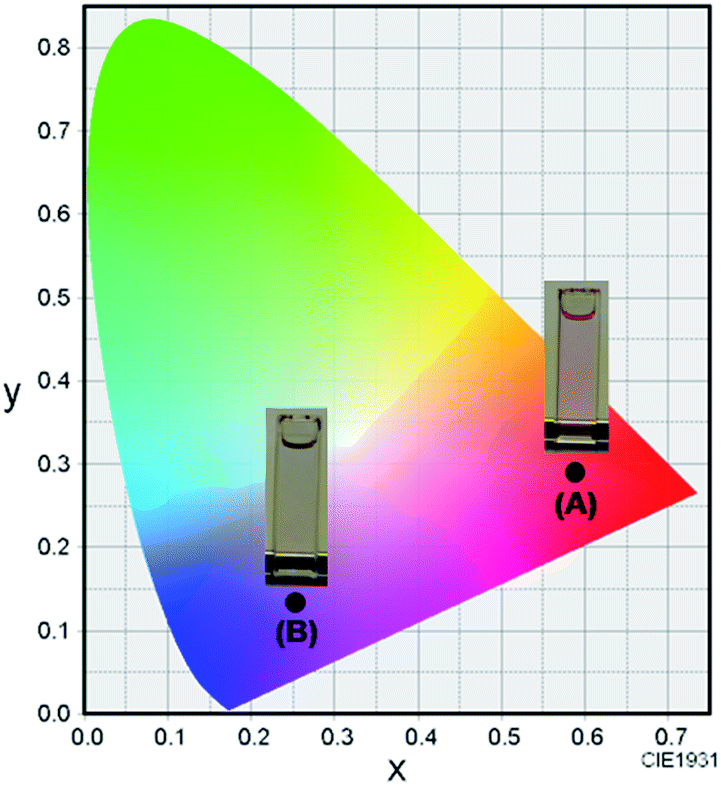
The synthetic route to 1 is shown in Scheme 1. First, the condensation reaction of 1-(2-(2-methoxybenzoyl)-phenyl)ethanone (2) with its bromo-derivative (3) in the presence of NH4OH gave the monobrominated and benzo-fused dipyrrin (4) in 25% yield after purification by column chromatography. This was followed by BF2-chelation with 4 using BF3·Et2O to give 5. A subsequent PdCl2(dppf)2-catalyzed pinacolborylation afforded target 1 in reasonable yield. Structural assignments of these compounds were conducted using spectroscopic data, those 1H NMR charts being shown in Fig. S1–S4 in ESI.† In this context, the 1H NMR spectra of 5 in DMSO-d6 at room temperature are somewhat complicated (Fig. S2 in ESI†), containing four sets of signals assignable to the anisole segments; for instance, proton resonances arising from the methoxy groups were discretely observed as singlets at 3.665 (A), 3.675 (B), 3.720 (C), and 3.732(D) ppm, respectively, (Fig. 1a). As inferred from our previous result,21c one set of higher-field shifted signals (A and B) and another set of signals (C and D) would be assignable to either of the anti- and syn-forms of 5, in which the anisole moieties are located on the opposite and same face of the dibenzopyrromethene core, respectively. Considering their unsymmetrical structures, each of anti- and syn-forms contains the corresponding unsymmetrical counterpart (Fig. 1b). To gain further insight into the structures, we recorded VT NMR spectra; with increases in the temperature of DMSO-d6 solution of 5, revealing a Tc value of 95 °C (ΔG≠ = ca. 78 kJ mol−1).28 These results indicate that rotation about the two Canisole–Cisoindole bonds may be restricted by both the isoindole ring and BF2 unit in 5. A similar result was obtained for 1 (Fig. S3, S5, and S6 in ESI†). The absorption and fluorescence spectra were measured in a THF solution of 1 at 25 °C (Fig. 2), which showed an absorption with a λmax value of 621 nm (ε = 8.74 × 104 M−1 cm−1) and fluorescence bands (λmax = 643 nm, λex = 550 nm). In accordance with the optical properties, the solution is bluein color, although a red emission can also be detected. In addition, the quantum yield (Φ) of 1 was evaluated to be 0.21 against Rhodamine B (ΦR = 0.97)29 when excited at 535 nm.30 Encouraged by its optical properties, we investigated how 1 responds to the addition of H2O2. First, the time-dependent responses of 1 toward H2O2 were examined by absorption and fluorescence spectrophotometries (Fig. 3). When adding H2O2 (500 μM) into an EtOH/H2O (1:1 v/v) solution of 1 (5 μM) at 25 °C, a slow kinetic change in the absorption spectra showed a bathochromic shift of 19 nm, with an isosbestic point at 635 nm. On the other hand, we observed a steep decrease in the fluorescent intensity at 643 nm, accompanied by a slight red shift in the band. The reaction product was successfully assigned as deboronated dye 6 (ref. 31) through H2O2-mediated oxidation (Scheme 2), with the 1H NMR spectrum showing proton resonance arising from Ar-OH at 9.60 ppm (Fig. S8 in ESI†). A linear relationship was observed between the exponential decrease in the fluorescence spectra and the reaction time, giving a pseudo-first-order rate constant (k) of 7.7 × 10−2 min−1. This means that its fluorescence was decreased by 80% within 22 min after adding H2O2. However, setting up basic conditions by adding TBAOH led to an acceleration of the reaction. The k value was calculated to be 1.69 min−1, being 21-fold larger than that under TBAOH-free conditions. As a result, it took only 2 min to attain 80% decrease in the fluorescence after adding H2O2.32 With this in mind, fluorometric titrations of 1 were carried out by adding incremental amounts of H2O2 in the presence of TBAOH (50 μM) in EtOH/H2O (1:1 v/v) at 25 °C, the data being collected within 2 min. Fig. 4 shows that the fluorescence intensities gradually reduced as a function of H2O2 concentration. No further change was observed when 80 μM of H2O2 was added to the solution. Given the relation between the fluorescence intensities and concentrations of H2O2 (Fig. 4b), this allowed us to determine a detection limit of 0.127 μM. Such a H2O2-induced change in the optical properties could be detected by the naked eye, which was followed by the trajectory of Commission Internationale de l'Éclairage (CIE) coordinates in the chromaticity diagram. When irradiated under UV (365 nm) under a natural white fluorescent lamp in a laboratory, a red emission with CIE coordinates x = 0.54 and y = 0.29 of the solution shifted to a blue emission with CIE coordinates x = 0.25 and y = 0.13 upon adding H2O2 under basic conditions (Fig. 5). The image data are also shown in Fig. S10 in ESI.†
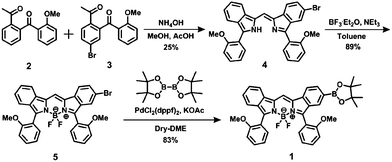 |
| | Scheme 1 Synthesis of 1. | |
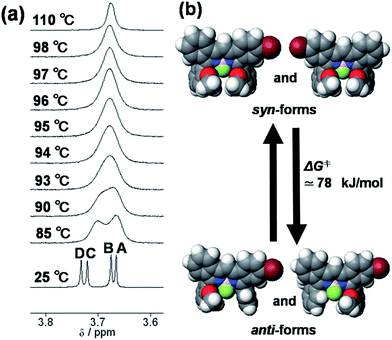 |
| | Fig. 1 (a) Variable temperature NMR of 5 in DMSO-d6. (b) Proposed isomers of 5. | |
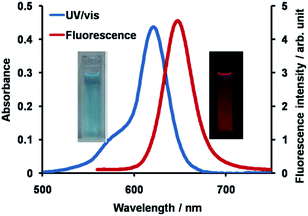 |
| | Fig. 2 Absorption and fluorescence spectra of 1 (5 μM) in THF at 25 °C. The excitation wavelength was 550 nm. | |
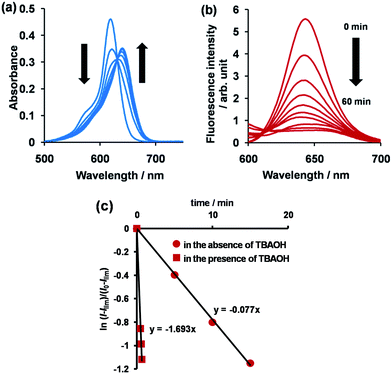 |
| | Fig. 3 Time course of absorption (a) and fluorescence (b) spectra of 1 (5 μM) in EtOH/H2O (1:1 v/v) at 25 °C, λex = 550 nm, and (c) a plot of ln[(l − Ilim)/(l0 − llim)] at 642 nm as a function of time in the presence or absence of TBAOH (50 μM). | |
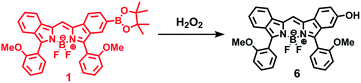 |
| | Scheme 2 H2O2-mediated oxidation of 1 to give 6. | |
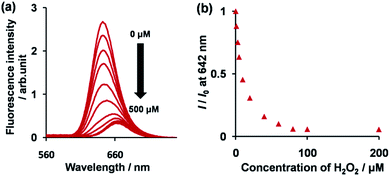 |
| | Fig. 4 (a) Change in the fluorescence of 1 (5 μM) upon adding incremental amounts of H2O2 in EtOH/H2O (1:1 v/v) in the presence of TBAOH (50 μM) at 25 °C. Each spectrum was acquired 2 min after the addition of H2O2. (b) A plot of fluorescent intensity at 642 nm as a function of H2O2 concentration. | |
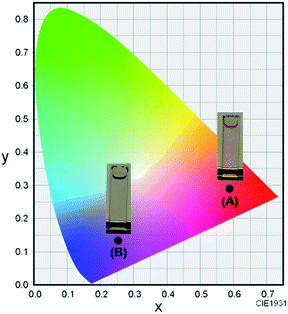 |
| | Fig. 5 The CIE coordinate diagram of the color of 1 (5 μM) (A) and 1 (5 μM) with H2O2 (80 μM) (B) in EtOH/H2O (1:1 v/v) in the presence of TBAOH (50 μM) at room temperature. | |
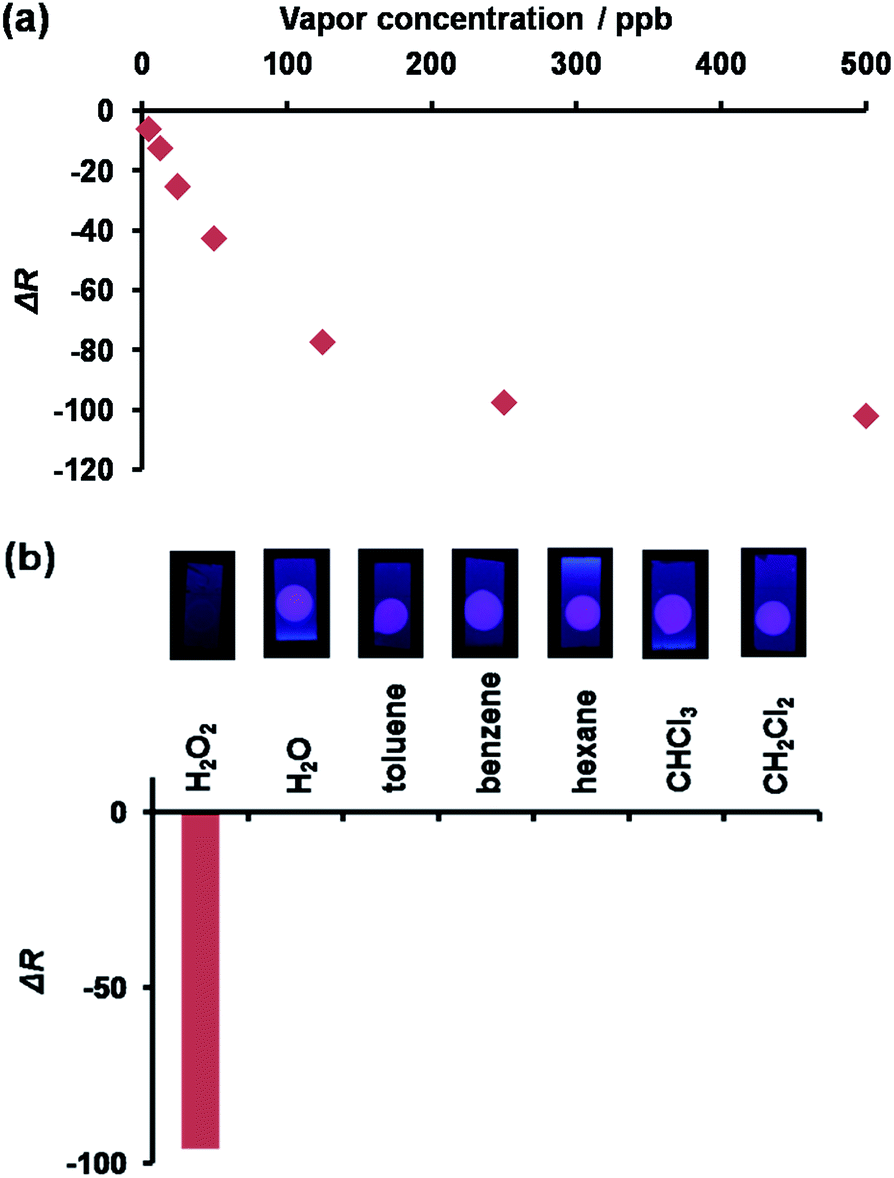
Our motivation in studying the possibility of using 1 as a fluorescent probe for vapor detection led us to test how 1 coated on a TLC plate could respond to the presence of H2O2 vapor. An EtOH solution of 1 (25 μM) and TBAOH (250 μM) was spin-coated on a RP-modified silica plate to fabricate a sensing plate, a bright red dot could be observed under UV light irradiation at 365 nm. As expected, when the plate was exposed to the H2O2 vapor, the red emission was quenched under UV light irradiation. The images upon H2O2 exposure were converted into red values (ΔR) using an image-processing program33 to evaluate the fluorescence response of 1 to various concentrations of H2O2. As inferred from Fig. 6a, the sensing plate could monitor H2O2 vapor at a level lower than 1 ppm. As such, the H2O2 vapor detection limit for the plate was evaluated to be 8.43 ppb, i.e., lower than the value of the permissible exposure limit (OSHA).34 For comparison, the 1-coated plate was tested against the saturated vapor of common solvents such as water (29000 ppm), toluene (34000 ppm), benzene (120000 ppm), hexane (180000 ppm), CHCl3 (230000 ppm), and CH2Cl2 (440000 ppm). Contrary to the results obtained with H2O2 (300 ppm), a negligible response was obtained (Fig. 6b) with the other solvents. Taken together, we confirmed that the 1-coated sensing plate exhibited a selective detection of H2O2 vapor over common solvents tested.35
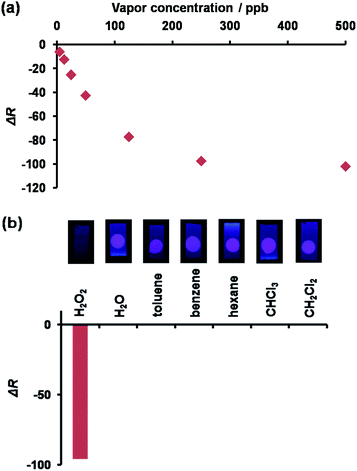 |
| | Fig. 6 (a) The emission response a 1-coated TLC plate exposed to varying amounts of H2O2 vapor. (b) The emissive response of a 1-coated TLC plate exposed to H2O2 and common solvents. | |
4. Conclusions
Our interest in exploring the functionality of a boron–dibenzopyrromethene dye led to the preparation of the corresponding pinacolboryl derivative 1. Taking advantage of the optical properties of 1, we investigated its response to the presence of H2O2, wherein the presence of H2O2 caused a significant decrease in the fluorescence of 1 due to the H2O2-mediated oxidation of the pinacolboryl unit. This change could also be detected visually. To the best of our knowledge, the 1-coated TLC plate represents the first demonstrated example of BODIPY analogues being applied to the detection of H2O2 vapor.
Notes and references
-
(a) T. D. James, P. Linnane and S. Shinkai, Chem. Commun., 1996, 281 RSC;
(b) R. Nishiyabu, Y. Kubo, T. D. James and J. S. Fossey, Chem. Commun., 2011, 47, 1106 RSC;
(c) Z. Guo, I. Shin and J. Yoon, Chem. Commun., 2012, 48, 5956 RSC;
(d) S. D. Bull, M. G. Davidson, J. M. H. Van den Elsen, J. S. Fossey, A. T. A. Jenkins, Y.-B. Jiang, Y. Kubo, F. Marken, K. Sakurai, J. Zhao and T. D. James, Acc. Chem. Res., 2013, 46, 312 CrossRef CAS PubMed;
(e) J. A. Peters, Coord. Chem. Rev., 2014, 268, 1 CrossRef CAS PubMed.
- X. Wu, Z. Li, X.-X. Chen, J. S. Fossey, T. D. James and Y.-B. Jiang, Chem. Soc. Rev., 2013, 8032 RSC.
-
(a) G. Springsteen and B. Wang, Chem. Commun., 2001, 1608 RSC; G. Springsteen and B. Wang, Tetrahedron, 2002, 58, 5291 Search PubMed;
(b) A. Nonaka, S. Horie, T. D. James and Y. Kubo, Org. Biomol. Chem., 2008, 6, 3621 RSC;
(c) S. Horie and Y. Kubo, Chem. Lett., 2009, 38, 616 CrossRef CAS.
-
(a) C. R. Cooper, N. Spencer and T. D. James, Chem. Commun., 1998, 1365 RSC;
(b) Y. Kubo, T. Ishida, T. Minami and T. D. James, Chem. Lett., 2006, 35, 996 CrossRef CAS;
(c) K. M. K. Swamy, Y. J. Lee, H. N. Lee, J. Chun, Y. Kim, S. –J. Kim and J. Yoon, J. Org. Chem., 2006, 71, 8626 CrossRef CAS PubMed;
(d) Z. Xu, S. K. Kim, S. J. Han, C. Lee, G. Kociok-Kohn, T. D. James and J. Yoon, Eur. J. Org. Chem., 2009, 3058 CrossRef CAS PubMed;
(e) J. K. Day, C. Bresner, N. D. Coombs, I. A. Fallis, L.-L. Ooi and S. Aldridge, Inorg. Chem., 2008, 47, 793 CrossRef CAS PubMed;
(f) W. Tan, D. Zhang, Z. Wang, C. Liu and D. Zhu, J. Mater. Chem., 2007, 17, 1964 RSC;
(g) W. Tan, D. Zhang and D. Zhu, Bioorg. Med. Chem. Lett., 2007, 17, 2629 CrossRef CAS PubMed.
-
(a) M. Jamkratoke, V. Ruangpornvisuti, G. Tumcharem, T. Tuntulani and B. Tomapatanaget, J. Org. Chem., 2009, 74, 3919 CrossRef CAS PubMed;
(b) R. Guan, H. Chen, F. Cao, D. Cao and Y. Deng, Inorg. Chem. Commun., 2013, 38, 112 CrossRef CAS PubMed;
(c) R. Badugu, J. R. Lakowicz and C. D. Geddes, J. Am. Chem. Soc., 2005, 127, 3635 CrossRef CAS PubMed.
- Current review, see: C. C. Winterbourn, Biochim. Biophys. Acta, 2014, 1840, 730 CrossRef CAS PubMed.
-
(a) L.-C. Lo and C.-Y. Chu, Chem. Commun., 2003, 2728 RSC;
(b) E. W. Miller, A. E. Albers, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2005, 127, 16652 CrossRef CAS PubMed;
(c) A. E. Albers, V. S. Okreglak and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 9640 CrossRef CAS PubMed;
(d) D. Srikun, E. W. Miller, D. W. Domaille and C. J. Chang, J. Am. Chem. Soc., 2008, 130, 4596 CrossRef CAS PubMed;
(e) F. He, F. Feng, S. Wang, Y. Li and D. Zhu, J. Mater. Chem., 2007, 17, 3702 RSC;
(f) D. Srikun, A. E. Albers and C. J. Chang, Chem. Sci., 2011, 2, 1156 RSC;
(g) C. Quin, L. Robertson, S. J. McQuaker, N. C. Price, M. D. Brand and R. C. Hartley, Tetrahedron, 2010, 66, 2384 CrossRef CAS PubMed;
(h) B. C. Dickinson, C. Huynh and C. J. Chang, J. Am. Chem. Soc., 2010, 132, 5906 CrossRef CAS PubMed;
(i) A. R. Lippert, G. C. Van de Bittner and C. J. Chang, Acc. Chem. Res., 2011, 44, 793 CrossRef CAS PubMed;
(j) W.-K. Oh, Y. S. Jeong, S. Kim and J. Jang, ACS Nano, 2012, 6, 8516 CrossRef CAS PubMed;
(k) S. W. Lee, H.-W. Rhee, Y.-T. Chang and J.-I. Hong, Chem.–Eur. J., 2013, 19, 14791 CrossRef CAS PubMed;
(l) R. Weinstain, E. N. Savariar, C. N. Felsen and R. Y. Tsien, J. Am. Chem. Soc., 2014, 136, 874 CrossRef CAS PubMed;
(m) X. Sun, Q. Xu, G. Kim, S. E. Flower, J. P. Lowe, J. Yoon, J. S. Fossey, X. Qian, S. D. Bull and T. D. James, Chem. Sci., 2014, 5, 3368 RSC.
-
(a) R. S. Ladbeck, M. Vogel and U. Karst, Anal. Bioanal. Chem., 2006, 386, 559 CrossRef PubMed;
(b) H. Lin and K. S. Suslick, J. Am. Chem. Soc., 2010, 132, 15519 CrossRef CAS PubMed.
- X. Li, Z. Zhang and L. Tao, Biosens. Bioelectron., 2013, 47, 356 CrossRef CAS PubMed.
-
(a) J. C. Sanchez and W. C. Trogler, J. Mater. Chem., 2008, 18, 5134 RSC;
(b) M. E. Germain and M. J. Knapp, Inorg. Chem., 2008, 47, 9748 CrossRef CAS PubMed;
(c) C. He, D. Zhu, Q. He, L. Shi, Y. Fu, D. Wen, H. Cao and J. Cheng, Chem. Commun., 2012, 48, 5739 RSC;
(d) M. Xu, J.-M. Han, Y. Zhang, X. Yang and L. Zang, Chem. Commun., 2013, 49, 11779 RSC.
-
(a) S. Tao and G. Li, Colloid Polym. Sci., 2007, 285, 721 CrossRef CAS PubMed;
(b) J. W. Grate, R. G. Ewing and D. A. Atkinson, TrAC, Trends Anal. Chem., 2012, 41, 1 CrossRef CAS PubMed.
-
(a) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS PubMed;
(b) G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem,. Int. Ed., 2008, 47, 1184 CrossRef CAS PubMed;
(c) M. Benstead, G. H. Mehl and R. W. Boyle, Tetrahedron, 2011, 67, 3573 CrossRef CAS PubMed;
(d) H. Lu, J. Mack, Y. Yang and Z. Shen, Chem. Soc. Rev., 2014, 43, 4778 RSC.
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130 RSC.
-
(a) C. Peters, A. Billich, M. Ghobrial, K. Högenauer, T. Ullrich and P. Nussbaumer, J. Org. Chem., 2007, 72, 1842 CrossRef CAS PubMed;
(b) Z. Li and R. Bittman, J. Org. Chem., 2007, 72, 8376 CrossRef CAS PubMed.
- Current review, see: Y. Ni and J. Wu, Org. Biomol. Chem., 2014, 12, 3774 CAS.
- L. Bonardi, H. Kanaan, F. Camerel, P. Jolinat, P. Retailleau and R. Ziessel, Adv. Funct. Mater., 2008, 18, 401 CrossRef CAS PubMed.
-
(a) S. Erbas, A. Gorgulu, M. Kocakusakogullari and E. U. Akkaya, Chem. Commun., 2009, 4956 RSC;
(b) Y. Cakmak, S. Kolemen, S. Duman, Y. Dede, Y. Dolen, B. Kilic, Z. Kostereli, L. T. Yildirim, A. L. Dogan, D. Guc and E. U. Akkaya, Angew. Chem., Int. Ed., 2011, 50, 11937 CrossRef CAS PubMed;
(c) S. G. Awuah and Y. You, RSC Adv., 2012, 2, 11169 RSC;
(d) A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77 RSC.
-
(a) O. A. Bozdemir, S. E.-Cakmak, O. O. Ekiz, A. Dana and E. U. Akkaya, Angew. Chem., Int. Ed., 2011, 50, 10907 CrossRef PubMed;
(b) R. Ziessel, G. Ulrich, A. Haefele and A. Harriman, J. Am. Chem. Soc., 2013, 135, 11330 CrossRef CAS PubMed.
-
(a) A. Bessette and G. S. Hanan, Chem. Soc. Rev., 2014, 43, 3342 RSC;
(b) S. P. Singh and T. Gayathri, Eur. J. Org. Chem., 2014, 4689 CrossRef CAS PubMed.
-
(a) Z. Ekmekci, M. D. Yilmaz and E. U. Akkaya, Org. Lett., 2008, 10, 461 CrossRef CAS PubMed;
(b) S. Madhu, R. Gonnade and M. Ravikanth, J. Org. Chem., 2013, 78, 5056 CrossRef CAS PubMed;
(c) Y. Fu, Q. He, D. Zhu, Y. Wang, Y. Gao, H. Cao and J. Cheng, Chem. Commun., 2013, 49, 11266 RSC;
(d) J. Xu, Q. Li, Y. Yue, Y. Guo and S. Shao, Biosens. Bioelectron., 2014, 56, 58 CrossRef CAS PubMed.
-
(a) Y. Kubo, Y. Minowa, T. Shoda and K. Takeshita, Tetrahedron Lett., 2010, 51, 1600 CrossRef CAS PubMed;
(b) Y. Kubo, K. Watanabe, R. Nishiyabu, R. Hata, A. Murakami, T. Shoda and H. Ota, Org. Lett., 2011, 13, 4574 CrossRef CAS PubMed;
(c) Y. Kubo, D. Eguchi, A. Matsumoto, R. Nishiyabu, H. Yakushiji, K. Shigaki and M. Kaneko, J. Mater. Chem. A, 2014, 2, 5204 RSC.
- K. Krzymiński, A. D. Roshal, B. Zadykowicz, A. Białk-Bielińska and A. Sieradzan, J. Phys. Chem. A, 2010, 114, 10550 CrossRef PubMed.
- Y. Kajiwara, A. Nagai, K. Tanaka and Y. Chujo, J. Mater. Chem. C, 2013, 1, 4437 RSC.
- G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991 CrossRef CAS.
- R. C. Reid, J. M. Prausnitz and B. E. Poling, Handbook of the properties of Gases and Liquids, McGraw-Hill, INC, 1987 Search PubMed.
- G. Scatchard, G. M. Kavanagh and L. B. Ticknor, J. Am. Chem. Soc., 1952, 74, 3715 CrossRef CAS.
- Computer Aided Book of Vapor Pressure, ed. S. Ohe, Data Book Publishing, INC., Tokyo, 2nd edn, 1976 Search PubMed.
- To assign each signal (A ∼ D) in more details, NOESY spectrum was measured. However, no cross peaks were obtained between methoxy and isoindole protons.
- G. Weber and F. W. J. Teale, Trans. Faraday Soc., 1957, 53, 646 RSC.
- An excitation spectrum associated with the emission was essentially the same as the absorption spectrum (Fig. S7 in ESI†).
- Integration values for the 1H NMR data were based on that (3H) of OMe signal of the anisole unit. 1H NMR (500 MHz, DMSO-d6) δ (ppm) 3.65 (s, 3H), 3.67 (s, 3H), 3.71 (s, 3H), 3.73 (s, 3H), 6.52 (t, 2H, J = 1.88 Hz), 6.99 (t, 1H, J = 7.33 Hz), 7.00 (t, 1H, J = 7.37 Hz), 7.03–7.07 (m, 2H), 7.08 (dd, 2H, J = 8.65 and 2.08 Hz), 7.15 (d, 1H, J = 8.40 Hz), 7.16 (d, 1H, J = 8.45 Hz), 7.17 (d, 1H, J = 7.85 Hz), 7.20 (d, 1H, J = 7.70 Hz), 7.21 (t, 2H, J = 7.28 Hz), 7.25 (d, 2H, J = 7.95 Hz), 7.35 (d, 1H, J = 6.80 Hz), 7.36 (d, 1H, J = 7.15 Hz), 7.42 (d, 1H, J = 6.00 Hz), 7.43 (d, 1H, J = 5.90 Hz), 7.44–7.49 (m, 6H), 7.99 (dd, 2H, J = 8.75 and 2.23 Hz), 8.10 (dd, 2H, J = 8.25 and 2.60 Hz), 8.47 (s, 2H), 9.60 (s, 2H); FAB MS: m/z 520 [M]+.
- As a control, time course of fluorescence spectra of 1 (5 μM) in the presence of TBAOH (50 μM) was measured in EtOH/H2O (1:1 v/v) at 25 °C. Although, when excited at 550 nm, the fluorescent intensity at 642 nm was somewhat decreased within 30s, the intensity remained at 72% of the initial value (Fig. S9 in ESI†).
- Image J was used as image-processing program.
- Internet website of US-Occupational Safety & Health Administration (OSHA).
- As a preliminary result, we found that saturated vapor of I2 led to a decrease in fluorescence intensity of 1, suggesting that 1 would be applicable to the detection of radioactive iodine-131.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data (Fig. S1–S11). See DOI: 10.1039/c4ra06061j |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.