Photoreduction of CO2 to methanol with hexanuclear molybdenum [Mo6Br14]2− cluster units under visible light irradiation†
Pawan Kumara,
Subodh Kumara,
Stéphane Cordierb,
Serge Paofaib,
Rabah Boukherroub*c and
Suman L. Jain*a
aChemical Sciences Division, CSIR-Indian Institute of Petroleum, Dehradun-248005, India. E-mail: suman@iip.res.in; Fax: +91-135-2660202; Tel: +91-135-2525788
bUniversité de Rennes 1, Institut Sciences Chimiques de Rennes, UR1-CNRS 6226, Equipe Chimie du Solide et Matériaux, Campus de Beaulieu, CS 74205, 35042 Rennes Cedex, France
cInstitut de Recherche Interdisciplinaire (IRI, USR CNRS 3078), Université Lille 1, Parc de la Haute Borne, 50 Avenue de Halley, BP 70478, 59658 Villeneuve d'Ascq, France
First published on 3rd February 2014
Abstract
Octahedral molybdenum clusters were found to be efficient visible light homogeneous photocatalysts for the reduction of carbon dioxide (CO2) to methanol. Photoreduction was carried out by using 20 watt white cold LED flood light in dimethyl formamide/water or acetonitrile/water solutions containing triethylamine as a reductive quencher. Among the two cluster-based compounds, Cs2[Mo6Br14] exhibited higher photocatalytic efficiency and afforded higher yield of methanol than (TBA)2[Mo6Br14] (TBA = tetrabutylammonium). After 24 h illumination, the yield of methanol was 6679.45 and 5550.53 μmol g−1 cat. using Cs2[Mo6Br14] and (TBA)2[Mo6Br14] cluster compounds as photocatalysts, respectively.
The continuously increasing concentration of carbon dioxide (CO2) in the atmosphere has become one of the most serious problems with regard to the greenhouse effect.1 Photocatalytic reduction of CO2 with water for the production of hydrocarbons could be one of the viable solutions to address the issues related to climate change as well as energy shortage.2 Over the past few decades, extensive efforts have been devoted to develop visible light active photocatalytic materials as visible light is ubiquitous, renewable and clean source of energy.3 Although semiconductors such as TiO2, ZnS, and CdS have widely been used as photocatalysts for CO2 reduction, their quantum yields and selectivities of products are low.4 Homogeneous photocatalysts, including transition metal complexes such as ruthenium(II) polypyridine carbonyl complex,5 cobalt(II) trisbipyridine,6 and cobalt(III) macrocycles7 have been widely investigated for this reaction. Enzyme catalysts have also been used for such photoreduction processes.8 Transition metal based molecular complexes are advantageous due to their high quantum efficiencies and high selectivity of products. However, in some instances, a weak absorption in the visible region makes the utility of these complexes limited. This limitation can be overcome by using high nuclearity transition metal cluster complexes having the basic formula, [M6(μ3-X)8L6]2−, which is also written as [Mo6Xi8La6]2− (i = inner; a = apical; X = halogen, L = halogen or functional organic ligand).9 In such face-capped metal atom clusters, the metallic electrons are delocalized on all the metal centers leading to particular intrinsic properties (magnetic, optical, redox processes). These nanosized molecular units exhibit a large absorption window from UV to visible as well as a large emission window from red to infrared due to the delocalization of valence electrons on all metal centers.10 Molybdenum halide based cluster complexes have been known since a long time and have been used for photochemical processes for energy storage, as singlet oxygen photocatalysts and as bio-imaging agents.11 However, to the best of our knowledge, these cluster complexes have not been investigated so far for the photocatalytic reduction of CO2.
In the present paper, we wish to report on the use of octahedral molybdenum cluster-based compounds Cs2[Mo6Br14] (i.e. Cs2[Mo6Bri8Bra6]) and (TBA)2[Mo6Br14] (i.e. (TBA)2[Mo6Bri8Bra6], TBA = tetrabutyl ammonium) as efficient visible light active photocatalysts for the photoreduction of CO2 to methanol by using water as the reaction medium and triethylamine as a sacrificial donor.
The synthesis and characterizations of Cs2[Mo6Br14] and (TBA)2[Mo6Br]14 have been fully described in the literature.12 The structures are based on [Mo6Xi8La6]2− cluster units depicted in Fig. 1 as anionic building blocks and Cs+ and (TBA)+ as counter cations.
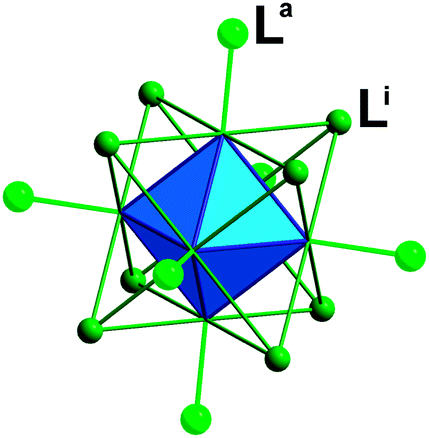 | ||
| Fig. 1 Schematic representation of the [Mo6Bri8La6]2− cluster unit. | ||
Initially, Mo6Br12 (Mo6Bri8Bra–a4/2Bra2) was prepared by the reaction of Br2 with Mo at 750 °C. Then an excision reaction was performed between a stoichiometric amount of MoBr2 and CsBr at 850 °C for two days. Afterwards, Cs2Mo6Br14 was dissolved in dry acetone, filtrated and recrystallized in pure form. (TBA)2[Mo6Br]14 was obtained by precipitation after dissolution of Cs2[Mo6Br14] in a mixture of ethanol and water followed by addition of a two-fold excess of (TBA)Br. Absorption spectra of the synthesized Mo-clusters are displayed in Fig. 2. They exhibit a strong absorption band near 300 nm originating from π–π* absorption, and the two bands at 360–390 and 400–450 nm corresponding to MLCT singlet and triplet absorptions, respectively.13 Furthermore, the Tauc plot was used to determine the bandgap as of the photocatalysts (Fig. 3). The value associated with the point of intersection of the line tangent to the plotted curve inflection point with the horizontal axis (hν axis) gives the band gap Eg. Eg values of 2.42 and 2.41 eV are deduced from the Tauc plots for Cs2Mo6Br14 and (TBA)2[Mo6Br]14, respectively, indicating that these clusters can absorb visible light.
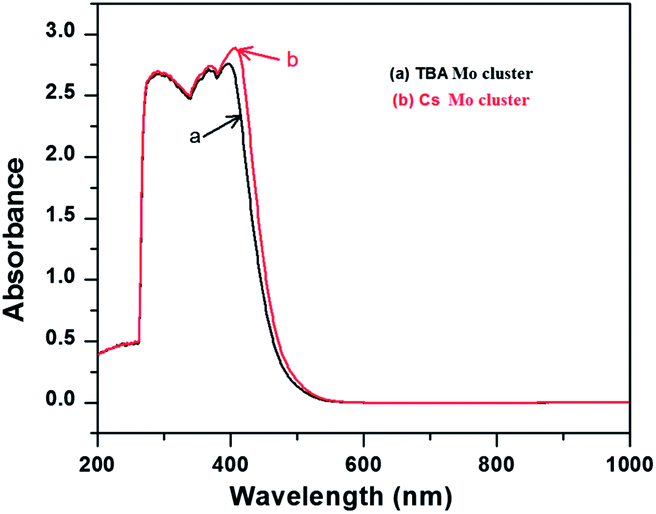 | ||
| Fig. 2 UV-Vis spectra of Mo clusters. | ||
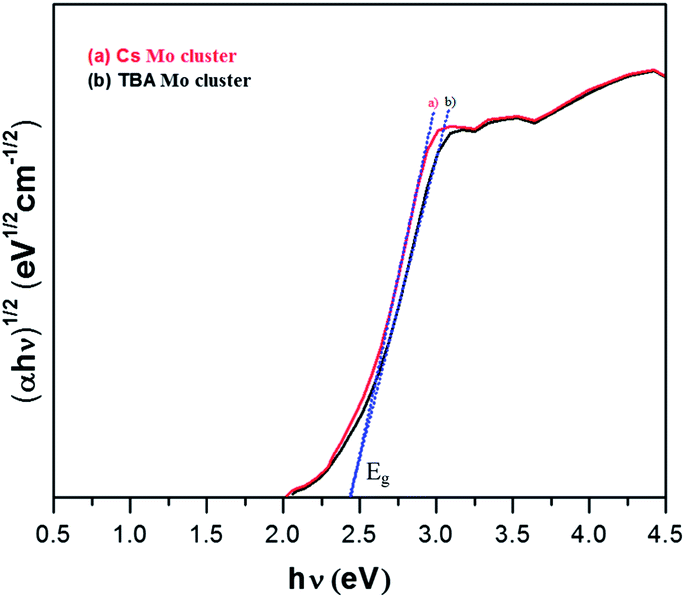 | ||
| Fig. 3 Tauc plot for band gap (Eg) determination of Mo-clusters. | ||
The as obtained catalysts were tested for the photoreduction of CO2. The photocatalytic reduction of CO2 using as synthesized Mo-clusters as catalyst was carried out at room temperature under visible light irradiation by using 20 watt white cold LED flood light in DMF, triethylamine as a sacrificial donor and water as a source of protons. After the photoreduction, 1 μL liquid sample was withdrawn and analyzed in a GC/FID equipped with a 30 m long Stabilwax® w/Integra-Guard® column. Methanol yield was used to evaluate the performance of the catalysts as it was obtained as the major reduction product. Methanol (MeOH) formation rate, RMeOH (μmol g−1 cat.) in the photocatalytic reduction of CO2 by using Cs2[Mo6Br14] and (TBA)2[Mo6Br14] are plotted in Fig. 4. The results suggest that molybdenum cluster Cs2[Mo6Br14] exhibits higher photocatalytic activity as compared to the (TBA)2[Mo6Br14]. After 24 h visible light irradiation, the yield of methanol using Cs2[Mo6Br14] and (TBA)2[Mo6Br14] as catalysts under identical experimental conditions was 6679.45 and 5550.53 μmol g−1 cat., respectively. The turnover numbers for MeOH formation were 13.1 and 12.1 using Cs2[Mo6Br14] and (TBA)2[Mo6Br14], respectively. The quantum yields of methanol formation were determined to be 0.061 and 0.051 for Cs2[Mo6Br14] and (TBA)2[Mo6Br14], respectively. The formation of methanol as the major reaction product was further confirmed by HRGC-MS and HPLC analysis (Fig. S2 and S3†).
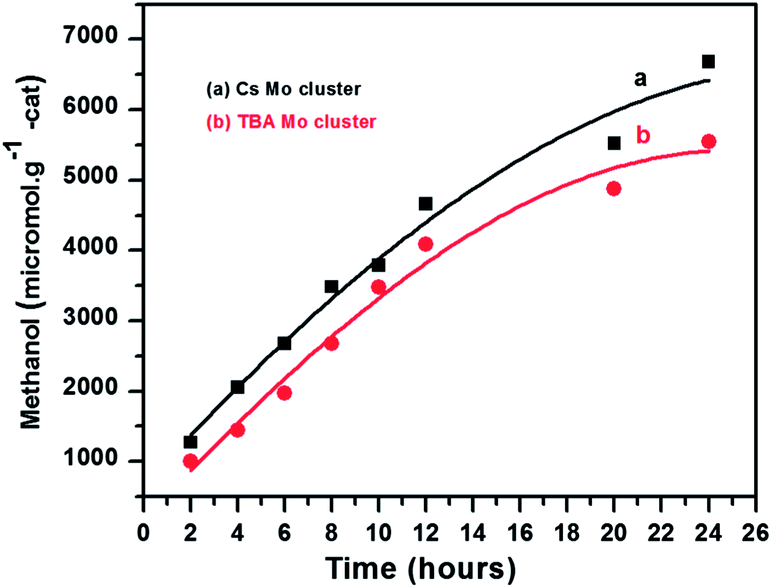 | ||
| Fig. 4 Methanol yield for CO2 photoreduction using (a) Cs2[Mo6Br14] and (b) (TBA)2[Mo6Br14]. | ||
Methanol was identified as the major reaction product using the developed catalytic system. The stoichiometry of the reaction is given in eqn (1).
| CO2 + 6H+ + 6e− → CH3OH + H2O | (1) |
Quantitative measurement of methanol was carried out by using GC-FID. A calibration curve was plotted by injecting an exact amount of methanol (1 μL) using auto samples (Fig. S4†). To evaluate the concentration of methanol produced as a result of the cluster catalyzed reaction, 1.0 μL of the final reaction solution was used for GC measurements. The concentration of methanol was calculated by integrating the peak area for the characteristic methanol band in the chromatogram (Fig. S1†).
Blank experiments, one under visible light irradiation without using photocatalyst and another under dark condition without irradiation using Cs2[Mo6Br14] cluster as catalyst under described experimental conditions, showed that there was no organic product formation for long periods of exposure (48 h).
To establish the formation of methanol from the photocatalytic reduction of CO2 and not deriving from DMF, we have performed the photoreduction of CO2 in another polar aprotic solvent i.e. acetonitrile under identical experimental conditions. The yield of methanol in acetonitrile was found to be comparable to that obtained in DMF (Fig. S3†). Furthermore, we carried out the photoreaction by using acetonitrile and catalyst in the absence of carbon dioxide under identical reaction conditions. No methanol formation was observed, confirming that the methanol is being formed from the photoreduction of CO2.
Importantly, the yield of methanol observed in the present reduction of CO2 is higher than the values previously reported for metal based complexes such as cobalt phthalocyanine (1032 μmol g−1 cat.), ruthenium phosphine (2210 μmol g−1 cat.)-catalyzed photoreductions of CO2 to methanol.14
The exact mechanism of the reaction is not clear at this stage. However, based on the literature reports it is assumed that the Mo-cluster molecule might be photoactivated to the triplet metal-to-ligand charge transfer (3MLCT) excited state, which is quenched by tertiary amine (triethylamine), as the first step of the photocatalytic reaction, to generate the one-electron reduced (OER) species of the molybdenum complexes.15,16 In the next step, the intermediate OER species react with CO2 to give radical anion of carbon dioxide (CO2−˙), which subsequently reacts with protons to produce methanol as depicted in Scheme 1.
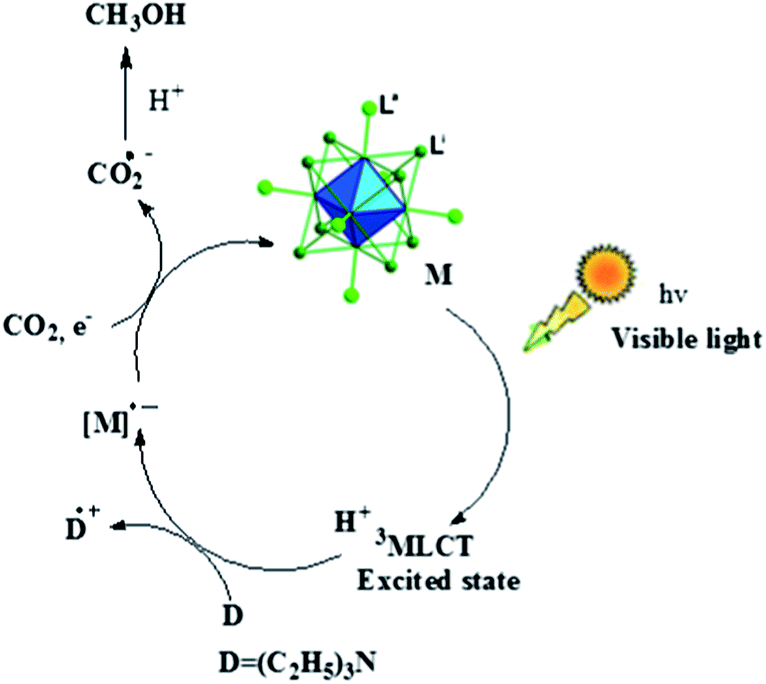 | ||
| Scheme 1 Possible mechanistic pathway. | ||
In summary, we have demonstrated that Cs2[Mo6Br14] and (TBA)2[Mo6Br14] cluster compounds exhibit excellent photocatalytic performance for the photoreduction of CO2 under visible light irradiation to give methanol selectively. Among the two cluster-based compounds, Cs2[Mo6Br14] exhibited higher photocatalytic efficiency and afforded higher yield of methanol than (TBA)2[Mo6Br14]. After 24 h visible light irradiation, the yields of methanol were 6679.45 and 5550.53 μmol g−1 cat. using Cs2[Mo6Br14] and (TBA)2[Mo6Br14] as photocatalysts, respectively. The results provided in this work hold promise in view of various photocatalytic applications of the Mo6 clusters under solar light irradiation.
We are thankful to the Director, CSIR-IIP for his kind permission to publish these results. PK acknowledges the CSIR, New Delhi, for the Research Fellowship. RB acknowledges financial support from the CNRS, the Université Lille1 and the Nord Pas de Calais region.
Experimental procedure for photocatalytic reduction of CO2
Photocatalytic reduction of CO2 was performed in a 60 mL borosil tube of 4 cm in diameter. Photoirradiation was carried out under visible light by using 20 watt white cold LED flood light (Model no.-HP-FL-20W-F-Hope LED Opto-Electric Co. Ltd). The reaction vessel was kept about 2 cm far away from the light source and the intensity of the light at the vessel was 85 W m−2 as measured by intensity meter. The vessel was initially charged with DMF (30 mL), triethylamine (10 mL) and deionized water (10 mL) and then the solution was degassed by purging nitrogen for 30 min under vigorous stirring. Then the photocatalyst (0.1 g) was added to the solution and the resulting mixture was saturated with carbon dioxide. The vessel was sealed, irradiated with light source and the samples were collected in every 2 h intervals. The samples were collected by using a long needle and the catalyst was removed with the help of a syringe filter (2 nm PTFE, 13 mm diameter). Quantitative determination was achieved by using Gas Chromatography FID using a flow rate of 0.5 mL min−1, injector temperature: 250 °C, and FID detector temperature: 275 °C. A calibration curve was used for quantification and for confirmation of linear response of GC-FID system.Blank reactions were conducted to ensure that methanol production was due to the photoreduction of CO2, and to eliminate surrounding interference. One blank was carried out by illuminating the solution in the absence of photocatalyst, and another was performed in the dark in the presence of photocatalyst under the identical experimental conditions. An additional blank test was performed by illuminating the reaction mixture in the presence of photocatalyst by filling N2 rather than CO2. No product was detected in the above three blank tests.
Notes and references
- M. M. Halmann and M. Steinberg, Greenhouse Gas Carbon Dioxide Mitigation Science and Technology, Lewis Publishers, 1999 Search PubMed.
- (a) G. A. Olah, Angew. Chem., Int. Ed., 2013, 52, 104–107 CrossRef CAS PubMed; (b) E. E. Benson, C. P. Kubiak, A. J. Sathruma and J. M. Smiejaa, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- (a) A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed; (b) S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS PubMed; (c) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kuhn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed.
- (a) H. Fujiwara, H. Hosokawa, K. Murakoshi, Y. Wada, S. Yanagida, T. Okada and H. Kobayashi, J. Phys. Chem. B, 1997, 101, 8270–8278 CrossRef CAS; (b) K. Ikeue, H. Yamashita, M. Anpo and T. Takewaki, J. Phys. Chem. B, 2001, 105, 8350–8355 CrossRef CAS.
- (a) H. Ishida, K. Tanaka and T. Tanaka, Organometallics, 1987, 6, 181 CrossRef CAS; (b) C. M. Bolinger, N. Story, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1988, 27, 4582 CrossRef CAS.
- J. Hawecker, J. M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1984, 328 RSC.
- (a) J. Grodkowski, D. Behar, P. Neta and P. Hambright, J. Phys. Chem. A, 1997, 101, 248–254 CrossRef CAS; (b) D. Behar, T. Dhanasekaran, P. Neta, C. M. Hosten, D. Ejeh, P. Hambright and E. Fujita, J. Phys. Chem. A, 1998, 102, 2870–2877 CrossRef CAS; (c) J. Grodkowski, T. Dhanasekaran, P. Neta, P. Hambright, B. S. Brunschwig, K. Shinozaki and E. Fujita, J. Phys. Chem. A, 2000, 104, 11332–11339 CrossRef CAS; (d) J. Grodkowski and P. Neta, J. Phys. Chem. A, 2000, 104, 1848–1853 CrossRef CAS; (e) J. Grodkowski, P. Neta and E. Fujita, J. Phys. Chem. A, 2002, 106, 4772–4778 CrossRef CAS.
- T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 132, 2132–2133 CrossRef CAS PubMed.
- (a) C. Mealli, J. A. López, Y. Sun and M. J. Calhorda, Inorg. Chim. Acta, 1993, 213, 199–212 CrossRef CAS; (b) L. F. Szczepura, B. A. Ooro and S. R. Wilson, J. Chem. Soc., Dalton Trans., 2002, 3112–3116 RSC.
- (a) J. A. Jackson, C. Turro, M. D. Newsham and D. G. Nocera, J. Phys. Chem., 1990, 84, 4500 CrossRef; (b) S. Cordier, K. Kirakci, D. Méry, C. Perrin and D. Astruc, Inorg. Chim. Acta, 2006, 359, 1705 CrossRef CAS PubMed.
- (a) A. Barras, M. R. Das, R. R. Devarapalli, M. V. Shelke, S. Cordier, S. Szunerits and R. Boukherroub, Appl. Catal., B, 2013, 130, 270–276 CrossRef PubMed; (b) A. Barras, S. Cordier and R. Boukherroub, Appl. Catal., B, 2012, 123, 1–8 CrossRef PubMed.
- K. Kirakci, S. Cordier and C. Perrin, Z. Anorg. Allg. Chem., 2005, 631, 411–416 CrossRef CAS.
- S. Sato, T. Morikawa, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 52, 988–992 CrossRef CAS PubMed.
- (a) S. Liu, Z. Zhao and Z. Wang, Photochem. Photobiol. Sci., 2007, 6, 695–700 RSC; (b) S. Wesselbaum, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 7499–7502 CrossRef CAS PubMed.
- (a) K. Koike, H. Hori, M. Ishizuka, J. R. Westwell, K. Takeuchi, T. Ibusuki, K. Enjouji, H. Konno, K. Sakamoto and O. Ishitani, Organometallics, 1997, 16, 5724–5729 CrossRef CAS; (b) P. Kurz, B. Probst, B. Spingler and R. Alberto, Eur. J. Inorg. Chem., 2006, 2966–2974 CrossRef CAS; (c) C. Kutal, M. A. Weber, G. Ferraudi and D. Geiger, Organometallics, 1985, 4, 2161–2166 CrossRef CAS.
- H. Takeda, K. Koike, H. Inoue and O. Ishitani, J. Am. Chem. Soc., 2008, 130, 2023–2031 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra47255h |
| This journal is © The Royal Society of Chemistry 2014 |