 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electric-field effects on the [FeFe]-hydrogenase active site†
Arndt R. Finkelmann, Martin T. Stiebritz and Markus Reiher*
ETH Zürich, Laboratorium für Physikalische Chemie, Wolfgang-Pauli-Strasse 10, 8093 Zürich, Switzerland. E-mail: markus.reiher@phys.chem.ethz.ch; Fax: +41 446331594; Tel: +41 446334308
First published on 11th July 2013
Abstract
The effect of a homogeneous electric field—as exerted by the protein environment and by an electrode potential—on the reactivity of the active site of [FeFe] hydrogenases is unravelled by density functional theory calculations.
Hydrogenases have been applied in electrochemical processes to produce molecular dihydrogen for clean-energy technologies.1 Electric fields affect the electronic structure and thus the reactivity of the active site. It is known that [NiFe] hydrogenases adsorbed on electrodes experience dispersion of reaction rates.2 This can be attributed to different orientations at the electron-transfer interface, though other effects, like double-layer potentials, might also be important.2,3 In general, little is known about the details of the interaction of stable and reactive intermediates with external fields in such situations. Here, we systematically investigate to what extent such fields can modify the energetics of key reaction steps at the active site of [FeFe] hydrogenases, which is the most desirable candidate for dihydrogen production. We should note that studies in a similar spirit have been performed for cytochrome P450 and proton transfer in a DNA base pair.4
[FeFe] hydrogenases catalyze the reversible formation of H2 with a high turnover frequency.5 The active site, the H-cluster, consists of a [4Fe–4S] cubane linked via a cysteine bridge to the so called [2Fe]H subcluster. The latter comprises two iron atoms, one proximal (Fep) and one distal (Fed) to the cubane, and an azadithiolate bridging ligand.6 H2 formation at the H-cluster comprises proton/electron transfer steps and proceeds via a H− species terminally bound to Fed.7,8 A crucial decomposition reaction, which one seeks to avoid, is the O2-induced inhibition that initiates the degradation of the enzyme starting with O2 coordination to Fed.9
Chemical processes at active sites of such metalloproteins are usually well described by a quantum mechanical (QM) model that considers only the active site and some important amino acid residues.10 While the electric field of a surrounding protein may be approximated by a polarizable dielectric continuum model in a sufficiently large QM model, the strength of electric fields exerted on proteins adsorbed on polarized electrodes is difficult to assess.11
In order to systematically screen electric-field effects on our 96-atom QM model (Fig. 1, left), we first need to understand the magnitude and direction of the field exerted by the protein itself on the active site. We extract this information from the electrostatic potential that we obtain by numerical solution of the Poisson–Boltzmann equation for the crystal structure of [FeFe] hydrogenase from Clostridium pasteurianum (pdb code: 3C8Y) and from Desulfovibrio desulfuricans (pdb code: 1HFE) (detailed information can be found in the ESI†).12 The field within a protein is then obtained as the derivative of the electrostatic potential at the position of interest; in our case at the position of the Fed atom. The resulting local field vectors at Fed, ![[E with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0045_20d1.gif) 0prot (
0prot (![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) Fed), are shown in Fig. 1 (red vectors) and were found to have a length of 0.0038 for C. pasteurianum and of 0.0026 for D. desulfuricans, both measured in Hartree atomic units (a.u.), in which the elementary charge and 4πε0 assume a value of one. Remarkably, these two field strengths are very similar and they also point in almost the same direction (see Fig. 1), although the sequence identity of the two enzymes is only 30%. Based on this information, a homogeneous electric field, 0prot() = 0prot (Fed), was derived to enter into the Kohn–Sham equations in the BP86-D3/def2-TZVP calculations on the 96-atom QM model of C. pasteurianum (see ESI† for details).
Fed), are shown in Fig. 1 (red vectors) and were found to have a length of 0.0038 for C. pasteurianum and of 0.0026 for D. desulfuricans, both measured in Hartree atomic units (a.u.), in which the elementary charge and 4πε0 assume a value of one. Remarkably, these two field strengths are very similar and they also point in almost the same direction (see Fig. 1), although the sequence identity of the two enzymes is only 30%. Based on this information, a homogeneous electric field, 0prot() = 0prot (Fed), was derived to enter into the Kohn–Sham equations in the BP86-D3/def2-TZVP calculations on the 96-atom QM model of C. pasteurianum (see ESI† for details).
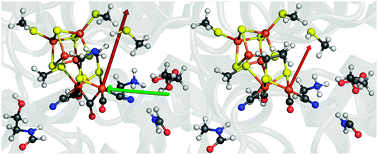 | ||
| Fig. 1 Left: 96-atom QM model of the active site extracted from Clostridium pasteurianum (pdb code: 3C8Y). The red arrow represents the effective field vector of the protein surrounding at Fed (0prot) and the green arrow indicates the arbitrarily chosen external field along the Fe–Fe bond (Fe–Fe). Right: corresponding presentation for Desulfovibrio desulfuricans (pdb code: 1HFE), where the red arrow again represents the effective field at Fed. Color code: C, grey; Fe, brown; H, white; N, blue; O, red; S, yellow. | ||
The external field in an electrochemical experiment would be screened by the protein and water surroundings of the H-cluster. Thus, in a sequence of calculations the field strength was varied in steps of ±0.001, ±0.002, ±0.003 a.u. around the reference of 0.0038 a.u. (denoted ±1prot, ±2prot and ±3prot) in order to investigate a possible external-field effect. In a second set of calculations, a field pointing from Fep to Fed, Fe–Fe, with a fixed strength of 0.0038 a.u. was applied to investigate the effect of a change in the field direction. Fe–Fe, which is perpendicular to the plane that separates the [2Fe]H subcluster and the cubane, is particularly interesting as it can induce charge transfer between both parts. Finally, calculations were performed with inverted field directions, denoted by a minus sign, i.e., −. Reactants (including barriers) investigated with respect to field effects are depicted in Fig. 2.
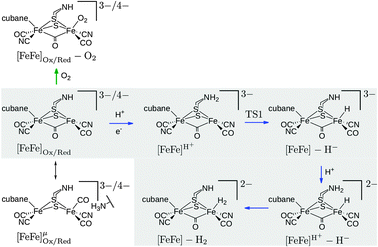 | ||
| Fig. 2 Investigated intermediates and reactions. The reactions in the grey box correspond to reaction steps of the catalytic cycle (blue arrows). O2 addition is indicated by a green arrow. The oxidized state corresponds to the formal oxidation state Fep(II)Fed(I), the reduced state to Fep(I)Fed(I). The oxidized species are doublets (S = 1/2), all other species are singlets (S = 0). Note that the Lewis structures shown do not represent the full QM model (cf.Fig. 1). The charge given is the charge of the full QM model. | ||
The change in partial charges on individual atoms induced by the electric field is small and discussed in the ESI.† A clear trend is only revealed when the partial charges of the [2Fe]H subcluster and the cubane (including sulphur atoms of coordinating cysteines) are separately added up. For all intermediates the field in the direction of 0prot induces a shift of electron density from the cubane to the [2Fe]H subcluster. The negative charge on the cubane decreases by +0.09e to +0.17e for different intermediates (and the strongest field +3prot). The negative charge on the [2Fe]H subcluster increases by −0.05e to −0.13e, accordingly. The decrease in negative charge on the cubane is always smaller than the increase in negative charge on the [2Fe]H subcluster because the charge is shifted to the amino acid environment of the [2Fe]H subcluster. Fe–Fe induces a transfer of negative charge from the cubane to the [2Fe]H subcluster. Inversion of the field direction leads to the expected inversion of the charge shifts. Hence, the protein field clearly polarizes the active site towards the [2Fe]H subcluster. The inverse field has a less pronounced effect. The polarization could be enhanced by a field in the Fe–Fe direction (or lowered by −Fe–Fe). The cubane serves as a charge reservoir and Fe–Fe and 0prot increase charge donation from the cubane and to the [2Fe]H subcluster.
The effect of the field on reaction energies of reaction steps in Fig. 2 of the catalytic cycle8,13 are summarized in Table 1 (see ESI† for data of inverted fields). In reaction [FeFe]H+ → TSI → [FeFe] − H− (rows 1–3 in Table 1) a proton is transferred from the bridgehead amine group to Fed to form the terminal hydride species. This reaction becomes less exothermic with increasing field in strength. For +3prot it is +10.7 kcal mol−1 less exothermic than in the field-free case. The reaction energy changes linearly with the field. For each increase in field strength the reaction becomes +1.6 kcal mol−1 less exothermic. By contrast, Fe–Fe leads to a −4.9 kcal mol−1 more exothermic reaction. For a less exothermic reaction the barrier is higher, while it is lower for a more exothermic reaction, but the change in barrier height is smaller than the change in reaction energy. The effect of 0prot, ±3prot and −0prot on the energy profile of the reaction [FeFe]H+ → TS1 → [FeFe] − H− (terminal hydride formation) is shown in Fig. 3. For the H2 formation reaction, [FeFe]H+ − H− → [FeFe] − H2, the reaction is up to +4.3 kcal mol−1 less exothermic with increasing field in prot direction. This decrease in exothermicity is linear as well. Importantly, both fields, Fe–Fe and −Fe–Fe, lead to a decreased exothermicity (+5.5 and +4.0 kcal mol−1, resp.) for H2 formation. Hence, 0prot disfavors both proton transfer reactions, while Fe–Fe favors hydride transfer but disfavors H2 formation.
−3prot| = 0.0008, |−2prot| = 0.0018, |−1prot| = 0.0028, |0prot| = 0.0038, |+1prot| = 0.0048, |+2prot| = 0.0058, |+3prot| = 0.0068, |Fe–Fe| = |−Fe–Fe| = 0.0038
| Intermediate | No field | −3prot | −2prot | −1prot | 0prot | +1prot | +2prot | +3prot | Fe–Fe | −Fe–Fe |
|---|---|---|---|---|---|---|---|---|---|---|
| H2 formation | ||||||||||
 | −20.7 | −19.5 | −17.9 | −16.3 | −14.8 | −13.2 | −11.6 | −10.0 | −25.6 | −17.5 |
| [FeFe]H+ → TS1 | 4.0 | 4.0 | 4.4 | 4.7 | 5.1 | 5.5 | 5.8 | 6.2 | 3.4 | 4.2 |
| [FeFe]H+ − H− → [FeFe] − H2 | −6.4 | −5.8 | −5.2 | −4.6 | −4.0 | −3.4 | −2.8 | −2.1 | −0.9 | −2.4 |
| O2 additions | ||||||||||
| [FeFe]Ox → [FeFe]Ox − O2 | −23.6 | −23.4 | −22.9 | −22.2 | −21.6 | −21.0 | −20.3 | −19.6 | −19.7 | −27.2 |
| [FeFe]Red → [FeFe]Red − O2 | −17.2 | −16.8 | −16.3 | −15.8 | −15.3 | −14.8 | −14.3 | −13.8 | −15.6 | −20.3 |
| Reduction | ||||||||||
| [FeFe]Ox → [FeFe]Red | 70.0 | 45.5 | 14.9 | −15.8 | −46.4 | −77.0 | −107.6 | −138.2 | 62.8 | 78.4 |
| [FeFe]μOx → [FeFe]μRed | 77.9 | 53.5 | 22.4 | −8.6 | −39.6 | −70.7 | −101.7 | −132.8 | 73.7 | 83.5 |
![Energy profiles for the reaction [FeFe]H+ → TS1 → [FeFe] − H− for different field strengths. The barriers are given in kcal mol−1.](/image/article/2013/CC/c3cc44112a/c3cc44112a-f3.gif) | ||
| Fig. 3 Energy profiles for the reaction [FeFe]H+ → TS1 → [FeFe] − H− for different field strengths. The barriers are given in kcal mol−1. | ||
The reduction of the H2-coordinated species and the species with a free coordination site in the μ-bridging position are, of course, most affected by the electric field (last two rows in Table 1). Surprisingly, Fe–Fe has only a small effect on the energies. Moreover, with +3prot, the O2 coordination energies become +4.0 and +3.4 kcal mol−1 less exothermic for the oxidized and reduced oxidation states, respectively, although the coordination reaction remains strongly exothermic.
To conclude, the polarization of the H-cluster induced by Fe–Fe is stronger than that induced by 0prot. Inversion of both fields leads to inversion of differential polarization but with smaller magnitude. This is due to the excess of charge on the cubane in most intermediates. 0prot leads to a reduced exothermicity of hydride and H2 formation which could be beneficial for the enzyme to work close to the thermodynamic equilibrium. Our results explicitly show that no crucial field-induced modulations of barrier heights and reaction energies are observed. The reversibility of the whole H2 formation cycle is not affected. Still, we might speculate that the [FeFe] hydrogenase from C. pasteurianum should feature an activation pattern that favors H2 oxidation and disfavors H2 formation compared to the one from D. desulfuricans because the former exerts a stronger field at the ligand-binding site. The measured activity data appear to indicate such a trend for H2 evolution.14
This work has been financially supported by the Schweizerischer Nationalfonds (Project no. 200021L_138536).
References
- K. A. Vincent, A. Parker and F. A. Armstrong, Chem. Rev., 2007, 107, 4366–4413 CrossRef CAS.
- C. Léger, A. K. Jones, S. P. J. Albracht and F. A. Armstrong, J. Phys. Chem. B, 2002, 106, 13058–13063 CrossRef.
- J. Hirst and F. A. Armstrong, Anal. Chem., 1998, 70, 5062–5071 CrossRef CAS.
- S. Shaik, S. P. de Visser and D. Kumar, J. Am. Chem. Soc., 2004, 126, 11746–11749 CrossRef CAS; J. P. Ceron-Carrasco and D. Jacquemin, Phys. Chem. Chem. Phys., 2013, 15, 4548–4553 RSC.
- C. Madden, M. D. Vaughn, I. Díez-Pérez, K. A. Brown, P. W. King, D. Gust, A. L. Moore and T. A. Moore, J. Am. Chem. Soc., 2012, 134, 1577–1582 CrossRef CAS.
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273–4303 CrossRef CAS; A. Silakov, B. Wenk, E. Reijerse and W. Lubitz, Phys. Chem. Chem. Phys., 2009, 11, 6592–6599 RSC; G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J.-M. Mouseca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CrossRef.
- W. Lubitz, E. Reijerse and M. van Gastel, Chem. Rev., 2007, 107, 4331–4365 CrossRef CAS; P. E. M. Siegbahn, J. W. Tye and M. B. Hall, Chem. Rev., 2007, 107, 4414–4435 CrossRef; D. W. Mulder, M. W. Ratzloff, E. M. Shepard, A. S. Byer, S. M. Noone, J. W. Peters, J. B. Broderick and P. W. King, J. Am. Chem. Soc., 2013, 135, 6921–6929 CrossRef; A. Adamska, A. Silakov, C. Lambertz, O. Rüdiger, T. Happe, E. Reijerse and W. Lubitz, Angew. Chem., Int. Ed., 2012, 51, 11458–11462 CrossRef.
- H.-J. Fan and M. B. Hall, J. Am. Chem. Soc., 2001, 123, 3828–3829 CrossRef CAS; Z.-P. Liu and P. Hu, J. Chem. Phys., 2002, 117, 8177–8180 CrossRef.
- M. T. Stiebritz and M. Reiher, Inorg. Chem., 2009, 48, 7127–7140 CrossRef CAS; M. T. Stiebritz and M. Reiher, Inorg. Chem., 2010, 49, 8645 CrossRef; S. T. Stripp, G. Goldet, C. Brandmayr, O. Sanganas, K. A. Vincent, M. Haumann, F. A. Armstrong and T. Happe, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17331–17336 CrossRef; M. K. Bruska, M. T. Stiebritz and M. Reiher, J. Am. Chem. Soc., 2011, 133, 20588–20603 CrossRef; M. T. Stiebritz and M. Reiher, Chem. Sci., 2012, 3, 1739–1751 RSC; M. K. Bruska, M. T. Stiebritz and M. Reiher, Dalton Trans., 2013, 42, 8729–8735 RSC; M. Bergeler, M. T. Stiebritz and M. Reiher, ChemPlusChem, 2013 DOI:10.1002/cplu.201300186.
- P. E. M. Siegbahn and F. Himo, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 323–336 CrossRef CAS; M. Podewitz, M. T. Stiebritz and M. Reiher, Faraday Discuss., 2011, 148, 119–135 RSC.
- C. P. Smith and H. S. White, Anal. Chem., 1992, 64, 2398–2405 CrossRef CAS; E. Gileadi, Physical Electrochemistry: Fundamentals, Techniques and Applications, Wiley-VCH, Weinheim, Germany, 2011 Search PubMed.
- A. S. Pandey, T. V. Harris, L. J. Giles, J. W. Peters and R. K. Szilagyi, J. Am. Chem. Soc., 2008, 130, 4533–4540 CrossRef CAS; Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Structure, 1999, 7, 13–23 CrossRef.
- G. Zampella, C. Greco, P. Fantucci and L. De Gioia, Inorg. Chem., 2006, 45, 4109–4118 CrossRef CAS; C. Greco, M. Bruschi, L. De Gioia and U. Ryde, Inorg. Chem., 2007, 46, 5911–5921 CrossRef; A. R. Finkelmann, M. T. Stiebritz, M. Reiher, 2013, submitted.
- E. C. Hatchikian, N. Forget, V. M. Fernandez, R. Williams and R. Cammack, Eur. J. Biochem., 1992, 209, 357–365 CrossRef CAS; M. W. Adams, Biochim. Biophys. Acta, 1990, 1020, 115–145 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, additional tables and structures. See DOI: 10.1039/c3cc44112a |
| This journal is © The Royal Society of Chemistry 2013 |