Hybridizing photoactive zeolites with graphene: a powerful strategy towards superior photocatalytic properties†
Z.
Ren
a,
E.
Kim
b,
S. W.
Pattinson
a,
K. S.
Subrahmanyam
c,
C. N. R.
Rao
c,
A. K.
Cheetham
a and
D.
Eder
*ad
aDepartment of Materials Science and Metallurgy, University of Cambridge, Pembroke Street, Cambridge, UK CB2 3QZ
bCavendish Laboratory, Department of Physics, University of Cambridge, Madingley Road, Cambridge, UK CB3 0HE
cChemistry & Physics of Materials Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur P.O, Bangalore, 560 064, India
dInstitut für Physikalische Chemie, Universität Münster, Corrensstrasse 28/30, 48149, Münster, Germany. E-mail: dominik.eder@uni-muenster.de; Fax: +49 (0) 251 83 29159; Tel: +49 (0) 251 83 23406
First published on 27th October 2011
Abstract
Research on zeolitic materials has recently taken off in a new direction, driven by the desire to design nanoscale hierarchical architectures with specific chemical functionalities. From both, a scientific and industrial perspective, photoactive zeolites, such as titanosilicates (TS-1), are among the most interesting candidates and research has focused on maximising their photocatalytic performance by means of nanostructuring and electronic modification. In this work, we employ graphene to create a new class of photocatalysts that truly advances semi-conductor photocatalysis to its next generation. We demonstrate that the photocatalytic activity of TS-1 for the degradation of organic dyes can be enhanced through graphene by more than 25 times. Responsible for this remarkable improvement are (1) a more efficient electron–hole separation via interfacial charge transfer processes and (2) surprising beneficial effects of graphene on the crystal morphology, the porosity and the adsorption behaviour of the zeolite.
Introduction
Within only a few years, graphene has established itself as one of the most intensely studied materials due to its exceptional physical properties, including high intrinsic mobility, high Young's modulus, excellent thermal conductivity and optical transmittance.1–3 These characteristics commend graphene as the material of choice in a variety of applications including transparent conducting electrodes, field emission/electric devices, and recently biosensing and electrochemical applications.4 Graphene has also attracted wide interest as a functional filler in nanocomposites, for its ability to improve the electronic and mechanical properties of polymers.5Nanocarbon–inorganic hybrids are a new class of composite materials,6 which are created by hybridizing carbon nanostructures, such as carbon nanotubes (CNTs) and graphene, with inorganic glasses or ceramics. In contrast to classical nanocomposites, where a low volume fraction of the carbon component is mixed into a matrix of conducting polymers or ceramics, hybrids are materials in which the nanocarbon is coated with a thin layer of the inorganic compound, so maximizing the interfacial area. The appeal of these hybrid materials arises especially from charge and energy transfer processes through this interface, which create synergistic effects that lead to enhanced and even novel properties, distinct from those of either building block. Although still at an early stage of research, nanocarbon–inorganic hybrids have shown superior performance as supercapacitors, batteries, fuel cells, photocatalysts and in field emission devices.6 Yet, there have been only very few reports on the hybridization of graphene with inorganic compounds, mainly focusing on electrochemical applications.7–13
Research on zeolitic materials has recently regained tremendous interest, driven by the desire to design nanoscale hierarchical architectures with specific chemical functionalities.14 Zeolites are generally classified as aluminosilicates with channels and cages of strictly regular dimensions in the nanometre or subnanometer length scale, termed micropores (0.5–2 nm). With very large surface areas, excellent adsorbent capacity and compositional variability, which allows control over the size and arrangement of the micropores as well as their internal surface chemistry (e.g. acidic/basic, hydrophobic/hydrophilic), zeolites have been used as molecular sieves, catalyst supports, and as membranes for gas separation and ion exchange. The incorporation of a heteroatom into the zeolitic framework, such as in titanium silicalite (i.e. TS-1),15 has expanded the range of applications by offering interesting new functionalities, such as photocatalytic activity.16,17 In this work, we demonstrate that hybridizing functional inorganic nanomaterials (i.e. the zeolitic TS-1) with CNTs and graphene creates a new generation of photocatalysts, due to (i) the potential of graphene to shape the size and morphology of TS-1, and (ii) synergistic effects based on interfacial heat and charge transfer processes.
Few-layer graphene (HG) was synthesized by direct current arc discharge of graphite in a water-cooled stainless steel chamber filled with a mixture of hydrogen and helium without using any catalyst18 and details are provided in the Experimental section. Scanning electron microscopy (SEM, JEOL 5800) and high-resolution transmission electron microscopy (HRTEM, FEI Tecnai F20 FEGTEM)) revealed that the as-grown graphene sample contained large (several μm) few-layer graphene flakes that were aggregated into loose flower-like structures (Fig. 1a and b). Almost all individual graphene flakes consisted of 2–3 layers (Fig. 1c). This was confirmed by atomic force microscopy (AFM). Fig. 1d shows a typical AFM line scan across a graphene flake with heights of about 0.7 and 1.1 nm, which correspond to two and three graphite layers, respectively. The sample was further characterized by Raman spectroscopy (Fig. 1e). As expected for an excitation at 633 nm, the D, G and 2D bands of graphene are positioned at 1315, 1564 and 2645 cm−1, respectively. The bands are rather narrow, which indicates good crystallinity, while a D/G ratio of about 0.4 is characteristic of relatively small (μm sized) graphene flakes with large numbers of edge atoms.19 Importantly, the 2D band has a width at half maximum of about 60 cm−1, which is typical of graphene with two layers,20 thus confirming both the HRTEM and AFM results. Finally, XRD diffraction (Fig. 3g) shows a 002 interlayer peak with a specific shape and width that is characteristic for few-layer graphene with 2–3 layers.21
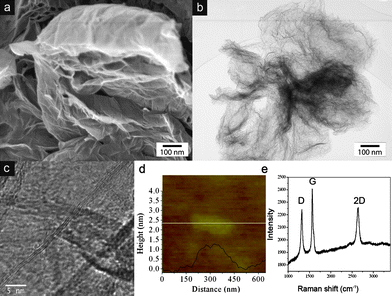 | ||
| Fig. 1 (a, b) SEM and TEM images of the as-grown graphene aggregates, (c) HRTEM and (d) AFM line scan of an individual graphene flake, and (e) Raman spectrum of a graphene aggregate. The combined characterisation indicates the presence of graphene with predominantly 2–3 layers. | ||
Graphene-TS1 and the TS-1 reference samples were synthesized with an in situ microwave-assisted solvothermal technique from metal–organic precursors.17 The synthesis parameters, concentrations and heat treatment conditions are detailed in the Experimental section. The key to this process is benzyl alcohol, which – as first demonstrated for TiO2 on CNTs22–24 – enables the deposition of continuous and uniform inorganic coatings onto the hydrophobic carbon surface without the need for chemical functionalisation. Benzyl alcohol works in three ways: (a) as a surfactant it supports the dispersion of the nanocarbon in aqueous solutions, (b) as a particle shaper it affects the shape and morphology of the inorganic compound, and (c) as an aromatic linking agent it adsorbs on graphene via π–π interactions and attracts the hydrophilic metal–organic precursors with its hydroxyl groups. This simple and versatile benzyl alcohol route has since been successfully applied for variety of metal oxide nanoparticles on CNTs.25
The addition of graphene during the synthesis had a remarkable effect on both the shape and dimensions of the TS-1 particles, as documented by TEM in Fig. 2. Without graphene (reference sample), TS-1 crystallized into large (>300 nm) particles with the common shape of hexagonal prisms (Fig. S1a, ESI†). With the addition of small amounts of graphene, their shape changed dramatically, first to ellipsoidal particles (<1 wt%, Fig. 2a), then to rectangular plates 1–5 wt%) and finally to spherical nanoparticles (>5 wt%). The final nanoparticles were considerably smaller than the other morphologies (i.e. 10 nm vs. 300 nm), and uniform in size and shape even up to high graphene concentrations (20 wt%). A dominant particle-shaping role of benzyl alcohol, similar to that observed for rutile-TiO2 crystals during phase transformation from anatase,23 can be ruled out since its concentration was identical for all samples. Thus, it seems that graphene is the main source for this effect.
 | ||
| Fig. 2 TEM images of graphene-TS1 hybrids synthesized with (a) 1 wt%, (b) 5 wt%, (c) 10 wt% and (d) 20 wt% graphene. The images document the effect of graphene on both the shape and size of the TS-1 particles. | ||
It is remarkable, however, that the particle size changed abruptly and not continuously, as recently demonstrated with CNTs for TS-117 and TiO2.26 It is important to note that, in the case of CNTs, the decrease in particle size was not accompanied by a change in morphology. The so-called heat-sink effect26 has been identified as the reason for the continuous change in particle size: local heat, released during crystallization and phase transformation treatments of the inorganic coating, is conducted away by the CNTs rather than consumed in a grain growth. This requires heat transfer from the inorganic coating to the CNTs. Hence, the thinner the coating, the more efficient the heat transfer and the smaller the particles. The thickness of the coating and the size of the final TS-1 particles thus decrease continuously with increasing CNT concentration. In contrast, the abrupt decrease in particle size and the simultaneous change in morphology in this work suggest a different mechanism for graphene, although some contribution through the heat-sink effect cannot be entirely excluded. We are further investigating the role of graphene in this effect using computer simulations. Preliminary results, however, suggest that the preferential adsorption of graphene on some TS-1 crystal surfaces at an early stage directs their growth in a way that favours rectangular plates over hexagonal prisms. In the process, the plates become thinner until they reach a thickness of 10–20 nm (Fig. 2b), which is roughly that of the small TS-1 nanoparticles. It is feasible that graphene, being adsorbed on the broadside of the plates, imposes some strain on the TS-1, which causes the plates to break apart into nanoparticles of uniform size and shape. This curious crystal-engineering effect of graphene clearly requires more detailed studies.
The presence of TS-1 was confirmed by X-ray powder diffraction (Fig. 3A). The TS-1 reference (Fig. 3A–a) and the composite (Fig. 3B–b) show sharp diffractions typical of TS-1 (ICSD: 92 536) with a dominant 101 diffraction at 2θ = 7.8°. It is important to note that no other reflections, i.e. from TiO2 and SiO2, are present in any sample. The 1 wt% hybrid (Fig. 3B–c) also shows the sole presence of TS-1, however, with a slightly enhanced 200 reflection at 2θ = 8.78° with respect to the 101 reflection. This can be understood as an elongation of the TS-1 particles into rectangular plates, which is in line with the TEM observations in Fig. 2. With increasing graphene concentration, the TS-1 reflections broaden, become weaker and finally disappear at concentrations above 5 wt%. It is generally difficult to identify very small, yet crystalline nanoparticles with XRD. A particle size of 5–6 nm is often cited as the detection limit, below which the material is considered amorphous. However, the presence of micropores in zeolites, such as TS-1 (volume fraction of ∼70%), can raise the detection limit to substantially larger particle sizes (i.e. >15 nm) compared with non-porous materials. Hence, the presence of very broad reflections (at the positions expected for TS-1) may still indicate the presence of crystalline, if very small, TS-1 particles. As expected, the spectra of these hybrids are dominated by the reflections of few-layer graphene.
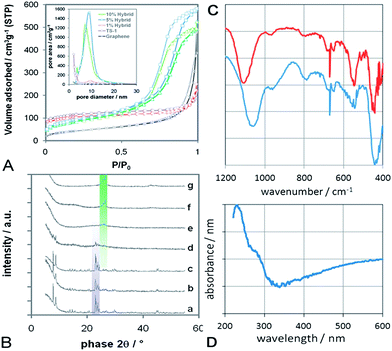 | ||
| Fig. 3 (A) BET nitrogen physisorption isotherms of graphene, TS-1 reference and various hybrids; Inset: pore size distribution, calculated according to BJH using the desorption branch of BET. Both sets of data indicate mesoporosity and narrow pore size distributions for the hybrids with 5–20 wt% graphene. (B) X-Ray diffraction of (a) TS-1 reference, (b) graphene/TS-1 composite with 10 wt% graphene, (c–f) graphene-TS1 hybrids with 1, 5, 10 and 20 wt% graphene, and (g) pure graphene. The violet-shaded diffractions indicate the presence of TS-1, while the green-shaded diffractions correspond to graphene, including the 002 diffraction at 26.2° associated with the presence of 2–3 layers. (C) FTIR of the composite (top) and the hybrid (bottom) with 10 wt% graphene, showing the major characteristics of tetrahedrally-coordinated Ti in the zeolitic MFI framework (i.e. Si–O–Ti vibrations at 960 cm−1, five-member ring of MFI at 550 cm−1) as well as the absence of extra-framework phases, such as TiO2 (i.e. bands at 850 and 1000 cm−1). (D) UV-Vis absorbance spectrum for the 10 wt% hybrid, showing a strong absorbance band at 230 nm due to charge transfer between O and isolated tetrahedral titanium species in the MFI framework. | ||
Another intriguing feature was revealed by nitrogen physisorption (Fig. 3A). The porosity and surface areas of the samples were determined according to Barret–Joyner–Halenda (BJH) and Brunauer–Emmett–Teller (BET), respectively. Single-layer graphene is known to possess Type II isotherms – the typical behaviour of nonporous solids.28 In our work, however, the isotherm of graphene is of Type IV with little, if any, hysteresis. This suggests the presence of few mesopores, presumably due to the aggregation of the graphene flakes into the flower-like structures shown in Fig. 1. The BET isotherms of both the TS-1 reference and the 1% hybrid show the Type I behaviour expected from purely microporous solids. Surprisingly, the isotherms of the hybrids with 5–20 wt% graphene show mixed Type I and IV behaviour with considerable H1-hysteresis. Such behaviour is typical for microporous materials that also contain a significant portion of mesopores.29 Indeed, the BJH pore size distribution in the inset of Fig. 3A confirms the presence of a considerable amount of mesopores with uniform pore sizes of 7–9 nm. Furthermore, the similar slopes of the adsorption and desorption branches indicate that these mesopores are well-ordered and of cylindrical rather than ink-bottle shape. If we assume that the presence of mesopores is due to an ordered packing of monodispersed spherical particles, then the presence of cylindrical pores suggests a simple cubic packing rather than the hexagonal ordering in the denser HCP and FCC packing. The void of a simple cubic packing - and thus the average pore size - can be calculated with d(√2 − 1). Taking d = 15 nm as the average diameter of the TS-1 particles from TEM studies (Fig. 2c), our calculations give an average pore size of 7 nm, which is in excellent agreement with the physisorption results.
Table 1 summarizes the surface areas according to the BET as well as the pore volumes and average diameters for the various samples according to BJH. The most surprising finding is that the BET surface areas of the hybrids were marginally smaller than those of TS-1 and the composite. This is not expected if one considers that a decrease in particle size should increase the specific surface area. For clarification, the samples were analysed by the t-plot method, which has been developed especially for mixed microporous–mesoporous materials.27,29 This method distinguishes between the internal surface area (micropores) and the external surface area (including mesopores). The t-plot data show that, in contrast to the total BET surface area, the external surface area was in fact larger in the hybrids (∼305 m2 g−1) than in the TS-1 reference and the composite (∼175 m2 g−1). Assuming spherical particles of SiO2 with a density ρ of 2.65 cm3 g−1 (corresponds to TS-1 without micropores), then the difference in the external surface area of particles with sizes d = 250 nm (TS-1 reference) and 15 nm (hybrids) would be roughly 140 m2 g−1, which is in agreement with our data. Hence, there is an increase in external surface area due to the decrease in the TS-1 particle size.
| Sample | S BET/m2 g−1 | S micropore/m2 g−1 | S external/m2 g−1 | V micropore/cm3 g−1 | V mesopore/cm3 g−1 | d mesopore/nm |
|---|---|---|---|---|---|---|
| Ts-1 | 384 | 199 | 175 | 0.09 | n/a | n/a |
| H-1% | 385 | 204 | 181 | 0.1 | 0.14 | 9.4 |
| H-5% | 334 | 31 | 303 | 0.01 | 0.91 | 8.7 |
| H-10% | 324 | 32 | 292 | 0.011 | 1.013 | 7.3 |
| H-20% | 311 | 29 | 282 | 0.01 | 0.823 | 6.7 |
| Graphene | 155 | n/a | 160 | 0.75 | n/a | |
| C-10% | 361 | 184 | 177 | 0.08 | 0.274 | n/a |
The situation, however, is completely different for the internal surface area, which was considerably larger in TS-1, the 1 wt% hybrid and the composite (200 m2 g−1) than in the hybrids with more than 5 wt% graphene (∼30 m2 g−1). This is a clear indication that part of the micropores was not accessible to N2 physisorption. One possibility, that the heat applied to these samples was not sufficient to completely remove the surfactant molecules from within the micropores, can be ruled out as this problem was not observed in CNT hybrids. Thus, we assume that graphene was wrapped around the TS-1 nanoparticles to form a core–shell structure and so prevented the oxidation of surfactants from within the micropores. As a consequence, the blocked micropores led to a decrease in the total surface area, despite the increase in external surface area (300 m2 g−1) as a result of the smaller particle size.
The samples were further analysed by FTIR and UV-Vis spectroscopy, shown in Fig. 2C and D for the hybrid and the composite with 10 wt% graphene, which is represented by the presence of sharp peaks around 670 cm−1.30 Both samples also show a significant peak centred at 965 cm−1. This band has been attributed to Si–O–Ti vibrations associated with the presence of isolated tetrahedral titanium in the well-ordered zeolitic MFI framework.31–33 Since this peak is generally absent in ZSM-5 and silicalite-1 (the all-silica MFI structure) as well as in extra-framework materials, such as TiO2 or SiO2, it is considered a “fingerprint band” of TS-1. The presence of TS-1 in both samples is further supported by the band around 550 cm−1, which is characteristic of the double five ring in crystalline MFI type zeolites.33 The optical density ratio of this band with respect to the band at 450 cm−1 is often taken as a measure for crystallinity.33 A I550/I450 ratio of 0.72 is considered highly crystalline, given that the optical density ratio of pure pentasil is 0.8.34 The ratios for the hybrid and the composite were 0.4 and 0.7, respectively, which correspond to 50–90% of the theoretical value. In contrast, amorphous SiO2-TiO2 gels (i.e. at the early stages of crystallisation) do not show any such band at 550 cm−1.35 A splitting of the band at 550 cm−1 into a doublet, similar to that observed in the hybrid, has been associated with the presence of nanoparticles.35 Interestingly, the band around 1100 cm−1 in the hybrid is considerably red-shifted with respect to the composite. Such a shift has been associated with surface strain, crystal size, and the presence of adsorbates and impurities,31,36,37 but has yet to be understood in more detail. Finally, the absence of bands at 850 and 1000 cm−1 confirms that no extra framework anatase-TiO2 was formed,38 which is in line with the XRD data.
The UV-Vis spectrum for the 10 wt% hybrid shows a strong absorption between 210 and 230 nm, which is characteristic for the pπ–dπ charge transfer between O and isolated tetrahedral Ti in the Ti–O–Si MFI framework.15,33,36 Neither crystalline nor amorphous silicalite-1 show this behaviour.31,39 The weak shoulder at 285 nm may be attributed to octahedrally-coordinated Ti species (typically found at 240–260 nm),31,36 or caused by graphene, which has an absorbance maximum at 270–280 nm.30 Furthermore, the absence of an absorption band at 330 nm verifies that the sample is free of extra-framework anatase-TiO2 phase,36 thus confirming both FTIR and XRD results.
The photocatalytic properties were evaluated for the degradation of 4-nitrophenol (4-NP) in an aqueous solution of H2O2 under UV irradiation. The dye concentration was followed throughout the reaction by UV-Visible spectroscopy (Perkin Ellmer Lambda 850) and a typical series of absorption spectra is shown in Fig. S2 (ESI†) for the 5 wt% hybrid. The spectrum without the catalyst contained only one peak with a maximum at 315 nm, which corresponds to 4-NP in solution, while a second peak at 400 nm emerged after adding the catalyst. This bathochromic shift can be attributed to charge-transfer absorption as a consequence of dye adsorption.40
To better represent dye adsorption and degradation, Fig. 4 plots the concentration of 4-NP corresponding to the peak areas over the reaction time. It is evident that the amount of adsorbed 4-NP (green curves) increased immediately after the addition of the catalyst at the expense of dissolved 4-NP molecules (blue curves). The time for the adsorbed 4-NP to reach saturation and thus equilibrium varied strongly for the different catalysts. In the case of graphene, equilibrium was reached after just a few minutes owing to its open structure und the strong interaction with the aromatic dye molecules. However, only 10% of the initial dye concentration was adsorbed on the graphene surface (Fig. 5a). In contrast, the percentages of dye adsorption in the TS-1 reference, the composite and the 1 wt% hybrid, were considerably higher (75–80%) owing to the presence of micropores. However, it took up to 90 min to reach equilibrium, which is a consequence of diffusion hindrance imposed by the small pore size.41
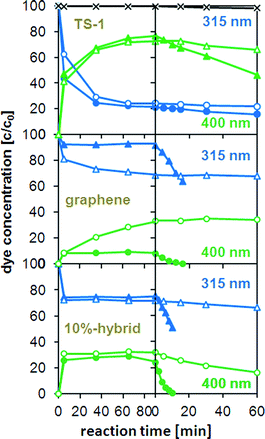 | ||
| Fig. 4 Change in absorbance of 4-nitrophenol (4-NP) with reaction time in the dark (left side of the dashed line) and under illumination at 365 nm (right side of dashed line) and normalized for the initial absorbance of 4-NP before adding TS-1, graphene, and the 10 wt% hybrid. The blue (△) and green (○) curves correspond to the absorbance at 315 and 400 nm, respectively. The light and dark curves follow the absorbance changes during the reaction in absence and in presence of H2O2, respectively, the black curve (×) represents the absorbance changes of 4-NP without any catalyst. | ||
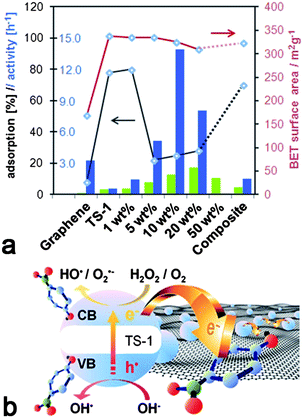 | ||
| Fig. 5 (a) Comparison of different catalysts containing graphene (blue) and CNTs (green) with regard to catalytic activity (columns), dye adsorption (black line) and BET surface area (red line); (b) Proposed scheme for the enhancement of photocatalytic performance in graphene-inorganic hybrids based on the charge transfer of photo-excited electrons from TS-1 into graphene. | ||
For comparison, the hybrids with high graphene concentrations showed similar adsorption behaviour as graphene: only 20–25% of the initial dyes were adsorbed and equilibrium was reached within minutes. Although the fast adsorption kinetics may be explained by the mesoporosity of the hybrids, the presence of micropores should account for considerably higher adsorption percentages. On the other hand, if we assume that the micropores are blocked by graphene, then the adsorption should depend solely on the accessibility of the graphene. Table 1 shows that the hybrids had twice as large surface areas (∼320 m2 g−1) compared to graphene (155 m2 g−1), which did indeed result in the adsorption of twice as many dye molecules (20% vs. 10%, see Fig. 1a, b). These results confirm our previous observations that the TS-1 nanoparticles in these hybrids are covered by graphene.
The photocatalytic activity is generally taken as the rate of decrease of the absorbance (i.e. dye concentration) over irradiation time. The changes in maximum absorbance (integrated peak area) with reaction time are also shown in Fig. 4. The reaction with H2O2, but without a catalyst, serves as a baseline, as the concentration of 4-NP did not change noticeably. It is evident that the concentration of 4-NP decreased exponentially upon turning on the UV light, indicating a first-order reaction. The values for the initial rate (from the slope in the log(C) vs. time t plot, i.e. activity) based on the number of Ti sites are shown in Fig. 5a and are compared with the corresponding hybrids with CNTs.17 For better understanding the photocatalytic behaviour, Fig. 5a also contains the BET surface areas of the catalysts and the relative percentage of dye adsorption. The most important result in this graph is the tremendous increase in activity for the graphene hybrids compared with the CNT hybrids. Also, the hybrids generally possessed greater activities compared with the corresponding composite and the individual components. For instance, by hybridizing with graphene, the activity of TS-1 increased by more than 25 times from k ∼0.5 h−1 to 13.8 h−1; this activity was also 6–7 times higher than for the corresponding CNT-hybrid. The dependence of the activity on the carbon concentration was similar for both hybrids, though the maximum activity in the graphene hybrid was reached at significantly lower concentrations (10 wt% for graphene vs. 25% for CNTs17). Most importantly, the new hybrids were also compared with the present state-of-the-art photocatalyst TiO2-P25 (Degussa). As shown in Fig. S3 (ESI†), the activity of P25 was about 50% higher than pure TS-1, in line with previous results;42 yet the hybrids were considerably more active than P25, which underlines the huge potential from hybridisation with nanocarbons. Finally, it is important to mention that the hybrids retained much of their high activity when reactivated for a consecutive reaction, only losing as much activity as expected from the loss of material upon recovering from the solution.
The reasons for this unusually large increase in activity for the hybrids are complex and a combination of various factors:
1. Larger interfacial area
A prolonged lifetime of photoactive electrons and holes as a consequence of charge separation at the interface has been most commonly cited as a major contribution to synergistic effects in nanocarbon–inorganic hybrids. In view of the importance for photochemical applications, photoactivated charge transfer processes have been studied by a great variety of computational and experimental techniques, including pump–probe spectroscopy, photoluminescence and Raman spectroscopy.6,13,43–45 Generally, a charge transfer of electrons from the inorganic semiconductor to the nanocarbon is feasible when the work function of the nanocarbon lies in between the band gap of the semiconductor.13 For instance, the work function of CNTs is known to range from 4.6 to 5.0 eV, depending on their size, chirality etc,46 while that of single layer graphene is calculated with 4.66 eV.2 These values are below the conduction band minima, yet above the valence band of most semiconductors, including TS-1.47 Thus, electrons that are photoexcited into the conduction band by means of irradiation can readily transfer through the interface of the hybrids into the nanocarbons, which are known to be efficient electron scavengers, while the transfer of holes from the valence band is impeded or hindered by the energy mismatch. A scheme for a possible charge-transfer model is shown in Fig. 5b. Recently, we demonstrated that a close proximity of the nanocarbon and the inorganic phase with a common interface, is crucial for enhancing the photocatalytic performance.17 Based on these findings, it is likely that a larger interfacial area – such as in graphene hybrids due to graphene's larger specific surface area – would provide more opportunities for interfacial charge transfer and thus lead to greater activities.The present data show that the enhancement in photocatalytic activity of TS-1 is about 6 times greater when hybridised with graphene (27–28 times of TS-1) than with CNTs (4–5 times of TS-1). However, the specific surface area of the graphene used in this work (155 m2 g−1) was only two and a half times larger than that of CNTs (60 m2 g−1).17 Also, if graphene was wrapped around the TS-1 particles, only half of its surface area would form an interface with the zeolite, hence the increase in interfacial area would be less than expected from the difference in the surface areas. Thus, it appears that the larger interfacial area in graphene hybrids is not alone responsible for the increase in activity.
2. Presence of large mesopores
In contrast to the CNT-hybrids, the graphene hybrids formed a mesoporous architecture with uniform pores sizes of about 8–9 nm. Such large pores can assist the diffusion of large molecules, such as 4-NP, towards and away from the active sites.41 The fast establishment of adsorption equilibrium in these hybrids is proof for the reduction of diffusion limitations, although its contribution to the photocatalytic reaction needs to be evaluated.3. Reactive edge atoms in graphene
An additional synergistic effect in graphene hybrids may arise from the edge atoms of graphene, which have been demonstrated to alter the electronic properties48 and significantly affect the adsorption of reactants.49 Sharma et al. also reported that edge atoms exhibited a twice as large reactivity toward electron transfer than bulk graphene.50Fig. 5a shows that the activity of uncoated graphene was well over two orders of magnitude greater than that of uncoated CNTs. Similarly, also the activity of the graphene-composite was considerably larger than the corresponding CNT composite, albeit to a lesser extent. These results are in line with previous studies and suggest that graphene may be a photo-active material on its own,51 due to the presence of reactive edge atoms. In general, the photocatalytic degradation of 4-NP over catalysts such as TS-1 and TiO2 occurs via oxidation, either by using hydroxyl radicals, i.e. with the aid of H2O2,42 or by directly consuming the holes in the photocatalyst.52 It is possible that, in contrast to CNTs, the dissociation of H2O2 into hydroxyl radicals is enhanced on graphene due to the reactive edge atoms. To support this hypothesis, a reaction carried out on graphene without H2O2 showed no significant activity for graphene or the composite. Thus, the degradation of 4-NP using the composites and the individual components proceeds predominantly via hydroxyl radicals. The beneficial contribution of graphene edge groups will require further in-depth investigation.4. Different reaction pathway with the graphene hybrids
In contrast to graphene and the graphene composite, the reaction without H2O2 resulted in a remarkably high activity for the hybrid with 10 wt% graphene, comparable to that of TS-1 with H2O2. Hence, the hydroxyl radicals from the dissociation of H2O2 are not the only reactive species in the degradation of 4-NP. Considering that the unique core–shell structure of graphene hybrids had a marked effect on the adsorption behaviour of 4-NP, it is possible that this morphology also provided a new photocatalytic pathway. We believe that photoexcited electrons, which are transferred from the TS-1 core into the graphene shell, reacted with adsorbed 4-NP via reduction to 4-aminophenol, similar to the mechanism reported by Ahn et al. for TiO2 particles modified with arginine.53 In this work, the terminal amine groups of arginine provided the required reduction, while the holes in TiO2 enabled the simultaneous oxidation of 4-NP. Despite using a less-powered UV source, the performance of our graphene hybrids matched the activity of their best catalyst, and that without H2O2 and arginine. We believe that by fine-tuning various process parameters, such as the pH of the solution, it will be possible to maximise the photocatalytic activity of the graphene hybrids, to understand the underlying reaction mechanisms and maybe even to channel the selectivity toward either the oxidation or reduction products.Conclusions
In this work, we hybridized a photoactive zeolite, TS-1, with various concentrations of graphene. Our results reveal some surprising effects of graphene on the crystal morphology, the porosity and the adsorption behaviour of the zeolite. Most remarkable is the beneficial effect on its photocatalytic activity for the degradation of organic dyes, which was enhanced through graphene by more than 25 times. Although we observed some beneficial effects, albeit to a lesser extent, also in TS-1 hybrids with CNTs,17 this work demonstrates that graphene is in many ways superior to CNTs as the carbon component in inorganic hybrids. The advantages of graphene are manifold, as it:(1) affects the nucleation and crystallisation of TS-1 during synthesis, changing its size and morphology from hexagonal-prisms to rectangular plates and finally to very small spherical nanoparticles;
(2) stimulates self-assembly of TS-1 nanoparticles into a mesoporous network with uniform pore sizes (8–9 nm), which can reduce diffusion limitations in solvent-based photocatalysis;
(3) shares a large interfacial area with the inorganic compound, which supports charge transfer processes during photocatalytic reactions, resulting in reduced charge recombination;
(4) provides reactive edge atoms that affect the adsorption of reactants and even contribute to the photocatalytic degradation of the organic dye;
(5) creates a new catalyst system, i.e. consisting of TS-1 nanoparticles encapsulated by graphene, which affects the adsorption of the dye molecules and alters the reaction mechanism for their photocatalytic degradation.
The hybridization of functional inorganic nanomaterials with CNTs and graphene does not simply improve their photocatalytic properties, but can be seen as a milestone in photocatalysis research. Future studies shall be dedicated to the role of edge atoms in graphene based materials, the nature and extent of interfacial charge and energy transfer processes, and to optimising the morphology of the photocatalytic system (i.e. by using single-layer graphene). Furthermore, this work provides new opportunities for research that exploits the synergism in similar graphene-based hybrid materials for a wide range of different applications, including chemical sensors, batteries/supercapacitors and solar cells.
Experimental
Synthesis of catalysts
Few-layer graphene (HG) was prepared by direct current arc discharge of graphite in a water-cooled stainless steel chamber filled with a mixture of hydrogen and helium without using any catalyst.18 The proportion of H2 and He used in our experiment is H2 (70 Torr)–He (500 Torr). In a typical experiment, a graphite rod (Alfa Aesar with 99.999% purity, 6 mm in diameter and 50 mm long) was used as the anode and another graphite rod (13 mm in diameter and 60 mm in length) was used as the cathode. The discharge current was 100 A, with a maximum open circuit voltage of 60 V. The arc was maintained by continuously translating the anode to keep a constant distance of 2 mm from the cathode. Typical synthesis time was 10 min. Soot material with web like appearance was formed on the inner walls of the reaction chamber and around the cathode after the evaporation.The graphene material was characterized using transmission electron microscopy (TEM), atomic force microscopy (AFM) and Raman spectroscopy. Raman spectra were recorded at different locations of the sample using Jobin Yvon LabRam HR spectrometer with 632 nm Ar laser. TEM images were obtained with a JEOL JEM 3010 instrument fitted with a Gatan CCD camera operating at an accelerating voltage of 300 kV. AFM measurements were performed using an Innova atomic force microscope. Surface area measurements were carried out in a QuantaChrome Autosorb-1 instrument.
TS-1 was synthesized via a sol–gel process, using tetrabutyl orthotitanate (TBOT) and tetraethyl orthosilicate (TEOS) as precursors, water as the gelator and tetrapropylammonium bromide hydroxide (TPAOH) as the template for the micropores.17 The final molar ratio was Ti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Si:TPAOH:BA = 1:20:2:6. In a typical experiment, the silicon precursor, TEOS, was added to a solution of TPAOH in isopropanol while stirring at room temperature. A solution of the titanium precursor, TBOT, in isopropanol was then dropped slowly into the now clear TEOS solution. After stirring for 30 min, water was added dropwise to stimulate gelation, and the mixture was kept stirring for another 2 h. The final suspension was subsequently treated in a Microwave reactor (Biotage Initiator 2.5, 400 W at 2.45 GHz) at 150 °C for 1.5 h. The product was washed with distilled water, dried at 90 °C overnight and further calcined at 550 °C for 5.5 h.
:Si:TPAOH:BA = 1:20:2:6. In a typical experiment, the silicon precursor, TEOS, was added to a solution of TPAOH in isopropanol while stirring at room temperature. A solution of the titanium precursor, TBOT, in isopropanol was then dropped slowly into the now clear TEOS solution. After stirring for 30 min, water was added dropwise to stimulate gelation, and the mixture was kept stirring for another 2 h. The final suspension was subsequently treated in a Microwave reactor (Biotage Initiator 2.5, 400 W at 2.45 GHz) at 150 °C for 1.5 h. The product was washed with distilled water, dried at 90 °C overnight and further calcined at 550 °C for 5.5 h.
The hybrids were synthesized in situ, i.e. in presence of graphene, using the above described sol–gel process. Graphene was first suspended in ethanol with the aid of benzyl alcohol (BA) and then mixed with TPAOH. The TEOS and the TBOT solutions as well as water were sequentially added to give a final molar ratio of Ti:Si:TPAOH:BA = 1:20:2:6. The concentration of graphene was varied from 1 wt% to 20 wt% with respect to the expected total mass of the hybrid.
Characterisation and photocatalytic testing
The morphology of the samples was studied by scanning electron microscopy (SEM), using a JEOL 6340F FEF-SEM and by normal/high resolution transmission electron microscopy (HRTEM), using a JEOL 200CX and a FEI TECNAI F20, both with an acceleration voltage of 200 kV. The structure of the samples was characterized by X-ray diffraction (XRD) using a Bruker D8 Advance, with Cu-Kα radiation, operated at 40 kV and 40 mA, λ = 1.5406 Å. All runs were done with 2θ from 5 to 60°, increment of 0.04 and scan speed of 0.8 s/step. Electron diffraction pattern were taken on selected samples using a JEOL 200CX. Specific surface area and porosity of the samples were measured using a TriStar 3000. All the samples were degassed at 180 °C for 3 h before measurements.The photocatalytic decomposition of organic compounds was tested for the decomposition of 4-nitrophenol (4-NP) under illumination with UV light (λ = 254 nm, 6 W), the UV source was fixed a 10 cm distance from the sample. In a typical photocatalytic testing experiment, 20 mg of a catalyst were added into a quartz beaker containing 50 ml distilled water with 40 mgL−1 4-NP and 30 mmol L−1 H2O2. The mixture was kept stirring in the dark for 90 min to ensure adsorption equilibrium, before the light was turned on. Throughout the whole experiment, an aliquot part of the solution was taken every 15 min, centrifuged at 4000 rpm for 3 min and analyzed by UV-VIS spectroscopy using a Perkin-Elmer LAMBDA 850. The absorption peaks were integrated and the changes over time were used to calculate the activity. Some of the catalysts were tested for recyclability and reusability.
Acknowledgements
D. E. is grateful to the Austrian Academy of Science (APART - Austrian Program for Advanced Research and Technology) for financial support. A. K. C. thanks the European Research Council for and Advanced Investigator Award. We thank H. Hoeing for the FTIR measurements.Notes and references
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS.
- M. Pumera, Chem. Soc. Rev., 2010, 39, 4146–4145 RSC.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS.
- D. Eder, Chem. Rev., 2010, 110, 1348–1385 CrossRef CAS.
- Y. Liang, H. Wang, H. Sanchez Casalongue, Z. Chen and H. Dai, Nano Res., 2010, 3, 701–705 CrossRef CAS.
- Y. Zhang, Z.-R. Tang, X. Fu and Y.-J. Xu, ACS Nano, 2010, 4, 7303–7314 CrossRef CAS.
- H. Wang, L.-F. Cui, Y. Yang, H. Sanchez Casalongue, J. T. Robinson, Y. Liang, Y. Cui and H. Dai, J. Am. Chem. Soc., 2010, 132, 13978–13980 CrossRef CAS.
- H. Wang, H. S. Casalongue, Y. Liang and H. Dai, J. Am. Chem. Soc., 2010, 132, 7472–7477 CrossRef CAS.
- D. Wang, D. Choi, J. Li, Z. Yang, Z. Nie, R. Kou, D. Hu, C. Wang, L. V. Saraf, J. Zhang, I. A. Aksay and J. Liu, ACS Nano, 2009, 3, 907–914 CrossRef CAS.
- Y.-L. Chen, Z.-A. Hu, Y.-Q. Chang, H.-W. Wang, Z.-Y. Zhang, Y.-Y. Yang and H.-Y. Wu, J. Phys. Chem. C, 2011, 115, 2563–2571 CrossRef CAS.
- Y. Lin, K. Zhang, W. Chen, Y. Liu, Z. Geng, J. Zeng, N. Pan, L. Yan, X. Wang and J. G. Hou, ACS Nano, 2010, 4, 3033–3038 CrossRef CAS.
- J. Pérez-Ramírez, C. H. Christensen, K. Egeblad, C. H. Christensen and J. C. Groen, Chem. Soc. Rev., 2008, 37, 2530–2542 RSC.
- B. Notari, Stud. Surf. Sci. Catal., 1991, 67, 243–256 CAS.
- S. Hashimoto, J. Photochem. Photobiol., C, 2003, 4, 19–49 CrossRef CAS.
- M. Krissanasaeranee, A. Wongkasemjit, A. K. Cheetham and D. Eder, Chem. Phys. Lett., 2010, 496, 133–138 CrossRef CAS.
- K. S. Subrahmanyam, L. S. Panchakarla, A. Govindaraj and C. N. R. Rao, J. Phys. Chem. C, 2009, 113, 4257 CrossRef CAS.
- Z. Ni, Y. Wang, T. Yu and Z. She, Nano Res., 2010, 1, 273–291.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401–187404 CrossRef CAS.
- C. N. R. Rao, K. Biswas, K. S. Subrahmanyam and A. Govindaraj, J. Mater. Chem., 2009, 19, 2457–2246 RSC.
- D. Eder, I. A. Kinloch and A. H. Windle, Chem. Commun., 2006, 1448–1450 RSC.
- D. Eder and A. H. Windle, Adv. Mater., 2008, 20, 1787–1793 CrossRef CAS.
- D. J. Cooke, D. Eder and J. A. Elliott, J. Phys. Chem. C, 2010, 114, 2462–2470 CrossRef CAS.
- S. Aksel and D. Eder, J. Mater. Chem., 2010, 20, 9149–9154 RSC.
- D. Eder and A. H. Windle, J. Mater. Chem., 2008, 18, 2036–2043 RSC.
- B. C. Lippens and J. H. deBoer, J. Catal., 1965, 4, 319 CrossRef.
- G. Chen, W. Weng, D. Wu, C. Wu, J. Lu, P. Wang and X. Chen, Carbon, 2004, 42, 753–759 CrossRef CAS.
- P. Schneider, Appl. Catal., A, 1995, 129, 157–165 Search PubMed.
- E.-Y. Choi, T. H. Han, J. Hong, J. E. Kim, S. H. Lee, H. W. Kim and S. O. Ki, J. Mater. Chem., 2010, 20, 1907–1912 RSC.
- B. Notari, Catal. Today, 1993, 18, 163–172 CrossRef CAS.
- B. Kraushaar and J. H. C. Hooff, Catal. Lett., 1988, 1, 81–84 CrossRef CAS.
- H. Xin, J. Zhao, S. Xu, J. Li, W. Zhang, X. Guo, E. J. M. Hensen, Q. Yang and C. Li, J. Phys. Chem. C, 2010, 114, 6553–6559 CrossRef CAS.
- J. C. Jansen, F. J. V.d. Gaag and H. V. Bekkum, Zeolites, 1984, 4, 369–372 CrossRef CAS.
- D. T. On, D. Lutic and S. Kaliaguine, Microporous Mesoporous Mater, 2001, 44–45, 435–444 CrossRef CAS.
- A. Tuel and Y. B. Taarit, Microporous Mater., 1993, 1, 179–189 Search PubMed.
- A. Tuel, Stud. Surf. Sci. Catal., 1997, 105, 261–268.
- A. Gamba, G. Tabacchi and E. Fois, J. Phys. Chem. A, 2009, 113, 15006–15015 CrossRef CAS.
- M. R. Boccuti, K. M. Rao, A. Zecchina, G. Leofanti and G. Petrini, Stud. Surf. Sci. Catal., 1989, 48, 133–137.
- T. Premkumar, K. Lee and K. E. Geckeler, Nanoscale, 2011, 3, 1482–1484 RSC.
- L. Li, M. Krissanasaeranee, S. W. Pattinson, M. Stefik, U. Wiesner, U. Steiner and D. Eder, Chem. Commun., 2010, 46, 7620–7622 RSC.
- G. D. Lee, S. K. Jung, Y. J. Jeong, J. H. Park, K. T. Lim, B. H. Ahn and S. S. Hong, Appl. Catal., A, 2003, 239, 197–208 CrossRef CAS.
- I. Robel, B. A. Bunker and P. V. Kamat, Adv. Mater., 2005, 17, 2458–2463 CrossRef CAS.
- F. Vietmeyer, B. Seger and P. V. Kamat, Adv. Mater., 2007, 19, 2935–2940 CrossRef CAS.
- A. Du, Y. H. Ng, N. J. Bell, Z. Zhu, R. Amal and S. C. Smith, J. Phys. Chem. Lett., 2011, 2, 894–899 Search PubMed.
- P. Avouris, M. Freitag and V. Perebeinos, Nat. Photonics, 2008, 2, 341–350 CrossRef CAS.
- N. U. Zhanpeisov, K. Tsujimaru and M. Anpo, Res. Chem. Intermed., 2004, 30, 121–132 Search PubMed.
- K. A. Ritter and J. W. Lyding, Nat. Mater., 2009, 8, 235–242 CrossRef CAS.
- T.-F. Yeh, J.-M. Syu, C. Cheng, T.-H. Chang and H. Teng, Adv. Funct. Mater., 2010, 20, 2255–2262 CrossRef CAS.
- R. Sharma, J. H. Baik, C. J. Perera and M. S. Strano, Nano Lett., 2010, 10, 398–405 CrossRef CAS.
- Y. Matsumoto, M. Koinuma, S. Ida, S. Hayami, T. Taniguchi, K. Hatakeyama, H. Tateishi, Y. Watanabe and S. Amano, J. Phys. Chem. C, 2011, 115(39), 19280–19286 Search PubMed.
- C. S. Turchi and D. F. Ollis, J. Catal., 1990, 122, 178–192 CrossRef CAS.
- W.-Y. Ahn, S. A. Sheeley, T. Rajh and D. M. Cropek, Appl. Catal., B, 2007, 74, 103–110 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–3. See DOI: 10.1039/c1sc00511a |
| This journal is © The Royal Society of Chemistry 2012 |