Hyperbranched polyethylenes by chain walking polymerization: synthesis, properties, functionalization, and applications
Zhongmin
Dong
and
Zhibin
Ye
*
School of Engineering, Laurentian University, Sudbury, Ontario P3E 2C6, Canada. E-mail: zye@laurentian.ca; Fax: +1 705 675-4862
First published on 31st October 2011
Abstract
Structurally different from existing polyethylene grades, hyperbranched polyethylenes (HBPEs) synthesized by Pd–diimine-catalyzed chain walking polymerization represent a novel class of polyethylene materials with unprecedented chain architectures and unique physical properties. A large number of research investigations have been carried out in the area in the past decade, and have led to the successful synthesis of a broad range HBPEs of various controlled chain topologies, molecular weights, and functionalities by tuning polymerization parameters and/or catalyst structures. Meanwhile, some new specialty applications of these materials have also been demonstrated in various fields ranging from lubricant additives to polymer building blocks to nanoencapsulation, with some interesting performance properties discovered. This review aims to summarize the developments in this area.
 Zhongmin Dong | Zhongmin Dong obtained his PhD in polymer chemistry and physics from Changchun Institute of Applied Chemistry, Chinese Academy of Science, on the subject of syntheses of hyperbranched vinyl polymer via RAFT polymerization of asymmetrical vinyl monomers in 2011. At present, he is undertaking his postdoctoral research on post-polymerization functionalizations of hyperbranched polyethylenes in Dr Zhibin Ye's group at Laurentian University. |
 Zhibin Ye | Zhibin Ye is currently an Associate Professor of Chemical Engineering and Canada Research Chair at Laurentian University (Sudbury, Canada). He received his BEng (1996) and MEng (1999) degrees from Zhejiang University (Hangzhou, China), and his PhD degree (2004) from McMaster University (Hamilton, Canada), all in chemical engineering. He started his independent academic career at Laurentian University in 2004. He is a recipient of Ontario Premier's Early Researcher Award (2007) and Canada Research Chair (2011). His research interests include polyolefins of complex chain architectures, transition metal catalysts, living polymerization techniques, polymer nanocomposites, and polymer rheology. |
1. Introduction
1.1 Hyperbranched polymers and synthesis strategies
As a novel family of polymers of unique chain architectures and some striking material properties, dendritic polymers, which include dendrimers and hyperbranched polymers, have been at the forefront of polymer research for more than two decades now. Compared with the traditional polymers with common linear, branched or crosslinked chain topologies, dendritic polymers possess distinctively a three-dimensional spherical architecture featured with extensive branch-on-branch structures, which endows them with many unique advantages including low melt/solution viscosity, good solubility and abundance of reactive sites or functionalities.1,2 In this family of polymers, hyperbranched polymers are the mimics of dendrimers having structural perfection, resembling their dendritic structures but with the presence of statistically distributed structural defects. Unlike dendrimers that often require tedious and sophisticated multi-step synthesis, hyperbranched polymers are often produced using the more convenient and cost-effective one-pot processes, which greatly encourages their large-scale production and industrial applications in various fields ranging from catalyst support to drug-delivery.1Research towards hyperbranched polymers blossomed in the late 1980s. Following Flory's theory that highly branched polymers can be synthesized without gelation by polycondensation of a monomer containing one A functional group and two or more B functional ones capable of reacting with A (i.e., ABx monomers whereas x ≥ 2),3 step-growth condensation polymerization of ABx monomers or A2 + Bx (x ≥ 3) monomers was first demonstrated for the synthesis of hyperbranched polyesters, polyimides, etc. Subsequently, addition polymerization of ABx monomers and self-condensation ring open polymerization of latent ABx monomers have also been developed to optimize the molecular weight (Mw) of the products, mainly due to the absence of the formation of small molecular byproducts during the polymerization progress.1 With these strategies, the resulting hyperbranched polymers are limited to those with carbonyl or ether groups in the backbones. In 1995, Fréchet and co-workers published the first seminal paper demonstrating the synthesis of hyperbranched vinyl polymers by self-condensation vinyl polymerization (SCVP) of AB* type monomers.4a,b Later on, it was discovered that the copolymerization of mono-vinylic monomer with multi-vinylic monomer (usually symmetrical divinyl monomers) and the homopolymerization of divinyl monomer can also lead to the formation of hyperbranched structure.4c,d Up to now, a large variety of hyperbranched polymers with different backbones and various functional groups have been successfully prepared via these strategies. Several recent review articles have summarized these synthetic strategies, along with the properties and applications of the resulting hyperbranched polymers.1
Despite the different polymerization mechanisms (polycondensation, ring opening, vinyl addition, etc.) involved in the aforementioned strategies, the design and use of multi-functional (co-)monomers have always been the key to the successful construction of the hyperbranched chain topology. Many of these multifunctional monomers are often not commercially available, and require special synthesis to suit each particular strategy. With regards to polyolefins (typically polyethylenes herein) synthesized from olefin stocks, the above common strategies are often not applicable for synthesis of hyperbranched polyolefins given the simple ethylene and olefin monomer stocks, which possess only a single olefinic double bond as the sole polymerizable functionality in each monomer unit. Alternative strategies are thus required in order to render hyperbranched polyolefins from these commercially abundant monomers.
1.2 Branched polyethylenes with branch-on-branch structures
Chain architecture or topology affects tremendously the properties and end applications of polyolefins. To this end, there are several elegant strategies that give rise to some typical branched polyethylenes with unique branch-on-branch structures and valuable materials and processing properties. One well known example is the low-density polyethylene (LDPE) produced via high-temperature high-pressure radical processes, which are featured with characteristic short chain branching (SCB), long chain branching (LCB), and branch-on-branch structures. In this case, the construction of the branch structures is achieved through the intermolecular and intramolecular chain transfer reactions ubiquitously present in the radical processes, which lead to the LCB and SCB structures, respectively.5 The presence of short branches and/or possible side long branches on the long branches renders branch-on-branch structures, which bear some similarity to dendritic structures. The control over these chain transfer reactions by tuning polymerization conditions provides the approach to adjust the branching structures in LDPE to some extent.5Another distinct example is metallocene linear low-density polyethylenes (mLLDPE) bearing LCB structures synthesized through metallocene-catalyzed ethylene coordination polymerization (e.g., with Dow's constrained geometry catalyst, 1 in Scheme 1).6 In these polymers, the formation of the unique LCB structures is accomplished through the tailor-designed single-site catalysts, which produce macromonomers and subsequently incorporate them to generate LCB structures. Tuning the polymerization conditions renders the adjustment of the densities of LCB and SCB. These mLLDPE with LCB are also featured with branch-on-branch structures but at much narrower molecular weight distribution and more uniform distribution of the short branches. The level or extent of branching and branch-on-branch structures in these polymers is, somehow, much reduced compared to those featured in dendrimers and hyperbranched polymers. For example, LDPE typically has 40–150 short alkyl branches per 1000 ethylene units and about one long branch per 10 short branches.7 In polyethylenes synthesized with constrained geometry catalyst, the level of LCB is even more sparse, with only about 0.44 branches per 5000 ethylene units.8 However, such low levels of LCB have been proven to have dramatic effects on physical properties of polyolefins.6 Meanwhile, the presence of LCB structures is often not uniform in these polymers, and is often more prevalent at high molecular weights, which is also the case in LDPE. Given these features, the architecture of these polymers with LCB still deviates greatly from those found in the commonly defined dendritic polymers having extensive branch-on-branch structures.
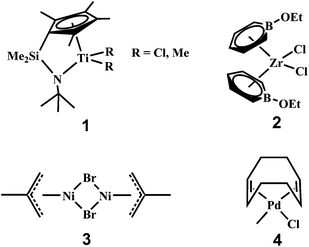 | ||
| Scheme 1 Representative catalysts rendering branch-on-branch structures in polyethylenes. | ||
In addition, the synthesis of some highly branched polyethylenes has also been reported. Bazan et al. first reported the synthesis of polyethylenes with SCB structures by employing tandem catalyst systems comprised of an oligomerization catalyst (2) and a polymerization catalyst (1), where the branching structures are formed by incorporation of 1-alkenes produced in situ with the oligomerization catalyst.9a Subsequently, many other tandem catalyst systems with different combinations of oligomerization and polymerization catalysts were reported for the synthesis of similar polymers of SCB structures through the same concept by Bazan et al.9b–d and other groups.10 Though having controllable high SCB contents (and the presence of sparse LCB structures in some cases), these branched polymers, in general, structurally mimic conventional linear low-density polyethylene (LLDPE) and have linear chain topology with few or no branch-on-branch structures.
Using another strategy, Sen et al. reported the synthesis of hyperbranched polyethylenes of various low molecular weights with the use of different catalysts (e.g., 3 and 4 in Scheme 1) based on Ni, Pd, Ta and Ti, respectively, in combination with Lewis acidic species (e.g., AlCl3 and AlEt3).11 The mechanism for the formation of the branching structures was suggested to result from two oligomerization processes, coordinative oligomerization of ethylene to form 1-alkenes and subsequent cationic oligomerization of the 1-alkenes. High branching contents (up to 0.64 methyl protons/total protons or 427 methyl ends per 1000 carbons) were found with the polymers, hinting at the presence of extensive branch-on-branch structures.11 However, detailed structural and property characterizations have not been reported further on this range of polymers. Meanwhile, the hyperbranched polyethylenes synthesized through this strategy generally have very low molecular weights (below 1000 g mol−1), which unavoidably restricts the broader applications of the polymers.
Conceptually different from the above existing strategies for designing branching structures in polyolefins, Guanet al. demonstrated in 1999 the first use of unprecedented chain walking polymerization (CWP) for the control of polyethylene chain architecture and synthesis of novel hyperbranched polyethylenes (HBPEs).12 Employing the highly versatile Brookhart's Pd–diimine catalysts, this catalytic polymerization technique has, since then, evolved to become a novel concept for the convenient yet sophisticated synthesis of a family of HBPE materials of unique properties from commercially abundant ethylene. Resembling dendrimers, these polymers are truly hyperbranched ones, possessing high branch density and extensive branch-on-branch structures. Extensive studies have been carried out on their synthesis, functionalization, structure and property characterizations, and applications.12 Several review articles have been published, summarizing some developments in this relevant area.13–15 Though excellent, these reviews are often summarized from a different perspective or have the featured focus on the research works done primarily by an individual group. This prompts us to organize this review herein, with the aim of providing an overall update of the developments and progress in this active research area on HBPEs.
2 Chain walking polymerization for synthesis of HBPEs
2.1 Pd–diimine catalysts and their unique features in olefin polymerization
The discovery of Pd–diimine catalysts by Brookhart et al.16 is key to the successful synthesis and developments of HBPEs. Scheme 2 shows the structures of two most commonly used Pd–diimine catalysts (5 and 6) in the literature, which have the same bulky diimine ligand but with different counter anions. In 1995, Brookhart et al. reported in their seminal paper a new class of Pd–diimine and Ni–diimine catalysts for the polymerization of ethylene and α-olefins.16 Compared to early transition metal catalysts (Ziegler–Natta and metallocene) and traditional late transition metal catalysts, this class of late transition metal catalysts show some striking unprecedented features in olefin polymerization. With sterically bulky α-diimine ligands, both Pd and Ni catalysts were found to give rise to high-molecular-weight polymers with unique chain microstructures from ethylene and α-olefins. In particular, the polyethylenes synthesized with Pd–diimine catalysts were shown to have high branching densities (ca. 103 branches/1000 carbons), which are far more highly branched than LDPE.16 On the other hand, Ni-diimine catalysts led to various polyethylenes, with the structures ranging from highly linear semicrystalline to moderately branched amorphous, depending on the ligand sterics and polymerization conditions (ethylene pressure and polymerization temperature), at high activities comparable to those of metallocene catalysts. Historically, late transition metal catalysts based on Ni and Pd often gave rise to low-molecular-weight oligomers when applied for olefin polymerization due to the prevalence of chain transfer reactions viaβ-hydride elimination, though with some exceptions.16 The high capability of these catalysts bearing bulky diimine ligands to render high-molecular-weight polymers is attributed to their unique square planar catalyst structures. In these catalysts, the bulky ortho-substituents on the aryl rings, which are nearly perpendicular to the metal–diimine plane, block the axial coordination sites of the metal center, hindering monomer access and thus suppressing greatly chain transfer reactions.16 Theoretical studies have confirmed this mechanism.17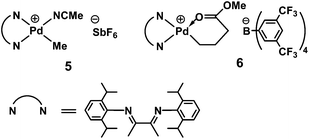 | ||
| Scheme 2 Typical Pd–diimine catalysts. | ||
The generation of branching structures in the polyethylenes is attributed to the most distinguished feature of this class of catalysts, their chain walking mechanism.16Scheme 3 depicts mechanistic steps involved in catalyst chain walking. Through nuclear magnetic resonance (NMR) studies, Brookhart et al. identified the catalyst resting state being the alkyl-ethylene complexes (7 in Scheme 3).16,18 At the ethylene-dissociated states (8), the metal center can undergo isomerization or chain walking through a sequential mechanistic process composed of β-H elimination (yielding olefin hydride complexes 9), bond rotation of the trapped olefin, and subsequent reinsertion to render a branched alkyl in 10. Ethylene trapping and insertion of 10 produces a methyl branch, while further chain walking via the same processes produces longer branches. This chain walking mechanism has been verified through mechanistic studies on model (diimine)Pd–n-propyl, Pd–isopropyl, Pd–n-butyl, and Pd–isobutyl systems.18 Theoretical studies have also confirmed the good agreement with the mechanism.19 Competing with chain propagation, fast chain walking of the Pd catalysts generates extensive branches with high branch densities, along with the branch-on-branch structures. The simplest form of branch-on-branch structures, iso-butyl group, is observed in the Pd–diimine polyethylenes, verifying the presence of branch-on-branch structures. The presence of the branch-on-branch structures also confirms that catalyst chain walking can go through tertiary carbons.12
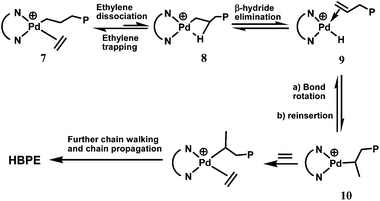
Chain walking also occurs in the polymerization of α-olefins with Pd–diimine and Ni–diimine catalysts, giving rise to unusual polymer chain microstructures and properties.20–22 Chain straightening, yielding reduced branching densities (1,ω-enchainment of α-olefins) and formation of methyl branches (2,ω-enchainment of higher α-olefins), is often found in the resulting polymers. The 1,ω-enchainment results from 2,1-monomer insertion followed with chain walking, and 2,ω-enchainment occurs through 1,2-monomer insertion followed with chain walking.20,21 Historically, the phenomenon of catalyst chain walking has been observed in the polymerization of ethylene and α-olefins with Ni(0)-bis(trimethylsilyl)aminobis(trimethylsilylimino) phosphorane complex, which led to short chain branched polyethylenes and chain straightened poly(α-olefin)s containing methyl branches, respectively.23 Compared to the Ni analogs, the Pd–diimine catalysts often show significantly higher chain walking capability with much longer walking distance (walking distance is defined herein as the number of carbons the catalyst walks through in each chain walking before entrapping and inserting the next monomer), rendering polyethylenes with high branching densities and extensive branch-on-branch structures. The Ni-diimine catalysts produce linear-structured polyethylenes containing short chain branches with methyl being the dominant one.13
Another outstanding feature of the Pd–diimine catalysts is their remarkable tolerance of polar functionalities and capability in incorporating polar functional monomers.24 It has been reported that ethylene polymerization can be undertaken in the presence of ethers, esters, organic acids, and alcohol. Emulsion polymerization and dispersion polymerization of ethylene in aqueous water phase have also been demonstrated with these catalysts. In addition, ethylene polymerization in supercritical CO2 has also been carried out successfully.25 These successful polymerizations in polar solvents/media confirm their great tolerance towards oxygen-containing polar functionalities. A range of polar functional monomers has been copolymerized with ethylene with Pd–diimine catalysts. The primary polar monomers include acrylates, functional α-olefins, acrylic acid, etc.25 With acrylates, a unique incorporation mechanism has been elucidated through NMR studies.24 In the copolymerization, acrylate insertion occurs primarily in a 2,1-fashion (see Scheme 4), followed with two isomerization steps to form a six-membered chelate structure (12). The chelate structure can be readily opened in the presence of ethylene for ethylene binding and insertion. With this mechanism, the incorporated acrylate ester groups are exclusively located at the end of branches. Given the commercial availability or easy synthesis of many functional acrylate monomers, this feature enables the convenient synthesis of functionalized polyethylenes containing desired functionalities through copolymerization.
 | ||
| Scheme 4 Mechanism for acrylate incorporation in copolymerization with ethylene with Pd–diimine catalysts.24 | ||
In addition, another valuable feature of Pd–diimine catalysts is their capability in initiating and catalyzing “living” polymerization of ethylene and α-olefins.26 At low temperatures (ca. 5 °C), successful “living” polymerization of both ethylene and α-olefins can be achieved, with a linear or close-to-linear increase of polymer molecular weight over monomer conversion while at maintained low polydispersity indices (PDIs). In particular, in ethylene polymerization at 5 °C and 400 psig, excellent livingness of the polymerization can be maintained up to 24 h with PDI below 1.1.26 This feature has enabled the successful synthesis of a range of well-defined polyethylenes of other new architectures, including star, block, and telechelic,27 which are out of the scope of this review.
This section summarizes the general features of Pd–diimine catalysts in ethylene and olefin polymerizations. More comprehensive details of this class of late transition metal catalysts, which are beyond the scope of this review, can be found in an existing review.13 As will be shown below, the use of these features and their combinations enables the successful synthesis of a new class of HBPE materials.
2.2 Synthesis of HBPEs via chain walking polymerization and tuning of polymer chain topology
Following the discovery of Pd–diimine catalysts and successful elucidation of their chain walking mechanism by Brookhart et al., Guanet al. demonstrated in 1999 the new concept of using catalyst chain walking to tune polymer chain topology and synthesize polyethylenes of hyperbranched chain topologies by ethylene polymerization with 6.12 In this concept, the control of polymer chain topology is achieved by mediating the competition between chain propagation and chain walking, the two elemental processes governing the nonlinear chain growth, via tuning polymerization condition (i.e., ethylene pressure). In the course of chain growth, the metal center walks randomly along the polymer chain between two consecutive monomer insertion events, with the chain walking distance following a statistic pattern. The next monomer unit is thus incorporated at any position on the polymer backbone, where the metal center walks to at the end of chain walking, instead of at the chain end, thus leading to nonlinear chain growth. At conditions where catalyst chain walking rate (Rwalk) is much faster than chain propagation rate (Rp), catalyst chain walking distance is long and the resulting polymer chain topology is hyperbranched with extensive branch-on-branch structures (Scheme 5). Differently, linear polymers with primarily short branches would result at conditions where Rp is significantly greater than Rwalk, due to the short catalyst chain walking distance.12,14
Ethylene pressure was demonstrated to be the simple polymerization parameter that can be easily changed to mediate the two competing rates due to their different dependencies on ethylene concentration. Through Brookhart's NMR studies, chain propagation rate is independent of ethylene concentration as the insertion of the bound ethylene of the alkyl ethylene complex is the turn-over limiting step.16,18 Differently, chain walking rate has an inverse first-order dependence on ethylene pressure since the catalyst undergoes chain walking only at the ethylene-dissociated state, whose concentration is inversely proportional to ethylene concentration.12,16,18 Reducing ethylene pressure can thus enhance catalyst chain walking rate and lead to longer chain walking distance. On the basis of their dilute solution properties (gyration radius and intrinsic viscosity), polymers synthesized at low ethylene pressures (e.g., 0.1 atm) were found to exhibit highly compact hyperbranched topology. Increasing ethylene pressure led to polymers of increasingly linearized topologies with increasing polymer gyration radius and intrinsic viscosity at equal molecular weights.12,28
Mathematical models based on probability theory have been developed to simulate the effect of ethylene pressure on polymer chain topology. The simulation results are in good agreement with the experimental evidence.29–31 With the decrease of ethylene pressure (i.e., increasing chain walking probability (Pw)), polymer chain topology changes from linear to globular hyperbranched structures while at nearly unchanged total branch density and branch distribution.29Fig. 1 shows sample conformations of polyethylene molecules formed at different chain walking probabilities as simulated by Escobedo et al.,29 which visualize the change of chain topology. Similar simulation studies have also been conducted to elucidate the dependence of short branch density on polymerization conditions in linear-structured polyethylenes synthesized with Ni–diimine catalysts.32
 | ||
| Fig. 1 Simulated conformations of polyethylene molecules formed at different chain walking possibilities. Pw refers to the probability of chain walking that controls the competition between chain-walking and monomer insertion. Reproduced from ref. 29 with permission from Wiley-VCH. | ||
Polymerization temperature has also been shown to affect polymer chain topology. At an ethylene pressure of 1 atm with catalyst 5, increasing polymerization temperature from 15 to 35 °C leads to increasingly enhanced chain compactness from the polymer intrinsic viscosity data, indicating increased catalyst chain walking distances at higher temperatures.33 Though both chain walking and propagation rate should increase with the temperature increase, chain walking rate appears to have a greater sensitivity. The window of temperature change for topology tuning is, however, often limited due to the increasingly severe catalyst deactivation with the temperature increase.
Guan and Popeney have also studied the effect of catalyst ligand electronics on polymer chain topology.34 A series of α-diimine ligands having the same sterics but different electron-donating or withdrawing substituents were screened. It was found that electron-deficient ligands tend to enhance catalyst chain walking capability and render polymers with more compact topologies at a fixed polymerization condition.34 However, the range of topology tuning solely through adjusting ligand electronics is often narrow and far restricted compared to that achievable by changing ethylene pressure.
2.3 “Blocking” strategy for topology tuning
Different from above topology-tuning strategies, Yeet al. have further discovered a unique alternative strategy—incorporating chain-walking blocking sites (ring structures) into the polymer backbone (i.e., chain walking passage)—for topology tuning in chain walking polymerization.35,36 Incorporated into polymer chains randomly at low contents (generally, below 5 mol%), some ring structures were found to act uniquely as chain-walking blocking sites, preventing or restricting catalysts from walking across them to prior chain segments.35,36 In the presence of these blocking sites, free long-range catalyst chain walking spanning over the whole chain is thus prohibited even at low ethylene pressures, and chain walking is mainly restricted locally within each chain segment between two neighboring blocking sites. Each chain is thus segregated into multiple hyperbranched segments with overall linearized chain topologies (Scheme 6), in contrast to the hyperbranched polymers resulting from unrestricted long-range chain walking. With the increase of the density of blocking sites, the level of topology linearization is enhanced without the need for a change in ethylene pressure or temperature. Unlike the other strategies above in which the topology tuning is achieved by essentially controlling catalyst chain walking time, herein it is achieved differently by restricting the catalyst walking region without exerting any control on the walking time. | ||
| Scheme 6 Blocking strategy for topology tuning in chain walking polymerization. | ||
The presence of branch-on-branch structures in HBPEs proves qualitatively that Pd–diimine catalysts can walk across tertiary carbons. However, whether the tertiary carbons affect catalyst chain walking was unknown. Considering this, Yeet al. first examined the potential of externally incorporated isolated tertiary carbons (not generated intrinsically through chain walking) as chain-walking blocking sites.37 To incorporate external tertiary carbons onto polymer backbones, chain walking copolymerizations of ethylene with sterically hindered 3,3-dimethyl-1-butene (DMB, 15) were designed with catalyst 5 at the ethylene pressure of 1 atm (Scheme 7).3715 was selected specifically as the comonomer given its possession of a quaternary carbon at the γ position, which can prevent the occurrence of chain straightening and the resulting loss of the tertiary carbon. Successful incorporation of 15 was achieved in the copolymers at various contents (0.20–3.0 mol%) through 1,2- or 2,1-enchainment, rendering externally incorporated tertiary carbons on backbone and t-butyl groups as side branches. Compared to corresponding HBPE homopolymers, these copolymers were found to exhibit very similar hyperbranched topologies on the basis of their nearly overlapping intrinsic viscosity curves in the Mark–Houwink plots. This thus evidences that tertiary carbons have negligible effects on catalyst chain walking and are thus ineffective as chain-walking blocking sites.
 | ||
| Scheme 7 Incorporation of DMB in ethylene–DMB copolymerization with catalyst 5.37 | ||
Subsequently, two five-membered aliphatic ring structures were demonstrated to be able to efficiently block catalyst chain walking. These ring structures are incorporated uniquely into polymer backbones by chain walking copolymerization of ethylene with special ring-forming comonomers using catalyst 5 at the ethylene pressure of 1 atm. In one case, 1,2-disubstituted cis-fused five-membered rings containing a quaternary carbon on the 4th position are incorporated through ethylene copolymerization with diethyl diallylmalonate (16), a substituted 1,6-heptadiene containing a central quaternary carbon bearing two ester substituents, as the ring-forming comonomer (Scheme 8).35 This diene and other similar ones primarily undergo cyclo-addition in Pd–diimine-catalyzed polymerizations through the consecutive insertion of both double bonds on the same monomer.38 The ring content in the copolymers was controlled in the low range of 0.26–3.6 mol%. In reference to the HBPE homopolymers, linearized topologies were clearly evidenced with these ring-containing copolymers on the basis of the increasingly raised intrinsic viscosity curves in the Mark–Houwink plots with the increase of ring content (Fig. 2). The topology is very sensitive towards ring incorporation such that significant linearization results even at the very low ring content of 0.26%. The linearization effects resulting from such ring incorporation were demonstrated to be as efficient as those achieved by increasing ethylene pressure or decreasing temperature. For instance, the topology of the copolymer with a ring content of 2.7% (synthesized at 1 atm and 25 °C) is nearly equivalent to that of the homopolyethylene synthesized at 30 atm and 25 °C (Fig. 2).35
 | ||
| Scheme 8 Schematic effects of ring incorporation on polymer chain topology in chain walking polymerization.35,36 | ||
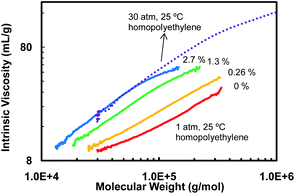 | ||
| Fig. 2 Effect of ring incorporation, with the use of 16 as the latent cyclic comonomer, on topology at the polymerization condition of 1 atm and 25 °C with catalyst 5. The percentages shown are the ring contents. Reproduced from ref. 35 with permission from the American Chemical Society. | ||
The blocking mechanism of the rings was attributed to the difficulty posed for the catalyst to walk across two consecutive tertiary carbons (CH) on the 1,2-disubstituted rings. To walk through the ring, the catalyst has to walk through the side of the ring consisting of the two consecutive CH carbons as the other side contains a quaternary carbon on the 4th position disallowing chain walking (see Scheme 8). Though the catalysts can walk across isolated tertiary carbons, walking through the two consecutive tertiary ring carbons is believed to be more difficult given the enhanced steric crowdedness, thus rendering the blocking effect.35
In another case, cyclopentene (17) was used as the ring-forming comonomer to generate primarily 1,3-enchained cis-fused five-membered rings on polymer backbone. For copolymers synthesized at ethylene pressure of 1 atm, increasing topology linearization was achieved with the increase of ring content (1–7.5 mol%), on the basis of raised intrinsic viscosity curves and enhanced zero-shear melt viscosity data upon ring incorporation. However, on the basis of their intrinsic viscosity curves in Mark–Houwink plots, the linearizing effect achieved with the cyclopentene rings appears to be weaker compared to that achieved with rings of 16 at similar ring contents, possibly due to the differences in the ring structures. While chain walking can only occur through one side of the rings of 16, it can occur through both sides of the cyclopentene rings. Meanwhile, due to the presence of a methylene carbon between the two methine carbons, the 1,3-enchained cyclopentene ring structures likely have lower steric resistance toward chain walking compared to the 1,2-enchained rings in copolymers of ethylene and 16. These two structural factors were proposed to reduce the blocking effect of cyclopentene rings.36
Unlike the backbone-incorporated rings, ring units incorporated at side groups were demonstrated to have no effect at all on catalyst chain walking. Ethylene copolymerizations with 2-allyl-2-methyl-1,3-cyclopentanedione (18) as the comonomer were undertaken with catalyst 5 to render copolymers containing pendant substituted 5-membered rings as side groups. The copolymers (ring content: 2.4 and 4.9%) were found to have overlapping identical intrinsic viscosity curves (i.e., identical topologies) as the corresponding HBPE homopolymer. This should be attributed to the unique presence of the quaternary carbon separating each ring from the polymer backbone. The catalysts thus cannot walk to the pendant rings and no blocking effect results. This further supports the blocking mechanism of the backbone-incorporated rings on catalyst chain walking.35
2.4 Molecular weight tuning of HBPEs
Besides chain topology, molecular weight is another important chain parameter affecting the properties and applications of HBPEs. Two approaches (i.e., catalyst and polymerization approaches) have been developed for tuning polymer molecular weight. The catalyst approach involves the design and use of Pd–diimine catalysts having different ligand steric crowdedness. In both Ni–diimine and Pd–diimine catalyst systems, ligand steric crowdedness has been demonstrated to have dramatic effects of polymer molecular weight and polymerization activity.16,39 The use of sterically crowded α-diimine ligands with bulky substituents on both diimine backbone and aryl rings (particularly those on the ortho-positions) often leads to significant increases in polymer molecular weight due to the reduced chain transfer reactions resulting from hindered monomer access to the sterically protected axial coordination sites.39Yeet al. have studied chain walking ethylene polymerizations with three additional Pd–diimine catalysts featured with various reduced ligand steric crowdedness (19–21, Scheme 9), besides 5, to investigate the effects of ligand sterics.40 In contrast to the high-molecular-weight HBPEs (weight-average molecular weight (Mw): about 150 kg mol−1) produced with 5 having the highest steric crowdedness among the four, mid-molecular-weight polymers (Mw: about 25 kg mol−1) were obtained with 21 and low-molecular-weight polymers (Mw: below 1 kg mol−1) were obtained with 19 and 20 having the least steric crowdedness at the polymerization condition of 1 atm and 15–35 °C. Like those obtained with 5, all the polymers synthesized with 19–21 are featured with high branching density and compact hyperbranched chain conformation on the basis of their NMR spectra and intrinsic viscosity data, despite their reduced molecular weights. This catalyst approach can thus offer HBPEs having molecular weight in a broad range from about 150 to below 1 kg mol−1.40 However, it is often not applicable for synthesis of polymers of higher molecular weights (much greater than 150 kg mol−1) as 5 is among the catalysts having the highest ligand steric crowdedness.
In the polymerization approach, the molecular weight tuning is achieved by controlling/varying polymerization parameters. Two parameters, polymerization time and the addition of crosslinker, can be changed to render molecular weight control to some extent. Other common parameters (such as temperature and ethylene pressure) often exert only marginal effects on polymer molecular weight and thus do not provide an efficient control of the molecular weight of HBPEs. As mentioned earlier, ethylene polymerization with 5 often exhibits a quasi-living feature as long as the polymerization temperature is sufficiently low (25 °C or lower). This “living” feature can thus be employed to obtain HBPEs of various lowered molecular weights by controlling polymerization time. Yeet al. have synthesized narrow-distributed HBPEs with various Mw values in the range of 9–80 kg mol−1 at 1 atm and 15 or 25 °C by changing polymerization time (1–6 h).27f This approach through “living” polymerization, however, suffers from low Pd economics given its one chain per metal feature.
The addition of crosslinkers containing dual or multiple polymerizable double bonds in chain walking polymerization is an efficient polymerization approach to render HBPEs of enhanced molecular weights. Yeet al. have shown ethylene copolymerization with 1,4-butanediol diacrylate (22) as a crosslinker for synthesis of HBPEs of various higher molecular weights using catalyst 5.4122 was chosen since Pd–diimine catalysts can incorporate efficiently acrylate comonomers. When 22 was used at small amounts (below critical gelation point), intermolecular crosslinking structures were successfully introduced into the polymers through dual insertion of 22. Though at low crosslinking contents, polymer molecular weight can be raised dramatically with Mw ranging from about 150 to about 3000 kg mol−1 depending on the feed concentration of 22.41
3. Structures and properties of HBPEs
3.1 Branching structures in HBPEs
The random, long-range catalyst chain walking imparts HBPEs with complex irregular branching structures and chain architectures, featured with high branch densities and numerous branch-on-branch structures.12,14–16,33 This complexity makes the structural characterization difficult. As the most common characterization technique for structural elucidation, NMR is herein only restricted to the determination of the overall branch density and the distribution of short branches in HBPEs. It cannot be used for the elucidation of chain topology and branch-on-branch structures as it only allows the identification of short branches with length below six carbons. From 1H NMR, HBPEs possess high overall branch densities, typically about 110 branches per 1000 carbons, which are significantly greater than those found in conventional LDPE and LLDPE.12,14–16,33 Some low-molecular-weight HBPEs synthesized with 19 and 20 having low ligand steric crowdedness have similar branching densities.4013C NMR facilitates the determination of the distribution of short branches. Fig. 3 shows representatively the 13C NMR spectrum of a polymer synthesized with catalyst 5 at 1 atm and 35 °C. Resulting from the random catalyst walking, the distribution of short branches follows generally a statistical pattern of decreasing branch number with increasing branch length, but with ethyl and butyl branches at disproportionally high numbers.13 The presence of the smallest branch-on-branch structure, sec-butyl branches, in the polymers is verified with 13C NMR (through the signals marked with asterisks (*) in Fig. 3).12 Its content increases with the decrease in ethylene pressure during polymerization, hinting at the increasing amount of branch-on-branch structures. However, the overall branch density and the branch distribution have no dependence on polymer topology and remain nearly unchanged when the topology is linearized upon the drastic increase in ethylene pressure.12,14–16 This is a unique characteristic found with all Pd–diimine polyethylenes, including both high- and low-molecular-weight ones synthesized with catalysts of different ligand steric crowdedness.40
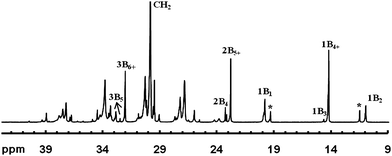 | ||
| Fig. 3 A representative 13C NMR spectrum of a HBPE synthesized with 5 at 1 atm and 25 °C. Short chain branching distribution (in the number of branches per 1000 carbons) of this polymer: methyl, 29.0; ethyl, 23.8; propyl, 2.4; butyl, 7.9; amyl, 3.0; hexyl+, 36.4; total, 102.5; Percentage of methyl from sec-butyl branches in the total methyl, 27%. Reproduced from ref. 35 with permission from the American Chemical Society. | ||
3.2 Topology elucidation in HBPEs and dilute solution properties
Though HBPEs had in fact been synthesized by Brookhart et al. in 1995 in their first seminal paper on Pd–diimine catalysts with the high branching densities demonstrated,16 the hyperbranched polymer topology and the topology tuning mechanism were not elucidated until in 1999 by Guanet al.12 To elucidate the chain topology information, Guanet al. characterized the topology-sensitive dilute properties of a range of polymers synthesized at different ethylene pressures (0.1–34.5 atm) with the use of triple-detection gel permeation chromatography (GPC) equipped with refractive index (RI), multi-angle light scattering (LS), and viscosity detectors.12 This technique enables the simultaneous determination of polymer molecular weight, dilute solution properties (including gyration radius (Rg) and intrinsic viscosity ([η])), and their correlations (such as the dependencies of Rg and [η] on molecular weight). It is often used in the topological characterization of polymers of complex chain topologies/architectures. Meanwhile, they also used dynamic light scattering (DLS) for the determination of polymer hydrodynamic radius (RH), which subsequently enables the calculation of the ratio of Rg/RH.12At equal molecular weights, dramatically reduced Rg and [η] data were found with the decreasing ethylene pressure, demonstrating conclusively the enhanced chain compactness (i.e., more branch-on-branch structures) resulting from longer chain walking.12 Meanwhile, the Rg/RH ratio was reduced from 1.7 for the polymer synthesized at 34.5 atm to 0.8 for the one synthesized at 0.1 atm. Reflecting polymer topology, the ratio is usually around 1.5–1.7 for linear polymers in good solvents and is predicted to be 0.78 for rigid spheres of uniform density. The low value of 0.8 found for the polymer synthesized at 0.1 atm confirmed its globular highly compact hyperbranched chain topology.12 Characterizations of polymer solutions with small angle neutron scattering (SANS) by both Lutz et al.42 and Guan and Cotts43 further evidenced the topological differences among the polymers synthesized at different ethylene pressures. A peak in the Kratky plot, characteristic of highly compact chain conformation, was found with the polymers synthesized at low ethylene pressures while it was absent in the polymers synthesized at high pressures.42,43 These unique dilute solution properties verify the spherical, highly compact hyperbranched chain topology of HBPEs. Meanwhile, the sensitive changes of these properties with the increase in ethylene pressure confirm the topology tuning mechanism in chain walking polymerization.
3.3 Melt rheological properties of HBPEs and dependence on topology
Besides dilute solution properties, rheological properties are also sensitively dependent on polymer chain topology and can be used inversely to infer polymer chain topology. Ye and Zhu carried out the first rheological study on HBPEs and demonstrated the dramatic effect of chain topology on polymer melt rheological properties.33 In their works, a range of polyethylenes having varying topologies was synthesized with 5 at different combinations of ethylene pressure (0.2, 1, 6.5, and 30 atm) and temperature (25 and 35 °C) and was characterized with melt rheometry technique. On the basis of their intrinsic viscosity curves in the Mark–Houwink plot (Fig. 4a), the produced polymers possessed various chain topologies while at similar molecular weights (Mw: ca. 150 kg mol−1) and polydispersity indices (PDI: ca. 2.1). Dramatically different melt rheological properties were found. Polymers synthesized at low ethylene pressure (0.2 and 1 atm) at 35 °C display typically Newtonian flow behavior with a low and constant complex viscosity (25 and 43 Pa s, respectively, at 25 °C) in the broad frequency range of 0.01 to 100 s−1 despite their high molecular weights (Fig. 4b).33a Creep-recovery rheological experiments further confirmed that these polymers appeared to be purely viscous with no or negligible elastic recovery.33b This ideal viscous flow behavior suggests the absence of chain entanglement in the polymer melts, which is characteristic of hyperbranched polymers whose highly compact chain topology and surface congestion prevent effectively chain entanglement.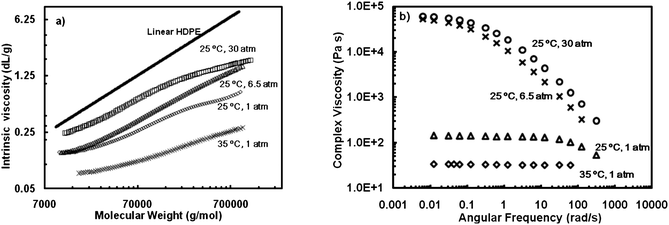 | ||
| Fig. 4 Mark–Houwink plot (a) and complex viscosity curve (b) of polymers produced at different polymerization conditions with 5. Reproduced from ref. 33a with permission from the American Chemical Society. | ||
As for polymers synthesized under higher ethylene pressure (6.5, and 30 atm) at 25 °C, non-Newtonian shear thinning behavior evolves and becomes more pronounced with the zero-shear viscosity values (η0) increased by up to more than three orders of magnitude (to 9.1 × 104 Pa s at 25 °C for the one synthesized at 30 atm) (Fig. 4b).33b These indicate the incrementally enhanced chain entanglements as a result of their increasingly linearized chain topology. These rheological data thus clearly demonstrate the topology change upon the variation in polymerization conditions in chain walking polymerization. Similarly, the rheometry technique has also been used to monitor the topology linearization upon the incorporation of ring units through the chain-walking blocking strategy.
Ye et al. further studied the relationships between η0 and molecular weight for polymers having different chain topologies, which allow the determination of another important rheological parameter, the critical entanglement molecular weight (Mc), and its dependence on chain topology.27f Traditionally, the relationship between η0 and molecular weight follows the power law η0 = KMb, where the value of the exponent b is about 1.0 for polymers with molecular weight below Mc and is about 3.4 above Mc.44 The transition around Mc is often sharp and represents the onset of chain entanglements. Generally, Mc is about twice the entanglement molecular weights (Me).44 It has been shown that some dendrimers do not have Mc due to the absence of chain entanglements as a result of their unique dendritic structures.45 In the investigation, Yeet al. synthesized four sets of narrow-distributed (PDI below 1.33) polymers having various molecular weights through “living” polymerization with 5 at four different combinations of ethylene and temperature (27 atm/5 °C, 3 atm/15 °C, 1 atm/15 °C, and 1 atm/25 °C, respectively).27f The intrinsic viscosity curves (Fig. 5a) constructed with the four respective sets of polymers clearly confirm their topological differences resulting from the different polymerization conditions. In each set, the intrinsic viscosity correlates to the molecular weight by following the Mark–Houwink equation though with different constants.
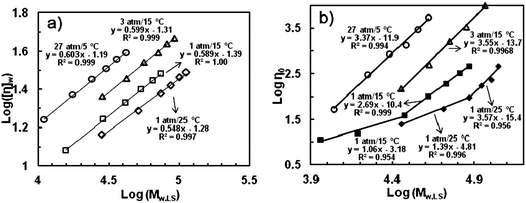 | ||
| Fig. 5 Intrinsic viscosity curves (a) and complex viscosity curves (b) of narrow-distributed HBPEs of various molecular weights synthesized via “living” polymerization with catalyst 5. Reproduced from ref. 27f with permission from the American Chemical Society. | ||
Fig. 5b shows their different dependencies of η0 at 25 °C on Mw. With the reduction of ethylene pressure and/or the increase of temperature, the η0 curve is shifted down continuously as a result of their increasingly compact topologies. For polymers synthesized at 27 atm/5 °C and 3 atm/15 °C, there is no transition found within their investigated molecular weight ranges with the value of b being 3.37 and 3.55, respectively, which agree well with the value of 3.4 found with many linear polymers. Their Mc values should thus be well below the corresponding investigated molecular weight ranges. For the sets of polymers synthesized at 1 atm/15 °C and 1 atm/25 °C, the unique transition is found with the Mc values being 28 and 87 kg mol−1, respectively. These Mc values are much greater than the typical value of about 2 kg mol−1 found for linear polyethylenes, demonstrating the compact hyperbranched chain topology in these polymers. Meanwhile, the greater value at the latter condition further confirms the more hyperbranched topology for those obtained at 25 °C. For those synthesized at 1 atm/15 °C, the b value is 1.06 below Mc, close to 1.0 found for linear polymers, and is 2.69 above Mc, which deviates somehow from 3.4 for linear polymers. For those obtained at 1 atm/25 °C, the b value is 1.39 below Mc and 3.57 above Mc.27f
Colby et al. have also studied the rheological properties of several Pd–diimine polyethylenes synthesized at different ethylene pressures (0.1–34 atm, room temperature).31 Similarly, η0 increases nearly a million times with the pressure increase while at similar polymer molecular weights (Mw about 400 kg mol−1). With the measured plateau modulus data, Me was calculated to be 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 2500 g mol−1 for the polymers synthesized at 10 and 34 atm, respectively. For polymers synthesized at low pressures (0.1–1 atm), the Me value could not be determined in their study due to the absence of rubber plateau.31
000 and 2500 g mol−1 for the polymers synthesized at 10 and 34 atm, respectively. For polymers synthesized at low pressures (0.1–1 atm), the Me value could not be determined in their study due to the absence of rubber plateau.31
3.4 Thermal properties of HBPEs
As a result of the hyperbranched structure, HBPEs are completely amorphous oil-like low-viscosity liquids at room temperature. From polymer characterization with differential scanning calorimetry (DSC), HBPEs synthesized with catalyst 5 at 1 atm and 15–35 °C typically have a glass transition at ca. −67 °C and a weak but broad crystal melting endotherm centered at ca. −34 °C with an enthalpy of about 9.5 J g−1 (Fig. 6).46 Consistently, a crystallization exotherm can be found in the DSC cooling cycle. This indicates that the polymers are still crystallizable, although at much lower temperatures with low crystallinity, despite their high branching density. Similar thermal behavior, with no distinct dependence on chain topology, is also featured in the polymers synthesized at higher ethylene pressures.36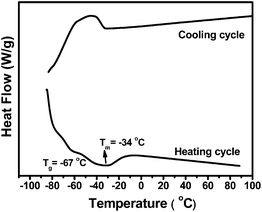 | ||
| Fig. 6 DSC curves for a HBPE synthesized with 5 at 1 atm and 35 °C.46 | ||
4 Synthesis of functionalized HBPEs by chain walking copolymerization
One of the most promising advantages of hyperbranched polymers is their abundance of functional or reactive groups at all branch ends, due to their construction from multifunctional monomers. Differently, HBPE homopolymers synthesized solely from ethylene do not contain any functionality, with branch ends being exclusively stable nonpolar methyl groups. Despite their compact hyperbranched chain topology, the absence of functionalities limits unavoidably their broader applications as functional materials. Given the addition polymerization nature of CWP, copolymerization of ethylene with other polar functional comonomers is the first option for the synthesis of functionalized HBPEs. Therein, the functionalities are designed specifically into the comonomers. Traditionally, such copolymerization is often challenging due to the sensitive poisoning of most early transition metal (Ziegler–Natta and metallocene) catalysts by polar functionalities. Owing to their remarkable low oxophilicity, these copolymerizations are possible with Pd–diimine catalysts, thus rendering a range of functionalized HBPEs containing various valuable functionalities while with the retained hyperbranched chain topology.Generally, two main classes of functional comonomers, acrylates and 1-alkenes, have been designed and employed for synthesis of functionalized HBPEs. Among them, the functional acrylates are particularly preferred as many of them are commercially available or can be conveniently synthesized. The functionalities that can be designed into these comonomers are often those containing oxygen, including ester, ketone, epoxide, ether, alcohol, etc., which Pd–diimine catalysts can tolerate.13 Some comonomers containing fluoro, phosphorous, and sulfone groups can also be copolymerized while comonomers with nitrogen-containing functionalities often inhibit the polymerization due to the binding affinity of nitrogen with Pd active sites. Generally, a longer distance between the double bond and the polar functionality helps the comonomer incorporation. In the case of functional 1-alkenes comonomers, it is often beneficial to have a blocking quaternary carbon between the double bond and the functionality, which can prevent the catalyst from walking to the functionality and being poisoned.13 The review by Brookhart et al.13 summarizes the polar comonomers that can be copolymerized with Pd–diimine catalysts, mostly from patent literature. We summarize below a brief overview of the functionalized comonomers reported for the synthesis of functionalized HBPEs after their review.
Guan et al. investigated chain walking copolymerization of ethylene with a range of functionalized 1-alkene comonomers (23–30 in Scheme 10) using catalyst 6 to synthesize functionalized polymers having the corresponding functionalities (i.e., hydroxyl, carboxylic ester, epoxide, saccharide, bromoester, and oligo(ethyleneglycol) sequences, respectively).47–49 These comonomers were designed to contain a quaternary carbon or a long spacing between the olefinic double bond and the functionality. Protection groups are used in 24, 29, and 30 to avoid the negative effects of hydroxyl groups on the catalyst. In particular, functionalized polymers containing 2-bromoisobutyryl groups were obtained with the use of 28 as comonomer. With 29, functionalized polymers containing a shell of hydrophilic oligo(ethyleneglycol) were obtained in one step.
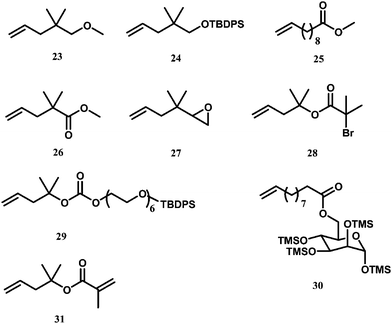 | ||
| Scheme 10 Functional 1-alkene comonomers used for synthesis of functionalized HBPEs viaethylene copolymerization. | ||
Differently, Yeet al. employed various functional acrylate comonomers (22, 32–34 in Scheme 11) for the synthesis of functionalized HBPEs containing different functionalities through ethylene copolymerization with catalyst 1.46,50–53 With 32 containing a polyhedral oligomeric silsesquioxane (POSS), they synthesized organic–inorganic hybrid materials with organic HBPEs tethered with inorganic POSS nanoparticles at contents up to 35 wt%. Due to the assembling feature of the tethered POSS nanoparticles to form small crystallites, these hybrid materials were demonstrated to have interestingly reinforced thermal and rheological properties.46 As an acrylate analogue of 28, 33 is a typical ATRP inimer for construction of hyperbranched polymers through SCVP. Copolymerization of ethylene with 33 also renders directly functionalized HBPEs containing 2-bromoisobutyryl groups.50 Though capable of incorporating acrylates and 1-alkenes, it was found that the Pd–diimine catalyst cannot copolymerize methacrylates.51 Utilizing this feature, Yeet al. designed and used two heterodifunctional comonomers (31 and 34) containing two different double bonds, one incorporable (1-alkenyl and acryloyl, respectively) and one incopolymerizable methacryloyl group, for chain walking copolymerization. Owing to the complete selectivity of the catalyst toward the acryloyl or 1-alkenyl group in the heterodifunctional comonomers, HBPEs tethered with methacryloyl groups at controllable contents were yielded.51
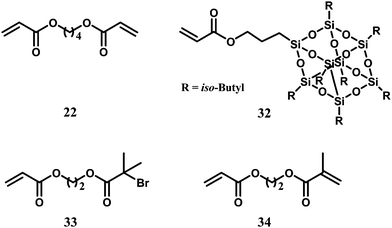 | ||
| Scheme 11 Functional acrylate comonomers. | ||
As mentioned earlier, chain walking copolymerization of ethylene with 22 at low concentrations leads to the synthesis of HBPEs of enhanced molecular weights due to the occurrence of intermolecular crosslinking.41Yeet al. further discovered that such crosslinking reactions can be suppressed or minimized kinetically when the feed concentration of 22 is much higher than the critical gelation concentration.52 This renders HBPEs containing predominantly pendant acryloyl groups with minimum crosslinking. This suppression of crosslinking results from the reduced concentration of low-reactivity pendant acryloyl groups relative to the high-reactivity monomeric acryloyl groups. This feature has thus been employed for synthesis of HBPEs tethered with acryloyl groups at different contents.52 Furthermore, through terpolymerization of ethylene with both 1,6-hexanediol diacrylate (a longer analogue of 22) and 33, Yeet al. further synthesized HBPEs containing dual acryloyl and 2-bromoisobutyryl functionalities at controllable ratios.53
In all functionalized polymers synthesized through copolymerization, the functionalities are all incorporated as side groups or branch ends, with their contents controlled by changing comonomer concentration in copolymerization. Their incorporation often has no or little effect on polymer chain topology and branching distribution as they are not located on the polymer backbone, i.e., the chain walking passage of the catalyst. However, their incorporation often leads to reduced catalyst activity and polymer molecular weight as the comonomers are generally less reactive compared to ethylene.50–53
In addition to the copolymerization strategy, Yeet al. have also demonstrated the synthesis of telechelic HBPEs containing different ω-end functionalities by quenching/end-capping ethylene “living” polymerization catalyzed with 5 using styrene derivatives.54 This employs the unique inhibitive reaction of styrene derivative with cationic Pd–diimine species to form stable π-benzyl intermediate complexes, which are inactive for ethylene polymerization. A range of narrow-distributed telechelic HBPEs containing various reactive ω-end functionalities, including benzyl chloride, 4-methylphenyl, and vinylbenzene groups, has been obtained.54
5 Applications of HBPEs and their functionalized polymers
5.1 Some applications of HBPEs
Due to their unique architectures and characteristic physical properties, hyperbranched polymers in general have received extensive interest for applications in various emerging fields, ranging from drug-delivery to coating to rheology modifiers.1 With HBPEs synthesized through CWP, Yeet al. have explored the following specialty applications: (1) shear-stable lubricant viscosity additives for high-molecular-weight HBPEs;55,56 (2) synthetic base stocks for medium- and low-molecular-weight HPBEs;40 (3) polymer processing aid (PPA) in the extrusion processing mLLDPE;57 (4) functionalization of multi-walled carbon nanotubes (MWCNTs) for solubilization in organic solvents and for formulation of polymer nanocomposites.58,59 Benefiting from their unique globular hyperbranched architectures, HBPEs have been demonstrated to exhibit some outstanding performance features in some of these specialty applications.To improve the viscosity thickening efficiency of HBPEs while at maximized shear stability, Yeet al. proposed, in a further study, to increase their molecular weights while keeping their advantageous highly compact hyperbranched topology.56 Traditionally, increasing polymer molecular weight often leads to enhanced viscosity thickening efficiency with deteriorated shear stability. They synthesized another range of HBPEs of various significantly enhanced molecular weights (Mw: 273–3720 kg mol−1) by chain walking ethylene polymerization in the presence of various small amounts of diacrylate as crosslinker. With the increase of polymer molecular weight, significant enhancements in viscosity thickening efficiency were achieved. For example, at a polymer dosage of 3 wt%, the VI value of the formulated lubricants increases from 131 to 165 when the Mw value increases from 116 to 3720 kg mol−1. This can thus reduce the polymer dosages in the formulation. Increasing polymer molecular weight, however, reduces the shear stability of the polymers. In contrast to the nearly zero shear degradation found with those having Mw values of 116 and 273 kg mol−1, significant shear-induced degradation was found with that having a Mw value of 2652 kg mol−1 with a SSI value of 36.4 despite its retained hyperbranched topology. Nevertheless, compared to the aforementioned linear polymer of much lower molecular weight (165 kg mol−1, SSI = 45), the HBPEs possessing such ultra-high molecular weights still possess significantly better shear stability, given the lower SSI values. This further demonstrates the advantage of hyperbranched topology in enhancing polymer shear stability.56
Subsequently, this functionalization approach was further found to improve the dispersion of MWCNTs in an ethylene–octene copolymer matrix.59 Compared to the severe aggregations found with unmodified MWCNTs, dramatically enhanced nanotube dispersion was achieved successfully in the nanocomposites after their surface functionalization with HBPE. The simple non-covalent surface functionalization makes the nanotubes more compatible with the polyolefin matrix.
5.2 Some applications of functionalized HBPEs
The presence of various functionalities in functionalized HBPEs further extends their applications as a class of functional polymeric materials. Two types of functional applications have been demonstrated, including (1) building blocks for construction of new polymers of more complex chain architectures; and (2) encapsulation and bioconjugation.Functionalized HBPEs containing various reactive or initiating groups have been employed as building blocks for designing polymers of more complex structures/architectures. Yeet al. have demonstrated the direct use of HBPEs containing acryloyl groups (synthesized by ethylene copolymerization with 22 at high feed concentrations using catalyst 5) as the homogeneous multivalent supports for binding catalyst 5.27b Serving as the specific catalyst binding sites, the tethered acryloyl groups react with 5, rendering ester chelate Pd–diimine complexes tethered covalently on the HBPE supports. Ethylene multifunctional “living” polymerization with these HBPE-supported catalysts leads to the construction of star polymers with multiple “living” arms (ca. 20–30 arms on average per star) growing from the HBPE core. The average arm number in the star polymers can be tuned by controlling the content of acryloyl groups in the functionalized HBPEs through changing the feed concentration of 22 in the first ethylene copolymerization step.27b In addition to serving as catalyst support as shown in this case, these double bond containing polymers (including the copolymers of ethylene with 31 and 34 as well) can also be employed as the macro-crosslinker in various thermosetting applications due to their possession of the polymerizable acryloyl or methacryloyl groups.
Guan and Sun synthesized dendritic polyethylenes containing multiple 3-butenoate groups by postpolymerization modification of hydroxyl-containing polymers (copolymer of ethylene and 24 followed with deprotection) and also employed them as the multivalent support for Pd–diimine catalysts.60Ethylene polymerization with these multivalent catalysts at 0.1 atm leads to the synthesis of large dendritic polyethylene nanoparticles bearing a dendrimer-on-dendrimer architecture.
Functionalized HBPEs containing 2-bromoisobutyryl groups, synthesized by copolymerizing ethylene with 28 or 33, have been used as polyfunctional ATRP macroinitiators for synthesis of core–shell structured star polymers containing a HBPE core and a shell of polymer arms from other monomers through ATRP.49,50 With the use of oligo(ethyleneglycol) methacrylate as the monomer for arms, the core–shell polymers can be rendered amphiphilic and exist as unimolecular micelles with temperature sensitivity when dispersed in water.48 In another case, telechelic HBPEs containing an ω-end benzyl chloride functionality, synthesized by polymer end-capping with vinyl benzyl chloride, have been used as monofunctional ATRP macroinitiators for the synthesis of block polymers containing HBPE blocks.54,61
In addition to their use as building blocks, some applications of the functionalized polymers as nanocarriers and bioconjugation have also been demonstrated. Guan and Chen demonstrated the nanoencapsulation of Nile Red using the functionalized dendritic polymers containing a shell of oligo(ethyleneglycol)s (copolymers of ethylene with 29) as the amphiphilic unimolecular micelles.48 Meanwhile, they further synthesized core–shell polymer nanoparticles with the shell containing N-hydroxysuccinamide functional end groupsviaATRP with the dendritic copolymer of ethylene and 28 as macroinitiators. The polymer nanoparticles showed good conjugation with small dye molecules and a protein (ovalbumin).49 Zhang et al. also showed the encapsulation of coumarin 153 in unimolecular micelles of amphiphilic core–shell polymers obtained by coupling chain walking copolymerization and ATRP.62
Conclusions
The discovery of the Pd–diimine catalysts and their outstanding catalytic features (chain walking mechanism, low oxophilicity, and “living” polymerization characteristics) in ethylene polymerization has rendered convenient one-pot synthesis of a broad range of HBPEs directly from ethylene stocks. By controlling polymerization parameters and catalyst structures, their chain parameters, including chain topology, molecular weight, and functionality, can be flexibly tuned in the polymerization as demonstrated in this review. Some applications of HBPEs have been also explored with some interesting performance properties demonstrated as a result of their beneficial chain topologies. Despite these desired elegant features, the commercial applications of HBPE materials are currently restricted primarily due to the high cost, low activity and stability of the existing Pd–diimine catalysts. The discovery of new, highly active, and cost-effective catalysts (for example, the Ni-based catalysts) with competing performance features thus remains the major challenge in the area.Acknowledgements
The authors thank the funding support from the Natural Science and Engineering Research Council (NSERC), Canada Research Chair (CRC), the Canadian Foundation for Innovation (CFI), the Ontario Ministry of Research and Innovation, Imperial Oil Ltd., the Center of Materials and Manufacturing of the Ontario Centers of Excellence (OCE), and Laurentian University.References
- (a) Some review articles on hyperbranched polymers are: Y. H. Kim, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1685 CrossRef CAS; (b) A. Hult, M. Johansson and E. Malmström, Adv. Polym. Sci., 1999, 143, 1 CrossRef CAS; (c) B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2505 CrossRef CAS; (d) M. Jikei and M. Kakimoto, Prog. Polym. Sci., 2001, 26, 1233 CrossRef CAS; (e) P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 457 CrossRef CAS; (f) C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183 CrossRef CAS; (g) C. R. Yates and W. Hayes, Eur. Polym. J., 2004, 40, 1257 CrossRef CAS; (h) B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2679 CrossRef CAS; (i) B. Voit and A. Lederer, Chem. Rev., 2009, 109, 5924 CrossRef CAS; (j) K. Inoue, Prog. Polym. Sci., 2000, 25, 453 CrossRef CAS.
- (a) Some review articles on dendrimers are: A. W. Bosman, H. M. Janssen and E. W. Meijer, Chem. Rev., 1999, 99, 1665 CrossRef CAS; (b) F. Vögtle, S. Gestermann, R. Hesse, H. Schwierz and B. Windisch, Prog. Polym. Sci., 2000, 25, 987 CrossRef; (c) S. M. Grayson and J. M. J. Fréchet, Chem. Rev., 2001, 101, 3819 CrossRef CAS; (d) S. Hecht, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1047 CrossRef CAS; (e) D. A. Tomalia, Prog. Polym. Sci., 2005, 30, 294 CrossRef CAS; (f) H. Frauenrath, Prog. Polym. Sci., 2005, 30, 325 CrossRef CAS; (g) M. A. Mintzer and M. W. Grinstaff, Chem. Soc. Rev., 2011, 40, 173 RSC.
- P. J. Flory, J. Am. Chem. Soc., 1952, 74, 2718 CrossRef CAS.
- (a) J. M. J. Fréchet, M. Henmi, I. Gitsov, S. Aoshima, M. R. Leduc and R. B. Grubbs, Science, 1995, 269, 1080 Search PubMed; (b) C. J. Hawker, J. M. J. Fréchet, R. B. Grubbs and J. Dao, J. Am. Chem. Soc., 1995, 117, 10763 CrossRef CAS; (c) Z. Guan, J. Am. Chem. Soc., 2002, 124, 5616 CrossRef CAS; (d) F. Isaure, P. A. G. Cormack, S. Graham, D. C. Sherrington, S. P. Armes and V. Bütünc, Chem. Commun., 2004, 1138 RSC.
- A. J. Peacock, in Handbook of Polyethylene, Marcel Dekker, New York, 2000, pp. 43–53 Search PubMed.
- P. S. Chum, W. J. Kruper and M. J. Guest, Adv. Mater., 2000, 12, 1759 CrossRef CAS.
- C. E. Carraher, Jr, in Seymour/Carraher's Polymer Chemistry, CRC Press, Boca Raton, FL, USA, 7th edn, 2008, pp. 155–156 Search PubMed.
- (a) W. Wang, D. Yan, S. Zhu and A. E. Hamielec, Macromolecules, 1998, 31, 8677 CrossRef CAS; (b) W. Wang, S. Zhu and S. J. Park, Macromolecules, 2000, 33, 5770 CrossRef CAS.
- (a) R. W. Barnhart, G. C. Bazan and T. Mourey, J. Am. Chem. Soc., 1998, 120, 1082 CrossRef CAS; (b) Z. J. A. Komon, X. Bu and G. C. Bazan, J. Am. Chem. Soc., 2000, 122, 1830 CrossRef CAS; (c) Z. J. A. Komon, G. M. Diamond, M. K. Leclerc, V. Murphy, M. Okazaki and G. C. Bazan, J. Am. Chem. Soc., 2002, 124, 15280 CrossRef CAS; (d) J.-C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001 CrossRef CAS.
- (a) Z. Ye, F. AlObaidi and S. Zhu, Macromol. Rapid Commun., 2004, 25, 647 CrossRef CAS; (b) F. AlObaidi, Z. Ye and S. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4327 CrossRef CAS; (c) Z. Ye, F. AlObaidi and S. Zhu, Macromol. Chem. Phys., 2005, 206, 2096 CrossRef CAS; (d) C. Bianchini, M. Frediani, G. Giambastiani, W. Kaminsky, A. Meli and E. Passaglia, Macromol. Rapid Commun., 2005, 26, 1218 CrossRef CAS; (e) J. Zhang, B. Li, H. Fan and S. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3562 CrossRef CAS; (f) M. Yang, W. Yan, X. Hao, B. Liu, L. Wen and P. Liu, Macromolecules, 2009, 42, 905 CrossRef CAS; (g) C. Bianchini, G. Giambastiani, A. Meli, I. G. Rois, A. Toti, E. Passaglia and M. Frediani, Top. Catal., 2008, 48, 107 CrossRef CAS; (h) E. D. Schwerdtfeger, C. J. Price, J. Chai and S. A. Miller, Macromolecules, 2010, 43, 4838 CrossRef CAS; (i) E. D. Schwerdtfeger, L. J. Irwin and S. A. Miller, Macromolecules, 2008, 41, 1080 CrossRef CAS.
- (a) J. S. Kim, J. H. Pawlow, L. M. Wojcinski, II, S. Murtuza, S. Kacker and A. Sen, J. Am. Chem. Soc., 1998, 120, 1932 CrossRef CAS; (b) S. Murtuza, S. B. Harkins, G. S. Long and A. Sen, J. Am. Chem. Soc., 2000, 122, 1867 CrossRef CAS.
- Z. Guan, P. M. Cotts, E. F. McCord and S. J. McLain, Science, 1999, 283, 2059 CrossRef CAS.
- S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169 CrossRef CAS.
- (a) Z. Guan, Chem.–Eur. J., 2002, 8, 3087 CrossRef; (b) Z. Guan, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3680 CrossRef CAS; (c) Z. Guan, Chem.–Asian J., 2010, 5, 1058 CAS.
- Z. Ye and S. Li, Macromol. React. Eng., 2010, 4, 319 CrossRef CAS.
- L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414 CrossRef CAS.
- L. Deng, T. K. Woo, L. Cavallo, P. M. Margl and T. Ziegler, J. Am. Chem. Soc., 1997, 119, 6177 CrossRef CAS.
- (a) D. J. Tempel, L. K. Johnson, R. L. Huff, P. S. White and M. Brookhart, J. Am. Chem. Soc., 2000, 122, 6686 CrossRef CAS; (b) L. H. Shultz, D. J. Tempel and M. Brookhart, J. Am. Chem. Soc., 2001, 123, 11539 CrossRef CAS.
- (a) A. Michalak and T. Ziegler, Organometallics, 1999, 18, 3998 CrossRef CAS; (b) A. Michalak and T. Ziegler, Organometallics, 2000, 19, 1850 CrossRef CAS.
- C. M. Killian, D. J. Tempel, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 11664 CrossRef CAS.
- E. F. McCord, S. J. McLain, L. T. J. Nelson, S. D. Ittel, D. Tempel, C. M. Killian, L. K. Johnson and M. Brookhart, Macromolecules, 2007, 40, 410 CrossRef CAS.
- Z. Ye, W. Feng, S. Zhu and Q. Yu, Macromol. Rapid Commun., 2006, 27, 871 CrossRef CAS.
- (a) W. Keim, R. Appel, A. Storeck, C. Krüger and R. Goddard, Angew. Chem., Int. Ed. Engl., 1981, 20, 116 CrossRef; (b) V. M. Möhring and G. Fink, Angew. Chem., Int. Ed. Engl., 1985, 24, 1001 CrossRef.
- (a) L. K. Johnson, S. Mecking and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 267 CrossRef CAS; (b) S. Mecking, L. K. Johnson, L. Wang and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 888 CrossRef CAS.
- See ref. 13 and relevant references cited therein.
- (a) A. C. Gottfried and M. Brookhart, Macromolecules, 2001, 34, 1140 CrossRef CAS; (b) A. C. Gottfried and M. Brookhart, Macromolecules, 2003, 36, 3085 CrossRef.
- (a) K. Zhang, Z. Ye and R. Subramanian, Macromolecules, 2009, 42, 2313 CrossRef CAS; (b) X. Xia, Z. Ye, S. Morgan and J. Lu, Macromolecules, 2010, 43, 4889 CrossRef CAS; (c) P. Liu, E. Landry, Z. Ye, H. Joly, W.-J. Wang and B.-G. Li, Macromolecules, 2011, 44, 4125 CrossRef CAS; (d) Y. Zhang and Z. Ye, Chem. Commun., 2008, 1178 RSC; (e) K. Zhang, Z. Ye and R. Subramanian, Macromolecules, 2008, 41, 640 CrossRef CAS; (f) Y. Xu, P. Xiang, Z. Ye and W.-J. Wang, Macromolecules, 2010, 43, 8026 CrossRef CAS.
- P. M. Cotts, Z. Guan, E. F. McCord and S. J. McLain, Macromolecules, 2000, 33, 6945 CrossRef CAS.
- Z. Chen, I. Gospodinov and F. A. Escobedo, Macromol. Theory Simul., 2002, 11, 136 CrossRef CAS.
- A. Michalak and T. Ziegler, Macromolecules, 2003, 36, 928 CrossRef CAS.
- R. Patil, R. H. Colby, D. J. Read, G. Chen and Z. Guan, Macromolecules, 2005, 38, 10571 CrossRef CAS.
- (a) L. C. Simon, J. B. P. Soares and R. F. de Souza, AIChE J., 2000, 45, 1234 CrossRef; (b) L. C. Simon, C. P. Williams, J. B. P. Soares and R. F. de Souza, Chem. Eng. Sci., 2001, 9, 4181 CrossRef.
- (a) Z. Ye and S. Zhu, Macromolecules, 2003, 36, 2194 CrossRef CAS; (b) Z. Ye, F. AlObaidi and S. Zhu, Macromol. Chem. Phys., 2004, 205, 897 CrossRef CAS.
- C. Popeney and Z. Guan, Organometallics, 2005, 24, 1145 CrossRef CAS.
- P. Xiang, Z. Ye, S. Morgan, X. Xia and W. Liu, Macromolecules, 2009, 42, 4946 CrossRef CAS.
- S. Morgan, Z. Ye, R. Subramanian, W. Wang and G. Ulibarri, Polymer, 2010, 51, 597 CrossRef CAS.
- P. Xiang and Z. Ye, Macromol. Rapid Commun., 2010, 31, 1083 CrossRef CAS.
- S. Park, D. Takeuchi and K. Osakada, J. Am. Chem. Soc., 2006, 128, 3510 CrossRef CAS.
- D. P. Gates, S. A. Svejda, E. Oñate, C. M. Killian, L. K. Johnson, P. S. White and M. Brookhart, Macromolecules, 2000, 33, 2320 CrossRef CAS.
- P. Xiang, Z. Ye and R. Subramanian, Polymer, 2011, 52, 5027 CrossRef CAS.
- J. Ye, Z. Ye and S. Zhu, Polymer, 2008, 49, 3382 CrossRef CAS.
- P. J. Lutz, M. S. Plentz, J. Kress, A. Lapp and M. Duval, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2000, 41, 1882 CAS.
- Z. Guan and P. M. Cotts, Polym. Mater. Sci. Eng. Prepr., 2001, 84, 382 CAS.
- W. W. Graessley, Adv. Polym. Sci., 1974, 16, 1 CrossRef.
- C. J. Hawker, P. J. Farrington, M. E. Mackay, K. L. Wooley and J. M. J. Fréchet, J. Am. Chem. Soc., 1995, 117, 4409 CrossRef CAS.
- J. Wang, Z. Ye and H. Joly, Macromolecules, 2007, 40, 6150 CrossRef CAS.
- G. Chen, X. Ma and Z. Guan, J. Am. Chem. Soc., 2003, 125, 6697 CrossRef CAS.
- G. Chen and Z. Guan, J. Am. Chem. Soc., 2004, 126, 2662 CrossRef CAS.
- G. Chen, D. Huynh, P. L. Felgner and Z. Guan, J. Am. Chem. Soc., 2006, 128, 4298 CrossRef CAS.
- K. Zhang, J. Wang, R. Subramanian, Z. Ye, J. Lu and Q. Yu, Macromol. Rapid Commun., 2007, 28, 2185 CrossRef CAS.
- J. Wang, K. Zhang and Z. Ye, Macromolecules, 2008, 41, 2290 CrossRef CAS.
- S. Morgan, Z. Ye, K. Zhang and R. Subramanian, Macromol. Chem. Phys., 2008, 209, 2232 CrossRef CAS.
- Y. Xu, P. Campeau and Z. Ye, Macromol. Chem. Phys., 2011, 212, 1255 CrossRef CAS.
- S. Li and Z. Ye, Macromol. Chem. Phys., 2010, 211, 1917 CrossRef CAS.
- J. Wang, Z. Ye and S. Zhu, Ind. Eng. Chem. Res., 2007, 46, 1174 CrossRef CAS.
- S. Morgan, Z. Ye, R. Subramanian and S. Zhu, Polym. Eng. Sci., 2009, 50, 911 Search PubMed.
- J. Wang, M. Kontopoulou, Z. Ye, R. Subramanian and S. Zhu, J. Rheol., 2008, 52, 243 CrossRef CAS.
- L. Xu, Z. Ye, Q. Cui and Z. Gu, Macromol. Chem. Phys., 2009, 210, 2194 CrossRef CAS.
- K. Petrie, A. Docoslis, S. Vasic, M. Kontopoulou, S. Morgan and Z. Ye, Carbon, 2011, 10, 3378 CrossRef.
- G. Sun and Z. Guan, Macromolecules, 2010, 43, 4829 CrossRef CAS.
- W.-J. Wang, P. Liu, B. Li and S. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3024 CrossRef CAS.
- L. Zhang, J. Su, W. Zhang, M. Ding, X. Chen and Q. Wu, Langmuir, 2010, 26, 5801 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |