
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoron–halide interactions for crystallization regulation of a 1.68 eV wide-bandgap perovskite prepared via a two-step method
Shizi
Luo
ab,
Daxiong
Liu
c,
Xiang
Deng
*c,
Zhuoneng
Bi
 b,
Shuguang
Cao
j,
Tongjun
Zheng
ab,
Liyao
Xiong
ab,
Hao
Li
d,
Ning
Li
d,
Lavrenty G.
Gutsev
*ef,
Nikita A.
Emelianov
e,
Victoria V.
Ozerova
e,
Nikita A.
Slesarenko
e,
Alexander F.
Shestakov
e,
Sergey M.
Aldoshin
e,
Gennady L.
Gutsev
g,
Pavel A.
Troshin
*eh,
Bochuan
Yang
*i,
Zhibo
Zhao
c and
Xueqing
Xu
*ab
b,
Shuguang
Cao
j,
Tongjun
Zheng
ab,
Liyao
Xiong
ab,
Hao
Li
d,
Ning
Li
d,
Lavrenty G.
Gutsev
*ef,
Nikita A.
Emelianov
e,
Victoria V.
Ozerova
e,
Nikita A.
Slesarenko
e,
Alexander F.
Shestakov
e,
Sergey M.
Aldoshin
e,
Gennady L.
Gutsev
g,
Pavel A.
Troshin
*eh,
Bochuan
Yang
*i,
Zhibo
Zhao
c and
Xueqing
Xu
*ab
aSchool of Energy Science and Engineering, University of Science and Technology of China, Hefei, 230026, P. R. China
bKey Laboratory of Renewable Energy, Guangdong Provincial Key Laboratory of New and Renewable Energy Research and Development, Guangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Guangzhou 510640, P. R. China. E-mail: xuxq@ms.giec.ac.cn
cGuangdong Mingyang Thin Film Technology Co., Ltd, Zhongshan, P. R. China
dInstitute of Polymer Optoelectronic Materials and Devices State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou, 510640, P. R. China
eFederal Research Center for Problems of Chemical Physics and Medicinal Chemistry of RAS, Semenov Prospect 1, Chernogolovka, 142432, Russia
fInstitute for Micromanufacturing, Louisiana Tech University, Ruston, LA 71272, USA
gDepartment of Physics, Florida A&M 711 University, Tallahassee, Florida 32307, USA
hZhengzhou Research Institute of HIT, 26 Longyuan East 7th, Jinshui District, Zhengzhou, Henan Province 450000, P. R. China
iRisen Energy Co., Ltd, Ningbo, Zhejiang Province 315609, P. R. China
jSchool of Materials Science and Engineering, Henan Polytechnic University, Jiaozuo, Henan 454000, P. R. China
First published on 11th November 2025
Abstract
An all-solution two-step method for preparing wide-bandgap perovskites has the advantages of low cost, good repeatability and scalability. However, achieving high-quality wide-bandgap perovskite films via an all-solution two-step method remains challenging due to uneven distribution of halogens and incomplete reactions between organic salts and inorganic salts. Herein, we introduced tris(pentafluorophenyl)borane (BCF) into an inorganic layer resulting in boron–halide bonding, which stabilized uniform halide distribution and regulated the porous structure of the lead halide films, facilitating the diffusion of organic salts. Additionally, the fluorine substituents formed hydrogen bonds with organic cations, making BCF a bifunctional additive that delayed the reaction between the organic ammonium salt and the inorganic precursor, which was conducive to the growth of large-grained perovskite crystals. During the perovskite crystallization process, BCF molecules migrated to grain boundaries and the film surface, achieving a highly positive regulation influence on the nanoscale morphology and structure of the perovskite absorber films, thus leading to a pinhole-free, stress-free and less defect perovskite films. Ultimately, the approach enabled single-junction 1.68 eV wide-bandgap perovskite solar cells with a champion efficiency of 23.49% (certified 22.73%) and a VOC of 1.291 V. Furthermore, the optimized perovskite films were pioneeringly and successfully integrated into monolithic perovskite/silicon tandem solar cells on textured silicon and achieved an efficiency of 31.12%, which is the highest value among tandem solar cells prepared by an all-solution two-step method, retaining >90% of initial performance after 500 hours of continuous operation.
Broader contextThe preparation of wide-bandgap perovskite solar cells via an all-solution two-step method offered potential pathways to regulate the crystallization rate of perovskite films. However, there are very few reports on the all-solution two-step sequential deposition of 1.68 eV wide-bandgap perovskite films. Therefore, it will become meaningful to investigate the crystallization kinetics of perovskite prepared by a two-step method. Herein, we regulated the crystallization kinetics of 1.68 eV wide-bandgap perovskite films by leveraging a simultaneous bonding of tris(pentafluorophenyl)borane (BCF) with halide anions and organic cations via the boron–halide and hydrogen bonding effects. These interactions stabilized halogen distribution, delayed and regulated the crystallization process of perovskite films, slowed down nonradiative recombination, and facilitated the rapid extraction of charges. As a result, we achieved a PCE of 23.49% and a VOC of 1.291 V. By integrating the optimized wide-bandgap PSCs on textured silicon, we pioneered an all-solution two-step sequential deposition method for preparing 1.68 eV perovskite/silicon tandem solar cells, which exhibited a PCE of 31.12%, which is the highest among those of two-step prepared tandem cells. This means that a huge breakthrough has been achieved in the preparation of wide-bandgap perovskite films using an all-solution two-step sequential deposition method. |
1. Introduction
Double-junction tandem solar cells (TSCs), particularly perovskite/silicon tandem solar cells, offer a promising path to exceed the Shockley–Queisser (SQ) efficiency limits of single-junction devices.1–3 By integrating two photoactive layers with complementary absorption spectra, TSCs can absorb photon energy to a greater extent, thus greatly improving the utilization of light.4–6 Theoretically, TSCs can show a power conversion efficiency (PCE) of up to 45%, far exceeding the ∼33% SQ limit of single-junction devices.7–9 In the most advanced TSC structures, wide-bandgap perovskite materials, which have tunable bandgaps and high absorption coefficients, are typically selected as the top sub-cell active layers of TSCs.10,11 The bandgap (Eg) of perovskites may be continuously tuned from 1.25 eV to over 2.0 eV by alloying different halides, such as iodide (I−), bromide (Br−), and chloride (Cl−). This tunability enables perovskite materials to achieve complementary absorption in a wide range of narrow-bandgap bottom cells, including crystalline silicon (c-Si), copper indium gallium selenide (CIGS), and Sn–Pb binary perovskites, making them highly suitable for constructing TSCs.12–17However, the high Br content (>20%) required to obtain a wide-bandgap perovskite generally leads to the well-known problem of increased ion migration, caused by a marked increase in the iodide vacancy concentration18,19 and phase segregation, resulting in a large number of defects that reduce PCE and compromise the long-term stability of the devices.20,21 These energy losses and instabilities are mainly due to the unbalanced nucleation rates between the I− and Br− components22 and the fact that iodide is preferentially oxidized in a mixed-halide perovskite.23 The perovskite films with uneven halide distribution show high defect density and low crystallinity.24 As such, large open-circuit voltage (VOC) losses are usually observed in wide-bandgap PSCs with an Eg of >1.65 eV.25,26 Generally speaking, improving the uniformity of halide distribution and the crystallinity of perovskite films by adjusting the crystallization kinetics of perovskite films is critical to further improve device performance.27 To address these issues, researchers have proposed various strategies to control the crystallization of mixed halide wide-bandgap perovskites, including component engineering, ligand engineering, Lewis's base additives, etc.20,28,29 Chen et al. developed a novel strategy using a cation alloy that not only tailors the lattice properties and crystallization but also effectively passivates the defects, enabling homogeneous halide distribution and substantially reducing defect density. These improvements have led to a remarkable power conversion efficiency (PCE) of 19.50% with a record open-circuit voltage of 1.35 V for 1.79 eV perovskite solar cells.30 Jiang et al. combined the rapid bromide crystallization with a gentle gas-quench method to prepare highly textured columnar 1.75 eV wide-bandgap perovskite films with reduced defect density, resulting in a PCE over 20% and an VOC of 1.33 V.27 Yang et al. reported a series of pyridine derivatives containing amino and carboxylic groups that were applied to modify the perovskite surface, which can interact with undercoordinated Pb2+ through coordination bonds and suppress halide ion migration via hydrogen bonding.24
Most strategies for preparing wide-bandgap perovskite films rely on the one-step spin-coating method, which often leads to non-uniform Br− distribution during the crystallization process.31 In contrast, there are limited reports on the fabrication of wide-bandgap PSCs using an all-solution two-step method. This method involves first depositing a lead halide film and then introducing an organic ammonium salt solution to form a perovskite film, offering potential pathways to control over the crystallization rate of wide-bandgap perovskites. For instance, Zhang et al. utilized a two-step method by incorporating formamidinium iodide (FAI) and rubidium acetate (RbAc) into a PbI2/PbBr2 solution (with a molar ratio of 0.83/0.17) to modify the crystallization kinetics of 1.63 eV perovskite films. This approach promoted the oriented growth of the perovskite layer, ultimately allowing the perovskite/silicon TSCs to exhibit a PCE of 27.64%.32
Herein, we presented a novel all-solution two-step method to regulate the crystallization kinetics of 1.68 eV wide-bandgap perovskite films by leveraging a simultaneous interaction of tris(pentafluorophenyl)borane (BCF) with halide anions and organic cations. Fourier-transform infrared (FTIR) spectroscopy, nuclear magnetic resonance (NMR) analyses and density functional theory (DFT) calculations revealed that the BCF molecules, with their electron-deficient boron center and multiple fluorine substituents, interacted with perovskite precursor components through boron–halide and hydrogen bonding effects. These interactions might regulate the reaction kinetics between the organic ammonium salt solution and the inorganic layer. In situ absorption spectroscopy studies demonstrated that the boron–halide interactions effectively stabilized uniform halide distribution within the lead halide film, while the chemical reaction and crystallization rates between the organic ammonium salt solution and the inorganic layer were delayed via boron–halide and hydrogen bonding interactions. This resulted in the growth of pinhole-free, stress-free and less defect perovskite films. The results of time-resolved photoluminescence and transient photocurrent measurement indicate that after the introduction of BCF, the electron–hole recombination time extended, slowing down nonradiative recombination and facilitating the rapid extraction of charges. As a result, the optimized single-junction PSCs exhibited a PCE of 23.49% and a VOC of 1.291 V, with a remarkably low VOC loss of only 0.39 V. The devices also exhibited exceptional long-term stability, retaining 90% of their initial efficiency after 750 hours of maximum power point (MPP) tracking. By integrating the optimized wide-bandgap PSCs with crystalline silicon (c-Si), we pioneered an all-solution two-step sequential deposition method for preparing 1.68 eV perovskite/silicon tandem solar cells on textured silicon, exhibiting a PCE of 31.12%, which is the highest among those of two-step prepared tandem cells, retaining >90% of initial performance after 500 hours of continuous operation.
2. Results and discussion
2.1. Interactions of BCF with the perovskite precursor components
The 1.68 eV wide-bandgap perovskite films were fabricated by dynamically dripping the organic salt solution onto a mixed inorganic layer, which was deposited from a 1.5 M PbI2/PbBr2 (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) precursor supplemented with 0.06 M CsI and 0.015 M RbCl. Tris(pentafluorophenyl)borane (BCF) was introduced into the inorganic layer as the strategic modifier, since it features highly electronegative fluorine substituents enhancing the electron-deficient boron center's interaction with halide anions (I−/Br−) via boron–halide bonding.33 Additionally, the fluorine substituents form hydrogen bonds with organic cations (e.g., methylammonium or formamidinium), making BCF a bifunctional passivator that modulates both halide anions and organic cations (Fig. S1). These interactions are anticipated to regulate halide distribution and crystallization kinetics, suppressing ion migration and yielding high-quality perovskite films with large grains and low defect density. For clarity, films processed without and with BCF are labeled control and target, respectively, while inorganic layers are denoted as PbX2 and PbX2 + BCF (X = I or Br).
:1 molar ratio) precursor supplemented with 0.06 M CsI and 0.015 M RbCl. Tris(pentafluorophenyl)borane (BCF) was introduced into the inorganic layer as the strategic modifier, since it features highly electronegative fluorine substituents enhancing the electron-deficient boron center's interaction with halide anions (I−/Br−) via boron–halide bonding.33 Additionally, the fluorine substituents form hydrogen bonds with organic cations (e.g., methylammonium or formamidinium), making BCF a bifunctional passivator that modulates both halide anions and organic cations (Fig. S1). These interactions are anticipated to regulate halide distribution and crystallization kinetics, suppressing ion migration and yielding high-quality perovskite films with large grains and low defect density. For clarity, films processed without and with BCF are labeled control and target, respectively, while inorganic layers are denoted as PbX2 and PbX2 + BCF (X = I or Br).
We firstly experimentally adopted Fourier-transform infrared spectroscopy (FTIR) spectroscopy measurements to elucidate the chemical interactions of BCF with the perovskite precursors. The FTIR spectra of BCF, BCF + PbI2, and BCF + PbBr2 are shown in Fig. 1a. The blueshift in the B–C stretching vibration of BCF molecules from 974 to 987 cm−1 is consistent with the boron–halide binding effect as supported by theoretical calculations. The shift was attributed to the increased electron cloud density of the B–C bond due to the contribution of the electron-rich halides, confirming the boron–halide interaction. Fig. S2a presents the FTIR spectra of the BCF + FAI composite and pure FAI. The characteristic peak corresponding to N–H bonds exhibited a redshift as compared to that of pure FAI, which was attributed to the formation of hydrogen bonds (N–H⋯F) between FA and the fluorine atoms in BCF. A similar N–H vibration redshift supporting the formation of hydrogen bonds (due to a decrease in the N–H bond strength) was observed when comparing the FTIR spectra of the BCF + MABr mixture and pure MABr (Fig. S2b). Thus, the observed spectral changes confirm the existence of strong interactions between BCF and the perovskite precursor components.
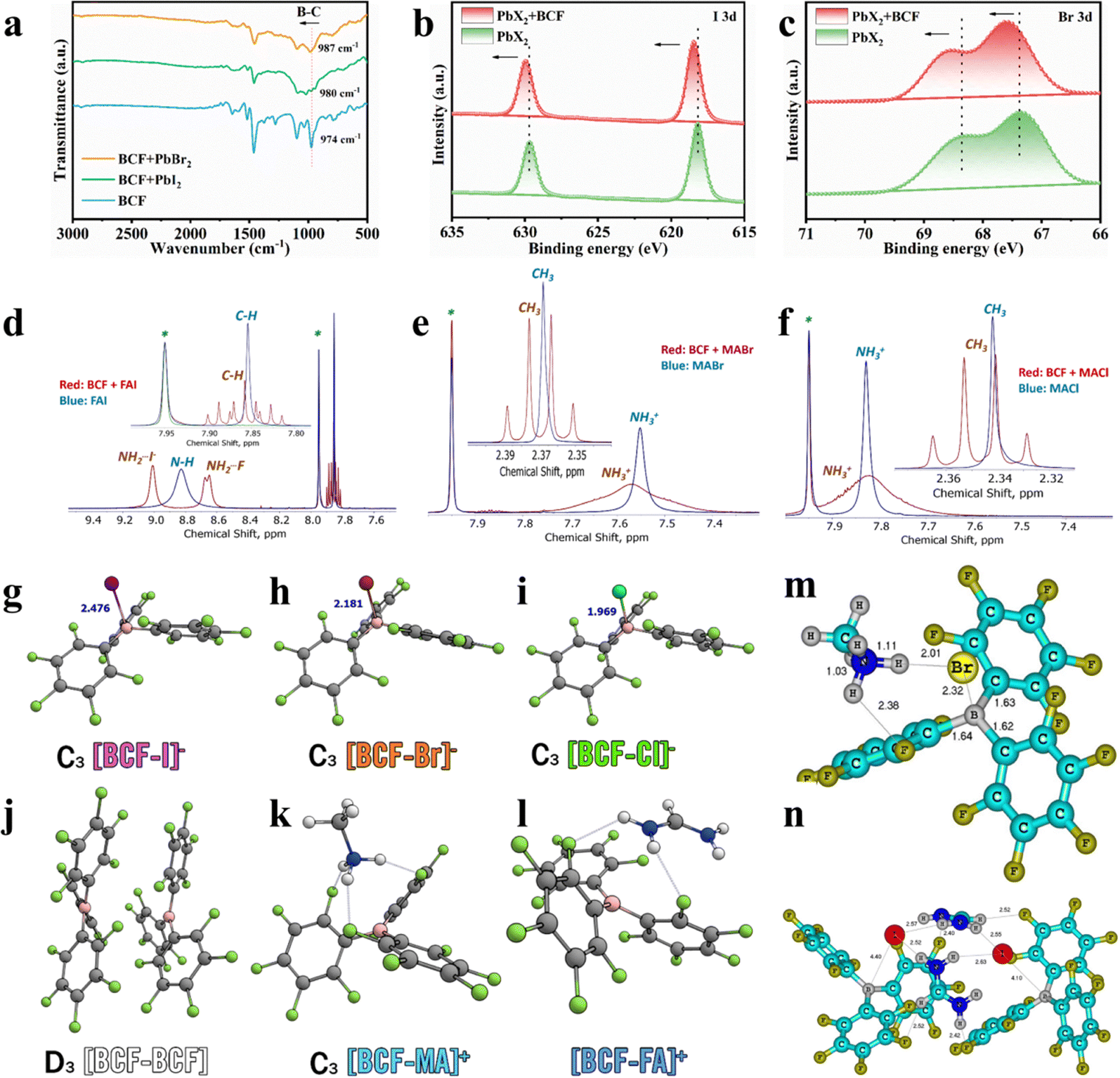 | ||
| Fig. 1 FTIR spectra of (a) BCF and BCF + PbX2 mixture. XPS spectra of (b) I 3d and (c) Br 3d for PbX2 and PbX2 + BCF films, respectively. The evolution of the 1 H NMR spectra of FAI (d), MABr (e) and MACl (f) upon interaction with BCF. The geometrical configuration of the [BCF–I] (g), [BCF–Br] (h), [BCF–Cl] (i) anionic complexes. The [BCF–BCF] neutral dimer (j), and the [BCF–MA] (k) and [BCF–FA] (l) cationic complexes, where symmetry is present it is listed before the complex name. The computed minimal-energy molecular structures of the MABr + BCF (1:1, m) and FAI–BCF (2:2, n) complexes. Short contacts are shown in the figures. | ||
Furthermore, the influence of BCF on the PbX2 films was investigated by X-ray photoelectron spectroscopy (XPS) measurements. As illustrated in Fig. S2c, the Pb 4f core-level XPS spectra revealed only a minor shift in the PbX2 + BCF film compared to the PbX2 film, suggesting that lead cations (Pb2+) and BCF likely do not interact directly. Instead, the electronic environment of the [PbX6]4− octahedra appears to be indirectly modified by the boron–halide interaction. This boron–halogen interpretations were further supported by the observed shifts in the XPS spectra of I 3d and Br 3d bands for PbX2 + BCF films due to the decrease in the electron cloud density of the halogen (Fig. 1b and c). These shifts provide additional evidence of the interactions between BCF and the halide anions (I− and Br−), confirming that BCF influences the electronic structure of the inorganic framework.
To further explore these interactions, nuclear magnetic resonance (NMR) spectroscopy was employed which provided additional insights into the chemical interactions of BCF with the perovskite precursor components. First, the 19F NMR spectra revealed some shifts of the signals corresponding to fluorine substituents in para- and meta-positions of the phenyl rings, which are the most distant from the boron center and, hence, they are sterically the most accessible for intermolecular hydrogen bonding and halogen–halogen interactions. These shifts were observed with comparable magnitudes when BCF interacted with PbI2, PbBr2, CsI, FAI, MABr and MACl (Fig. S3).
The 1H NMR spectra have revealed spectacular changes for FAI, MABr and MACl when they were mixed with BCF. Firstly, one could notice that the CH group signal of FAI undergoes splitting to a quintet, which implies that all four neighboring N–H protons become symmetrically non-equal and they do not exchange on the timescale of the NMR measurement (Fig. 1d). Furthermore, the broad peak at 8.83 ppm corresponding to all four NH protons of the pristine FAI undergoes splitting in two distinct signals: a singlet at 9.01 ppm and a doublet at 8.66 ppm, thus further confirming the loss of symmetry of the FA+ cation. Similarly, the peak corresponding to the –NH3+ protons of the methylammonium cation in MABr and MACl becomes much broader upon interaction with BCF (Fig. 1e and f). At the same time, the singlet corresponding to the methyl group (CH3–) of the methylammonium cation shows a spectacular splitting to a quartet, which is only possible if all three protons of –NH3+ attain some fixed geometry and become symmetrically non-equal. All these findings taken together provide univocal evidence for the occurrence of hydrogen bonding interactions mediated by BCF. Very similar situation happens with CsI, which also shows a notable shift in the 133Cs NMR spectra evidencing the change of the coordination environment for the Cs+ cation (Fig. S4).
To theoretically analyze the interactions between BCF and the key perovskite precursor components the density functional theory (DFT) calculations were performed (see the SI for details). According to the DFT calculations, BCF covalently binds halides X− which leads to the formation of complex anions [BCF–X]− (X− = I−, Br−, Cl−), named boron–halide interactions. The bond enthalpies corresponding to the gas-phase binding enthalpy between the halide and BCF are ΔH° = −1.54 eV, −2.01 eV, −2.23 eV and the binding Gibbs free energy under standard conditions are ΔG° = −1.16 eV, −1.62 eV and −1.85 eV for I, Br and Cl, respectively (Fig. 1g–i). The bonds between BCF and MA as well as FA are weaker, with the gas-phase binding enthalpy values ΔH° = −0.67 and −0.53 eV under standard conditions and relatively modest Gibbs free binding energies ΔG° = −0.24 eV and −0.15 eV, respectively, which indicate that the cations would be much less restricted in their motion in the presence of BCF. It is also worth pointing out that the dimerization of BCF with itself has ΔH° = −0.88 eV and ΔG° =–0.17 eV, which means that some energy is required to break apart these noncovalent interactions when mixing it with the salts (Fig. 1j–l). Thus, the organic counter-ions FA+ and MA+ become confined to the corresponding bulky organoboron anions but can likely move relatively freely among them, which results in the formation of hydrogen-bonded complexes with a fixed unsymmetrical geometry. Furthermore, the minimal-energy computed molecular structures of BCF adducts with MABr and FAI were also calculated (Fig. 1m and n). In particular, the complex formation between MABr and BCF is thermodynamically favorable and proceeds with the energy gain of 6.4 kcal mol−1. The Br− anion in this complex forms a short contact with the B center of BCF, which confirms the halide-boron bonding type of interactions. Furthermore, the CH3NH3+ cation forms hydrogen bonds with the Br− counterion and F-substituent of BCF (Fig. 1m). The simulated FTIR spectra (Fig. S5a) showed that the most intense N–H bond stretching peak shifts from 3404 to 3377 cm−1 upon MABr complexation with BCF, which is consistent with the experimental data (Fig. S2b). The computed 1H NMR chemical shifts for the MABr–BCF complex also align well with the experimental results. Thus, the CH3 group is predicted to appear as a quartet with a chemical shift of 2.57 ppm (exp. 2.37 ppm), whereas the NH3+ group protons have a computed chemical shift of 7.72 ppm (exp. 7.57 ppm). The computed difference in the chemical shifts of C–H and N–H protons almost matches the experimental value (5.15 vs. 5.21 ppm, respectively).
The interaction patterns of BCF with FAI appeared to be more sophisticated as compared to the MABr + BCF system. DFT calculations predicted very small energy gain upon 1:1 complex formation between BCF and FAI, which cannot compensate for the decreased entropy of the system. However, the association of 2 FAI with 2 BCF molecules produces a very stable complex (Fig. 1n) with the energy gain of 46 kcal mol−1. The I− anions form short contacts with boron atoms of BCF, which confirms the boron–halide bonding effect. Two NH2 groups of the FA cations become strongly non-equivalent in the complex. Two protons of one NH2 group form short contacts with the I− anions, while the protons of another NH2 group participate in the hydrogen bond with the F-substituent of BCF. This arrangement first results in the distinct chemical shifts for protons of two NH2 groups: ca. 9.01 ppm (NH2 bonded to I−) and ca. 8.65 ppm (NH2 bonded to F). For the latter peak, the 1H–19F interactions lead to the signal splitting to the doublet (8.676 and 8.646 ppm). As a consequence of these amine proton bonding effects, the CH proton of the FA cation appears in the NMR spectra as a triplet of triplets. The computed chemical shifts for two types of N–H (9.09 and 8.25 ppm) and CH (7.70 ppm) protons agree reasonably well with the experimental values (9.01, 8.65 and 7.86 ppm, respectively). Finally, the simulated FTIR spectrum of the FAI–BCF interaction product (Fig. S5b) reveals the major N–H stretching band at ∼3256 cm−1, which is shifted to lower wavenumbers with respect to its position for pristine FAI modeled by its tetramer (3315–3325 cm−1), which is consistent with the experimental FTIR spectra shown in Fig. S2a.
The theoretical calculation results perfectly match the experimental FTIR and NMR spectral data, thus establishing reliable structures for the complexes of BCF with FAI, MABr and MACl. These findings collectively confirmed the existence of boron–halide interactions and hydrogen-bonding within the BCF complexes with the perovskite precursor components, which represents the strategic mechanism to regulate the two-step process of crystallization and growth of a wide-bandgap perovskite, thereby producing high-quality absorber films.
2.2. Crystallization regulation of wide-bandgap perovskite
To elucidate the crystallization kinetics of PbX2 (Fig. 2a and c) and perovskite (Fig. 2b and d) films during two-step sequential deposition, we performed in situ ultraviolet-visible (UV-vis) absorption spectroscopy studies. During the spin-coating process, after depositing the PbX2 solution onto the hole transport layer (HTL) film, the UV-vis spectra for both the PbX2 and PbX2 + BCF films exhibited similar features. The absorption edge of both PbX2 intermediate films underwent a rapid blueshift due to the large amount of solvent being removed, and then remained unchanged during the spin-coating process. However, during the annealing process at 70 °C, the absorption edge of the PbX2 film gradually redshifted, approaching 500 nm, while the absorption edge of the PbX2 + BCF film remained stable, staying close to 480 nm. This means that the boron–halide interactions effectively stabilized the uniform distribution of iodide and bromide ions within the lead halide film. This stability is attributed to the formation of stable intermediates between BCF and lead halide via the boron–halide interaction, which regulates the crystallization process.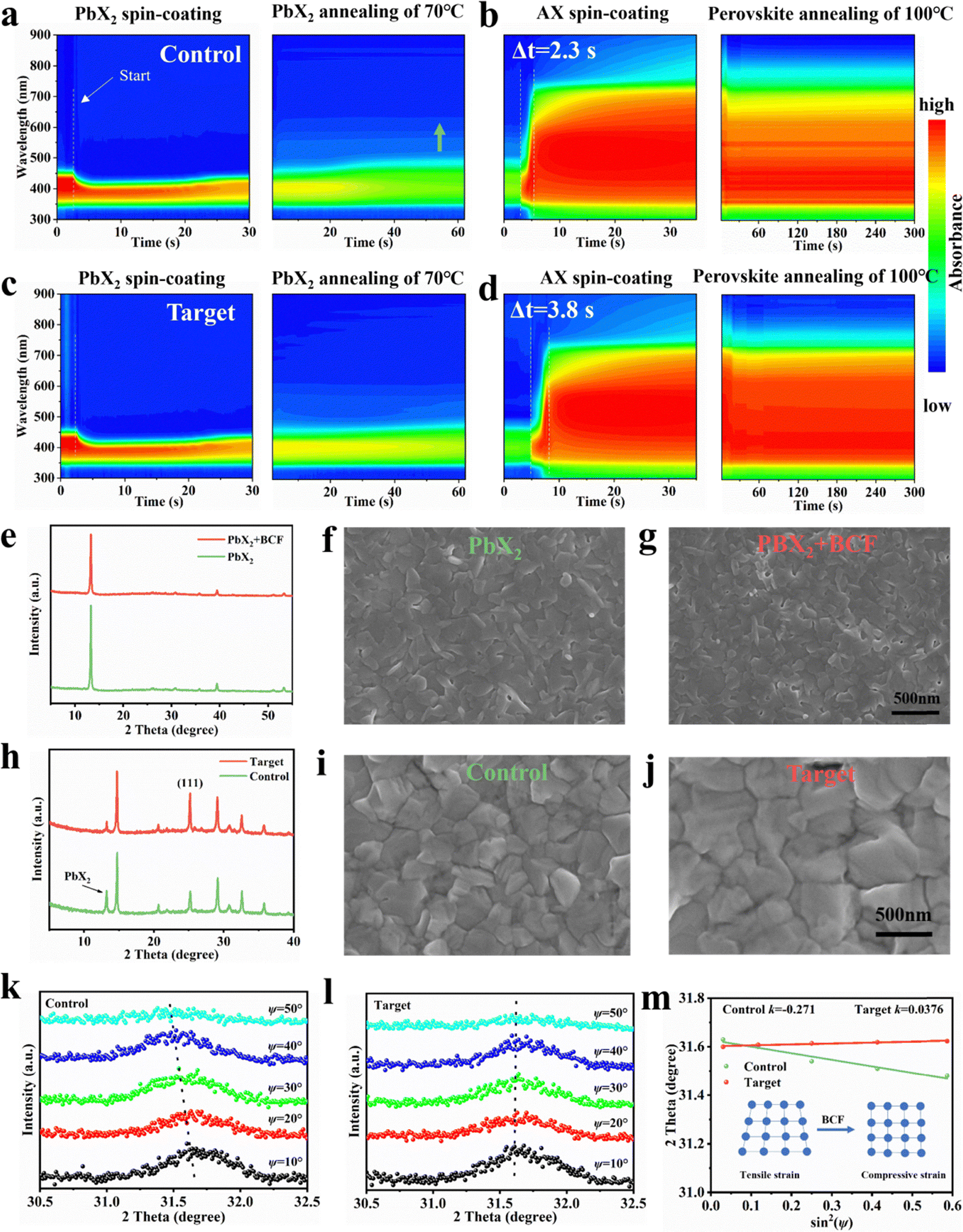 | ||
| Fig. 2
In situ UV-vis spectra of the differently processed PbX2 (a), (c) and perovskite (b), (d) films during spin-coating and annealing process. XRD patterns and top view SEM images of differently processed PbX2 (e)–(g) and perovskite (h)–(j) films. GIXRD patterns at different Ψ angles of the control (k) and target (l) perovskite films at an incident angle of 0.5°. (m) Linear fit of 2θ-sin2ψ of the control and target perovskite films. | ||
Upon dynamically dripping the organic ammonium salt solution onto the lead halide film, rapid nucleation and initial crystal growth were observed in the control perovskite films, as evidenced by an absorption shift to approximately 738 nm (the characteristic absorption edge of the 1.68 eV perovskite) within 2.3 seconds. In contrast, the absorption edge of the target perovskite film redshifted to approximately 738 nm more slowly, taking 3.8 seconds. This delay indicated that the BCF–PbX2 intermediates and hydrogen-bonded complexes slowed the intermolecular exchange process between the organic salt and lead halide, allowing for better controlled crystallization.
During the subsequent thermal annealing process at 100 °C, the target perovskite film displayed a uniform and stable absorption spectrum, whereas the control perovskite film showed slightly weaker absorption features. This discrepancy is attributed to the presence of residual lead halide and Pb0 in the control film as proved by XRD and XPS measurements, which hindered its light absorption efficiency. It is worth noting that a slight redshift from 738 nm to 743 nm occurred at the absorption edge of the control film, which is attributed to the formation of an iodine-rich phase on the film. The in situ UV-vis absorption spectroscopy results demonstrated that the boron–halide interaction and hydrogen-bonded complexes effectively stabilized the uniform distribution of iodide and bromide ions to suppress phase separation, and delays the reaction and crystallization kinetics between the organic ammonium salt and lead halide. This controlled delay was expected to promote the formation of larger grains in the perovskite films with fewer grain boundaries and enhance the overall quality of the perovskite films.
Fig. 2e presents the X-ray diffraction (XRD) patterns of the PbX2 and PbX2 + BCF films. The intensity of the diffraction peaks corresponding to the PbX2 crystal planes decreased with the introduction of BCF, attributed to the formation of a fluffy, porous morphology upon incorporating the BCF additive into the PbX2 film. To further investigate the morphological changes induced by BCF, the scanning electron microscopy (SEM) and atomic force microscopy (AFM) measurements were carried out. The top-view SEM showed that the addition of BCF results in a significant decrease in the grain size of PbX2, which consequently leads to an increase in the areal density of grain boundaries and voids (Fig. 2f and g). Furthermore, the cross-sectional SEM images clearly demonstrated that the addition of BCF resulted in the formation of numerous voids and interlaced patterns within the PbX2 + BCF layer (Fig. S6), which is not observed in the PbX2 films. These findings confirm the strong influence of BCF addition on the morphology of the PbX2 films, which facilitated the sufficient diffusion of the organic ammonium salts during the perovskite formation process. This observation was supported by the increase in root mean square (RMS) roughness from 6.7 nm for the PbX2 film to 7.8 nm for the PbX2 + BCF film, as measured by AFM, further confirming the morphological changes induced by BCF (Fig. S7). Infrared scattering-type scanning near field optical microscopy (IR s-SNOM) was applied to further characterize the PbX2 + BCF films. This technique enables simultaneous AFM topography measurement with the collection of local spectral data from the area comparable to the tip radius of the cantilever (20–30 nm).34,35 Thus, IR s-SNOM could reveal the distribution of the BCF molecules within the target PbX2 + BCF films. The major part of BCF was accumulated at the grain boundaries, and also observed on the surface of the PbX2 grains (Fig. S8), which confirms that the boron–halide interactions control the growth of the PbX2 + BCF films.
To investigate the influence of the BCF modifier within the PbX2 precursor on the perovskite films crystallinity, we analyzed the X-ray diffraction (XRD) patterns of the perovskite film. Both types of the perovskite films processed without and with BCF revealed a peak at ≈13°, attributed to residual unreacted PbX2 (Fig. 2h). The intensity of this PbX2 peak was significantly lower for the target perovskite film as compared to the control film. This reduction suggested that the incorporation of BCF facilitated PbX2 conversion in the reaction with the organic ammonium salts and improved interdiffusion of the organic and inorganic components. This optimization promoted the growth of high-quality perovskite crystals. Notably, trace amounts of lead halide remained in the target perovskite film, which was expected to passivate surface and grain boundary defects, thereby improving device performance. Furthermore, the intensity of the (111) diffraction peak was notably stronger in the target perovskite film, indicating that BCF promoted the growth of the (111) crystal plane.
To further evaluate the effects of BCF on perovskite film quality, surface morphological characterization was conducted using scanning electron microscopy (SEM) and atomic force microscopy (AFM). The SEM images (Fig. 2i and j) revealed that the target perovskite films consist of larger grains with fewer grain boundaries (GBs) compared to the control films, which was attributed to the slower chemical reaction and crystallization kinetics between the organic ammonium salts and the lead halides due to the boron–halide and the hydrogen bonding interactions.36,37 This improvement of the perovskite grains was corroborated by cross-sectional SEM images (Fig. S9), which showed no distinct grain stacking in the vertical direction in the case of target films, facilitating efficient charge carrier transport. Additionally, the target perovskite film exhibited a higher water contact angle (72.40°) than the control film (53.47°) (Fig. S10), demonstrating that BCF treatment enhanced the film's ability to resist water intrusion, thereby improving its stability under ambient conditions and thus enhancing its potential for real-world applications.
AFM measurements (Fig. S11) further confirmed that the surface roughness (RMS) of the target perovskite films decreased from 27.8 nm to 20.3 nm, promoting better interfacial contact and charge transfer with the subsequently deposited ETL. Kelvin probe force microscopy (KPFM) was employed to explore the alignment of the interface energy levels. As shown in Fig. S12 and Fig. S13, the surface contact potential distribution (CPD) of the target perovskite film is more uniform, within a range of ±10 mV, compared to the control film, which exhibits a broader range of ±15 mV. This uniformity was attributed to the more homogeneous surface structure and a more controllable crystallization and growth process of the target perovskite films. The KPFM results also suggested that the target perovskite film exhibited a more positive surface potential, indicating that the Fermi level on the perovskite surface is closer to the conduction band to form a more n-type surface electric potential of the film. This alignment is favorable for efficient electron extraction and transfer, further enhancing device performance.
In order to evaluate the influence of BCF additives on the residual stress of the perovskite films, the surface residual stress from 100 nanometers depth below the surface of the perovskite film was measured by deep-resolution grazing incidence X-ray diffraction (GIXRD) combined with the 2θ-sin2ψ method (Fig. 2k and l). As is well known, the negative slope of the 2θ-sin2ψ method linear fitting indicated the residual tensile stress in the perovskite film, and the positive slope of the 2θ-sin2ψ method linear fitting indicated the compressive strain in the perovskite film. Obviously, due to the formation of the boron–halide interactions and hydrogen-bonded complexes between BCF molecules and perovskite precursor components, the slope of 2θ-sin2ψ variation changed from negative to positive, indicating that stress changed from tensile stress (183.1 MPa) of the control perovskite film to compressive stress (−25.4 MPa) of the target sample. These findings suggested that BCF released the strain during thermal annealing and exhibited an excellent ability to regulate grain growth and morphological structure, and improved device performance and mechanical stability (Fig. 2m and Fig. S14).
An additional insight into the effects of the BCF additive on the nanoscale structure of the grown perovskite films was gained using IR s-SNOM. Using this technique, we could individually follow the nanoscale distribution of organic cations (MA+, FA+) and the BCF additive in the perovskite films. The obtained results are presented in Fig. 3. For the control perovskite film, we observed very uniform distribution of the methylammonium and formamidinium cations in the film. In contrast, the target perovskite films showed some minor local gradients in the concentrations of MA+ and FA+ cations thus creating a “perovskite bulk heterojunction” system, which we have shown recently is highly beneficial for the PSC performance.38 Furthermore, we demonstrated that BCF effectively covered the perovskite film surface and showed the highest abundance at the grain boundaries, thus contributing to the passivation of defects and suppressing the nonradiative trap-assisted recombination of charge carriers.
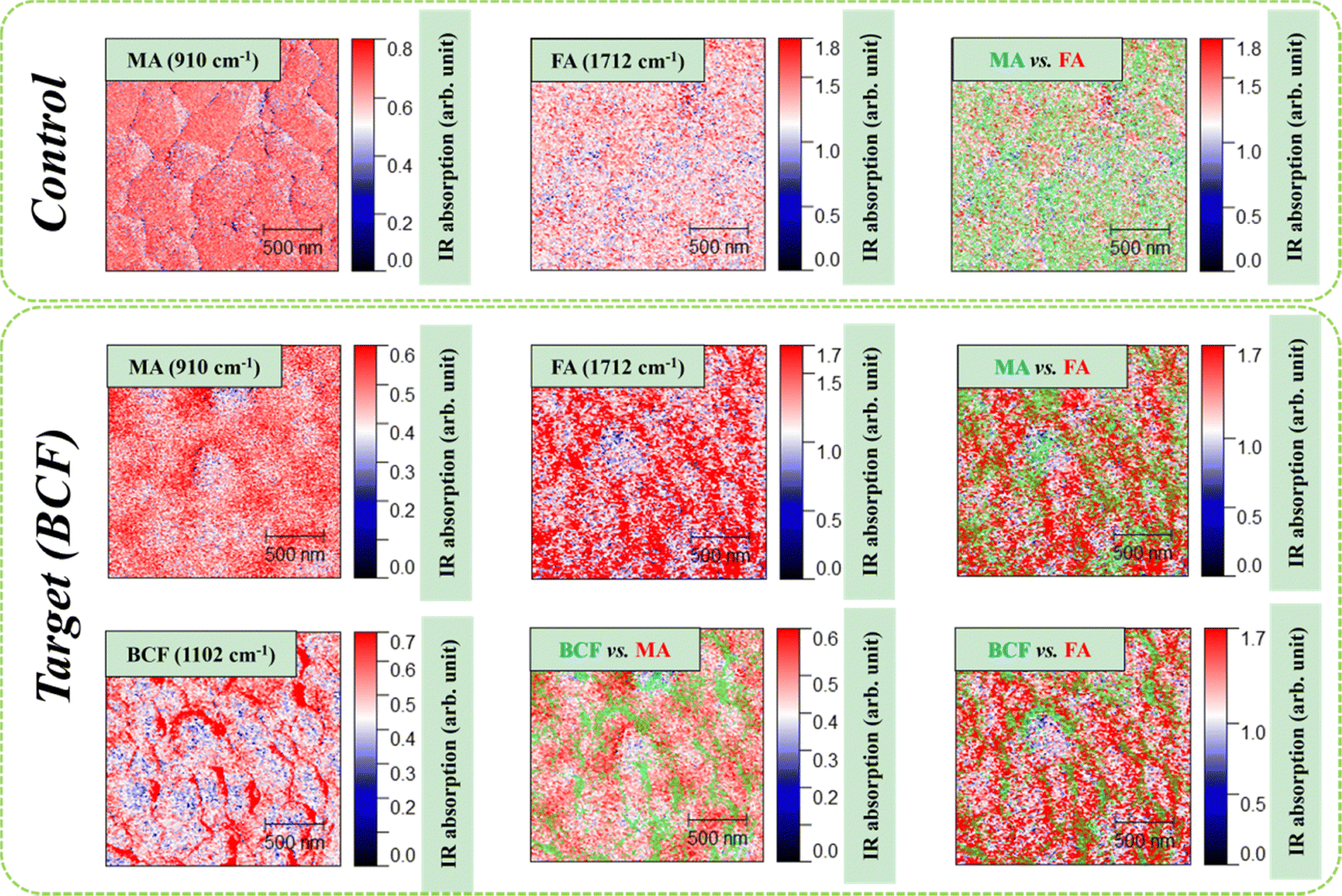 | ||
| Fig. 3 IR s-SNOM imaging data for control and target perovskite films. The red color in s-SNOM maps corresponds to a higher concentration of the analyzed species. Superposition maps are shown in green and red color code as specified in the legends. | ||
Thus, the obtained IR s-SNOM imaging data corroborated with the results provided by other techniques and proved unambiguously that the introduction of BCF had a highly positive influence on the nanoscale morphology and structure of the perovskite absorber films grown using the two-step deposition method.
2.3. Nonradiative recombination analysis of perovskite films
XPS analysis was conducted to further investigate the interactions between the perovskite films and BCF. The Pb 4f peaks exhibited a shift toward lower binding energy, indicating strong interactions between BCF and the perovskite film. This shift can be attributed to the synergistic effects of the fluorine-substituted aromatic rings in BCF bonding with the organic ammonium ion and boron center bonding with halide anions. These interactions could lead to a significant change in the electron environment of lead cations Pb2+ on the film surface (Fig. 4a). Additionally, the peak intensity of Pb0, which acts as a nonradiative recombination center, was effectively suppressed in the target perovskite film, suggesting that the introduction of BCF stabilized the octahedral coordination environment of [PbX6]4− on the perovskite film surface, which is consistent with the results obtained from in situ UV-vis absorption spectroscopy. Similar shifts were observed in the I 3d and Br 3d peaks, further supporting the effects of halide binding on the boron center (Fig. 4b and c). Survey XPS spectra and N 1s core level spectra of the perovskite films are shown in Fig. S15a and b, respectively. The shift in the N 1s peak after BCF addition was consistent with the hydrogen bonding interactions revealed for FA+/MA+ cations by NMR spectroscopy, as discussed above. The XPS results collectively demonstrated that the BCF additive interacted with the perovskite to passivate defects within the film.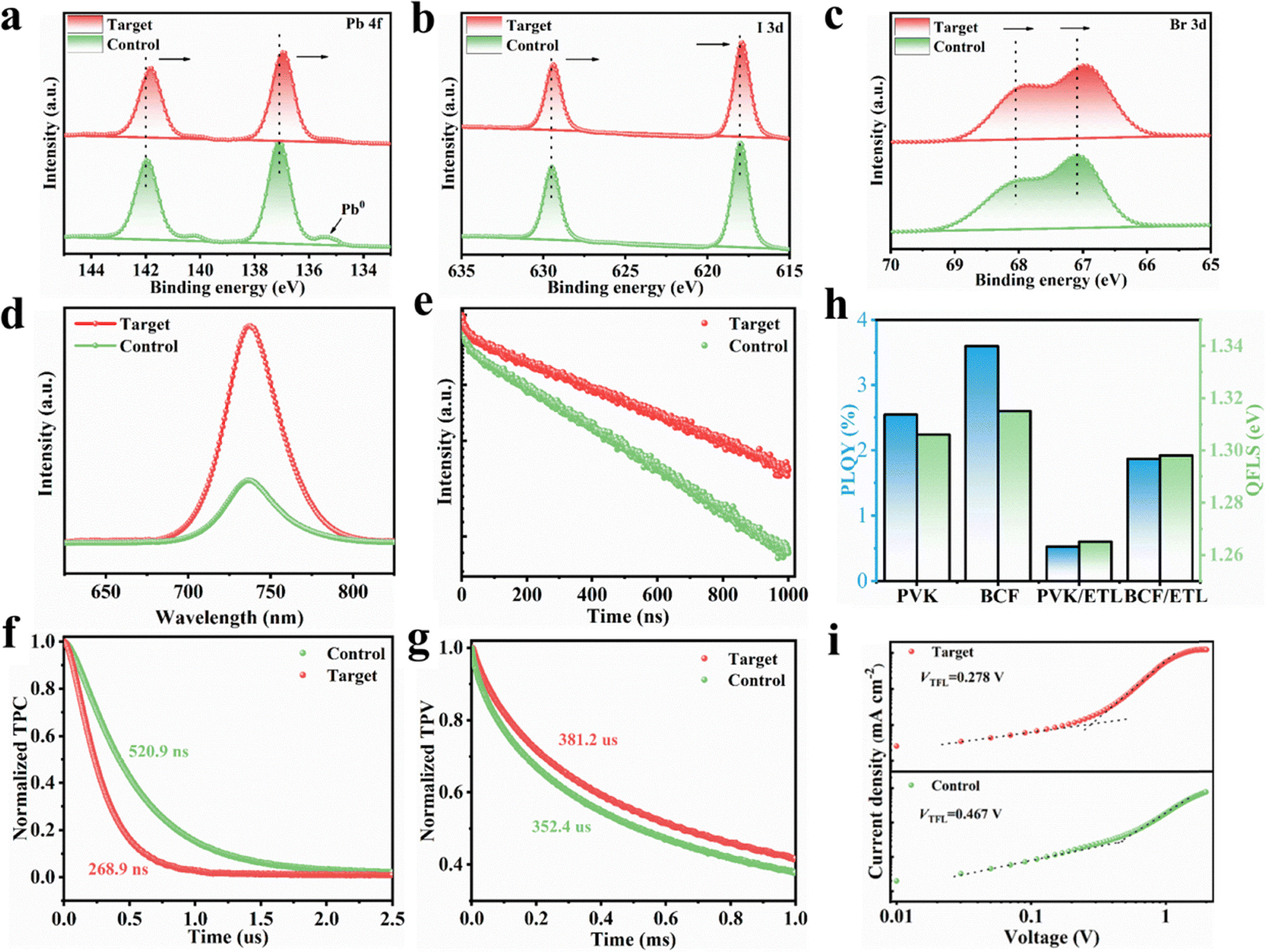 | ||
| Fig. 4 Core-level XPS Pb 4f (a), I 3d (b) and Br 3d (c) spectra of control and target perovskite films, respectively. Steady-state PL spectra (d) and TRPL spectra (e) of the control and target perovskite films. (f) TPC decay curve and (g) TPV decay curve of the control and target perovskite solar cell devices. (h) PLQY and QFLS results of control and target perovskite films without or with ETL on glass, respectively. (i) Dark J–V curves of the hole-only devices based on the control and target perovskite films. | ||
The charge transfer and recombination kinetics of the perovskite films were investigated using steady-state photoluminescence (PL) and time-resolved photoluminescence (TRPL) measurements. The target perovskite film exhibited higher PL intensity compared to the control film, indicating reduced density of defect states and improved film quality due to effective defect passivation (Fig. 4d). TRPL results (Fig. 4e) were fitted using eqn (S1) and S2, revealing an increase in the average carrier lifetime from 731.25 ns to 1180.70 ns with a slightly enhanced rapid decay lifetime (τ1) and a significantly enhanced slow decay lifetime (τ2), which indicates that the introduction of BCF not only significantly passivated the bulk defects in the perovskite film but also inhibited non-radiative recombination at the interface (Table S1). On the bare glass substrate without the charge extraction pathway, the perovskite films only exhibited the characteristic slow PL decay due to the dominance of bulk defect-mediated recombination.39
Transient photocurrent (TPC) decay testing was used to characterize the carrier extraction and transport capabilities within perovskite solar cells. As shown in Fig. 4f, the target device exhibited a shorter decay lifetime of 268.9 ns, compared to 520.9 ns for the control device. The faster TPC decay indicated that the addition of BCF can facilitate the transport and transfer of photogenerated charges, demonstrating the stronger carrier extraction ability of the target device. The device transient photovoltage (TPV) decay test was employed to further reveal the nonradiative recombination of charge carriers within and at the interface of perovskite films when the device is in an open-circuit state (Fig. 4g). The decay lifetime of TPV for the target device is 381.2 μs, longer than that of the target device (352.4 μs). The addition of BCF effectively suppresses nonradiative recombination within and at the interface of perovskite films, thereby prolonging the carrier lifetime in the target devices. These findings collectively affirm that the introduction of BCF molecules can promote carrier extraction and reduce nonradiative recombination losses, thereby realizing high-performance photovoltaic devices.40
To quantify the nonradiative recombination loss at the perovskite film interface, we measured the photoluminescence quantum yield (PLQY) to estimate the quasi-Fermi level splitting (QFLS) relevant to the theoretically feasible VOC (Fig. 4h and eqn (S3)). The PLQY increased from 2.544% to 3.602%, indicating an improvement in the efficiency of treating perovskite with BCF on glass substrates. The results showed that the average QFLS increased from 1.306 eV to 1.315 eV, which was directly related to the increment of VOC in PSCs. Significantly, the PVK/ETL films exhibited a remarkable PLQY drop, which was attributed to additional defects introduced by the ETL. After the addition of the ETL, the QFLS of the control film decreased to 1.265 eV, indicating a 41 mV increase in nonradiative recombination loss, while the QFLS of the BCF/ETL remained stable at 1.298 eV, decreasing by only 17 mV (Table S2). These results emphasized that BCF effectively reduced VOC loss by suppressing nonradiative recombination.
Furthermore, the defect density of states (Nt) in the perovskite films was quantitatively evaluated using eqn (S4). Hole-only devices (ITO/NiOx/Me-4PACz/Perovskite/Spiro-OMeTAD/Ag) were fabricated for space charge-limited current (SCLC) measurements. As can be seen in Fig. 4i, Nt of the target film decreased from 6.73 × 1015 cm−3 to 4.01 × 1015 cm−3 compared to the control film (Table S3). This reduction in Nt enhanced charge mobility, further supporting the role of BCF in improving perovskite crystal quality.
Photoinduced phase segregation, primarily driven by ion migration through halide vacancies, is a critical factor which limits the performance and longevity of wide-bandgap perovskite films. The evolution of PL spectra under one-sun illumination was analyzed to evaluate halide segregation. In particular, the control perovskite film exhibited a significant redshift in its PL spectra, indicating the formation of narrow bandgap I-rich defect states due to phase segregation (Fig. S16a). In contrast, the BCF-treated film showed suppressed photoinduced phase segregation (Fig. S16b), suggesting that BCF contributes to the formation of more stable wide-bandgap perovskites. To quantify these changes, we calculated the PL peak shifts after 90 minutes of illumination. The BCF-treated wide-bandgap perovskite films showed a smaller redshift by 9 nm, compared to the 21 nm of the control film. This smaller shift demonstrated BCF's ability to inhibit photoinduced phase segregation, which was likely due to defect passivation and halide immobilization on the perovskite grain surface by boron–halogen interactions.
Strong evidence for the remarkable stabilizing effect of BCF was obtained using unique in situ IR s-SNOM measurements that to the best of our knowledge have been performed for lead halide perovskite films for the first time. The tested samples were irradiated with blue laser light (460 nm, ca. 100 mW cm−2) directly at the microscope stage. Running a continuous measurement produced a series of IR s-SNOM images for control and target samples, which could be compared on the same timescale. From the Video S1, it is clearly seen that the target perovskite film demonstrates much higher stability under light exposure than the control sample. Indeed, a strong depletion of organic formamidinium cations is observed in control films after 2 h of light-induced aging, while the target films remain almost intact under the same conditions. These findings directly prove a strong stabilization effect of BCF and its ability to suppress light-induced ion migration and degradation of inorganic cations.
The enhanced stability of wide-bandgap perovskites must correlate with the ability of BCF to suppress the photoinduced segregation of Br-rich and I-rich phases. Mechanistically, the halide phase segregation process involves the formation of Pb0 and I3− species, their accumulation at the grain boundaries, and subsequent recombination (Pb0 + I3− → [PbI3]−) with the formation of an I-rich phase.41,42 We have shown that BCF significantly improves the photostability of PbX2 films (I:Br = 3:1) and suppresses the formation of metallic lead in the samples exposed to 100 mW cm−2 illumination at a temperature of 77 ± 3 °C in a pure nitrogen atmosphere (Fig. S17). This finding provides clear evidence that BCF controls the behavior of PbX2 films, suppresses Br–I segregation, and enhances inorganic phase photostability, which finally leads to the stabilization of wide-bandgap perovskites.
To gain a further mechanistic insight, we modelled the behavior of the PbI2–BCF system using DFT calculations for a simple cluster model composed of four interconnected ribbons of PbI2 (Fig. S18). This cluster has a series of coordinationally unsaturated Pb2+ ions surrounded by 3, 4 or 5 iodide anions, which can be considered as a realistic model for the PbI2 grain surface. DFT calculations demonstrate that the Pb0 atom appearing within the PbI2 structure tends to pair with one of the Pb2+ cations and forms a [Pb–Pb]2+ molecule-like fragment. The maximal energy gain of 46.2 kcal mol−1 is reached when the [Pb–Pb]2+ fragment is localized on the surface of the PbI2 cluster (Fig. S18a). Interestingly, the presence of more Pb0 atoms within PbI2 makes them clustering together forming Pb2, Pb4 and even bigger species, which forms the initial stages of the metallic lead phase nucleation process.
However, the presence of BCF in the system drastically changes its dynamics since BCF was shown to bind to Pb0 with the energy gains varied from 4 to 14 kcal mol−1 depending on the Pb0 localization. The most realistic structure shown in Fig. S19 is characterized by the surface localization of metallic lead with Pb0 and BCF binding energies of 44.2 and 13.1 kcal mol−1, respectively.
In the further modelling, we introduced an I2 molecule into the system, forming a triiodide (I3−) fragment on the surface of the PbI2 cluster. Thus, the set of structures presented in Fig. S19 represents the intermediate state with the photogenerated I3− and Pb0 species, which could evolve in several ways. First, I3− could release I2 as a vapor into the gas phase and then the material decomposition becomes irreversible with the accumulation of metallic lead as a distinctive feature. Second, Pb0 could nucleate as small metallic clusters, then react with I3− and form a separate iodide-rich phase, thus leading to the I–Br phase segregation. However, BCF effectively binds to Pb0 and suppresses the metallic lead nucleation process. Furthermore, our calculations show that when the distance between I3− and Pb0 species reduces, as illustrated by the transition from the structure A to structure B in Fig. S19, the BCF binding energy also decreases dramatically from 11.6 to 5.4 kcal mol−1. Simultaneously, the I2 binding energy also becomes twice smaller (20.2 and 10.3 kcal mol−1 for structures A and B), which provides ideal conditions for BCF desorption from the surface and recombination of Pb0 with I2 leading to the regeneration of PbI2.
Thus, our modelling unravels the unique mechanism, which involves reversible binding of BCF to Pb0 on the PbX2 surface (blocking metallic lead phase nucleation) and subsequent release of Pb0 when I3− diffuses closer, thus promoting their recombination and PbI2 formation. This extraordinary BCF behavior makes it a strategic stabilizer of perovskite absorber films with potential of showing tremendous improvements in the PSC operational lifetime.
2.4. Device performance and stability of single junction wide-bandgap PSCs
To evaluate the impact of the BCF additive on photovoltaic performance of the modified absorber material, single-junction wide-bandgap perovskite solar cells (PSCs) were fabricated by the all-solution two-step method in an inverted configuration: ITO/NiOx/Me-4PACz/Perovskite/PCBM/BCP/Ag. The schematic diagram of the device is illustrated in Fig. 5a, and the detailed fabrication process is provided in the Experimental section of the SI. We assembled p–i–n PSCs and revealed that the optimal BCF concentration in perovskite precursor solution is 1 mg mL−1, which enables the best device performance (Fig. S20, S21 and Table S4). The power conversion efficiency (PCE) of the champion PSCs increased considerably from 21.74% (control device) to 23.49% (target device), accompanied by an improvement in open-circuit voltage (VOC) from 1.257 V to 1.291 V and in the fill factor (FF) from 79.7% to 83.2%, as demonstrated in Fig. 5b and Table S5. Furthermore, one of our best single-junction devices employing BCF was sent for certification by an authorized organization and yielded a certified PCE of 22.73%, with a VOC of 1.278 V (see in Fig. S22). This represents one of the highest PCEs reported for 1.68 eV wide-bandgap PSCs to date and the highest PCE achieved for 1.68 eV PSCs fabricated using the two-step method (Table S6 and S7). Furthermore, we purchased PbI2 from three different companies for the experiments of the target group in order to evaluate the impact of the PbI2 origin and potentially present impurities on the performance of the devices. From Fig. S23 and Table S8, it can be observed that the performance of the three different batches of devices prepared using PbI2 from different companies was basically the same. However, the perovskite solar cells prepared using PbI2 from C company with lower purity have smaller standard deviations for all performance indicators, which better replicate the device efficiency. These findings indicate that the trace impurities in PbI2 have a negligible effect on the performance of the devices. This is consistent with previous studies, which indicate that a water content below 15 ppm can minimize its impact on the performance of devices to the greatest extent.43 Instead, the different preparation processes of the PbI2 material used in different companies will ultimately determine the reproducibility of the device characteristics. Also, there are relevant studies indicating that the role of water in perovskite solar cells has little or even beneficial impact.44,45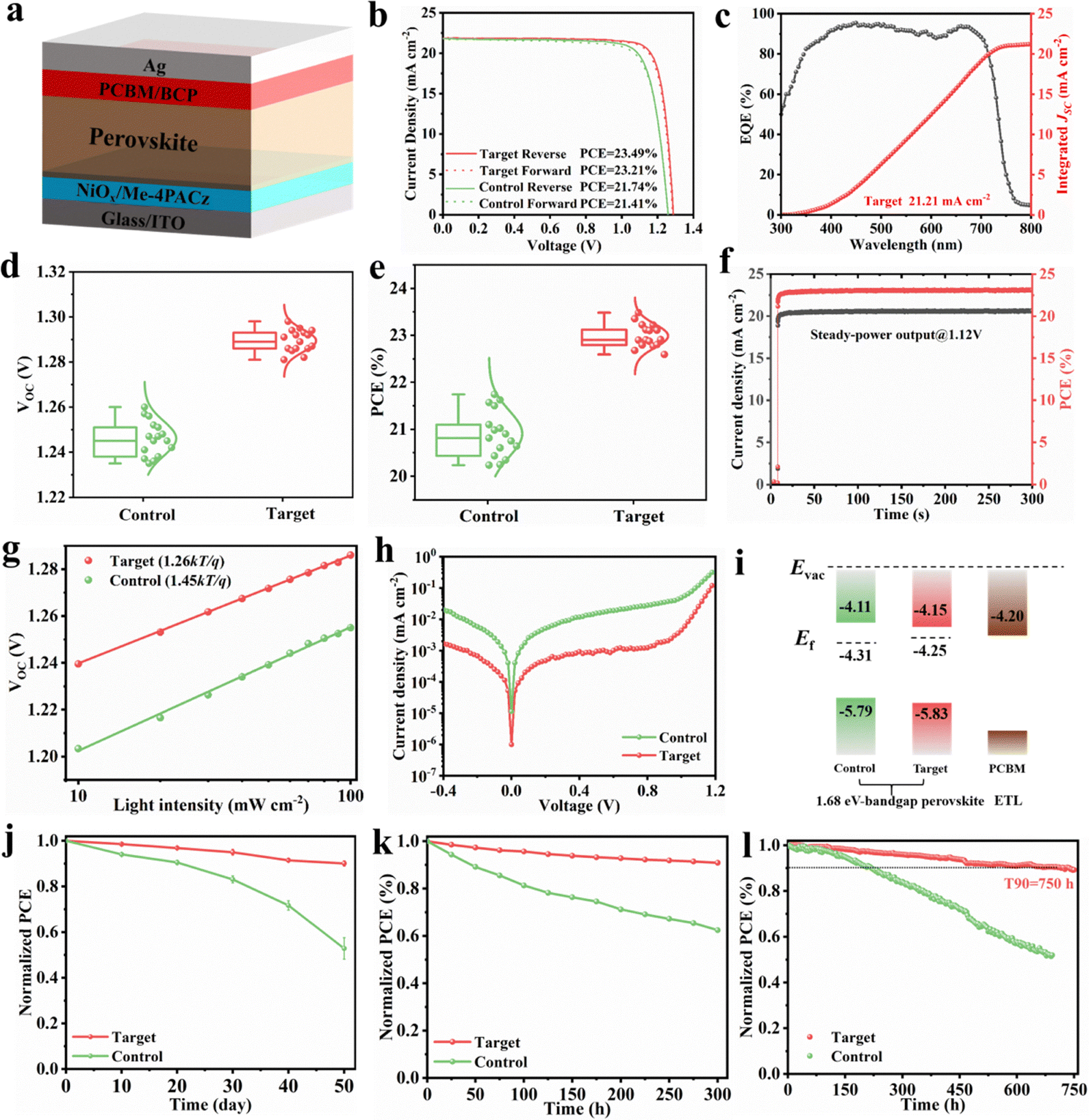 | ||
| Fig. 5 (a) Schematic diagram of the inverted 1.68 eV wide-bandgap PSC device structure. (b) The forward and reverse sweep J–V curves of the best-performance devices with the differently processed perovskite absorber films. (c) EQE spectra of the target perovskite absorber films. (d) and (e) The VOC and PCE distributions for the different series of devices. (f) The maximum power point steady-state power output for target devices under 1 sun illumination. (g) Light intensity dependence of VOC for control and target devices. (h) Dark J–V curves of PSCs fabricated using differently processed perovskite films. (i) Energy level diagram of differently processed perovskite films. (j) The long-term stability test for the unencapsulated devices stored in air with a relative humidity of 50% at 25 ± 5 °C. (k) The heat stability test of unencapsulated PSCs with and without BCF upon heating at 85 °C in N2. (l) Operational stability of PSCs in the MPPT regime under 100 mW cm−2 white LED illumination in N2 at a temperature of approximately 50 °C. | ||
Additionally, the integral short circuit current density (JSC) derived from external quantum efficiency (EQE) measurements for the target PSCs was 21.21 mA cm−2, closely matching the JSC value of 21.87 mA cm−2 obtained from J–V measurements under simulated AM 1.5G illumination (Fig. 5c). Statistical analysis of the photovoltaic parameters (Fig. 5d and e) confirms the reproducibility of these BCF-optimized devices. The performance enhancement is attributed to improved trap passivation and crystallinity facilitated by the BCF additive. The target device exhibited a stable power output (SPO) efficiency of 23.2% when measured at the maximum power point (Vmax = 1.12 V) (Fig. 5f), consistent with the PCE obtained from the J–V curve.
Furthermore, the relationship between VOC and the logarithm of the illumination intensity yielded fitted slopes of 1.45kT/q for the control device and 1.26kT/q for the target device, as calculated using eqn (S5) (Fig. 5g). Dark J–V measurements (Fig. 5h) revealed that the BCF-treated devices exhibited a lower dark saturation current and reduced leakage current compared to the control device, indicating suppressed leakage pathways and improved diode ideality. This suggests a reduction in trap-assisted recombination, further corroborating the higher PCE of the BCF-modified device.
To investigate the influence of the BCF additive on the optical properties of the perovskite photoactive layer, UV-vis absorption spectra were measured. Fig. S24 demonstrates that the target perovskite film exhibited enhanced light absorption intensity, primarily due to improved perovskite crystal quality. Tauc plots derived from the UV-vis absorption data confirmed a bandgap (Eg) of 1.68 eV for the perovskite absorber layer. Ultraviolet photoelectron spectroscopy (UPS) was also employed to analyze the optoelectronic properties of the perovskite films (Fig. S25). The UPS results indicated a decrease in work function (WF) from 4.31 eV (control) to 4.25 eV (target), consistent with Kelvin probe force microscopy measurements (Table S9). The upward shift in WF suggests that the perovskite surface exhibits n-type doping characteristics after BCF incorporation, which is expected given that BCF is a Lewis base. The energy level diagram for the PSC functional layers is presented in Fig. 5i. The introduction of BCF optimizes the energy level alignment at the perovskite/ETL interface, enhancing carrier extraction and contributing to the higher PCE achieved in the photovoltaic devices. These findings underscore the significant role of BCF in improving the performance of wide-bandgap PSCs through enhanced crystallinity, trap passivation, and optimized energy level alignment.46
Comprehensive stability studies were then carried out in accordance with the stability protocols for PSCs. In ambient stability tests, the unencapsulated devices were stored in air with a relative humidity of 55% at 25 ± 5 °C and their performance was regularly monitored, as can be seen in Fig. 5j. After ambient storage for approximately 50 days, the control devices maintain, in average, only 50% of their initial PCE, while the target series of PSCs maintain, in average, 90% of their initial PCE, respectively, which is attributed to the high-quality perovskite films and a higher exposure ratio of the (111) crystal plane of target devices. The heat stability of unencapsulated devices at 85 °C with day/night cycles was assessed. Remarkably, PSCs based on BCF retained above 90% of their initial efficiency after 300 h, compared with 60% retention (Fig. 5k). Operating stability was assessed under harsh conditions in N2 at a temperature of approximately 50 °C (unencapsulated, continuous 1 sun illumination, and negative electric bias) (Fig. 5l). The PSCs employing BCF as a modifier retain 90% of their initial PCE value after 750 h, in sharp contrast to the low 50% PCE retention observed for the control devices.
2.5. Perovskite/Si tandem solar cells
We utilized BCF-modified perovskite films to fabricate monolithic perovskite/c-Si TSCs based on c-Si solar cells in Fig. 6a, which is the first time that the 1.68 eV wide-bandgap perovskite prepared by the all-solution two-step method has been applied in a perovskite/silicon TSC device. The sputtered indium zinc oxide (IZO) was used as a transparent electrode with a thickness of 40 nm. It is worth noting that in order to ensure the complete coverage of the perovskite film on the silicon pyramids, we increased the concentration of the PbX2 precursor solution from 1.5 M to 1.8 M. The cross-sectional SEM images of the PbX2/silicon stack are shown in Fig. S26. The detailed fabrication method of the tandem device can be found in the experimental section of the SI. Compared with the single-junction PSCs preparation process, the top perovskite sub-cell of the TSCs was optimized due to the light trapping effects enabled by covering the entire surface of the silicon with pyramidal structures. Fig. 6b shows the cross-sectional SEM image of the Si/NiOX/Me-4PACz/perovskite/C60/SnOX/IZO/Ag/MgFX structure. The best-performing perovskite/c-Si TSCs of 1 cm2 using BCF modifier achieved a champion PCE of 31.12%, VOC of 1.940 V, JSC of 20.265 mA cm−2, and FF of 79.2% under reverse scan with a negligible hysteresis (Fig. 6c and Table S10 and S11). We also verified the reproducibility of the processes, exhibiting a narrow statistical distribution, acquired from 16 devices of multiple processing batches (Fig. 6d). Fig. 6e shows the EQE spectra of the two sub-cells in the high-performance BCF-based perovskite/c-Si TSCs devices. The integrated JSC values for the perovskite and silicon sub-cells are 20.41 and 20.28 mA cm−2, respectively, which closely match the values extracted from the J–V scan curve. The stabilized PCE of 30.81% is recorded for the BCF-modified TSCs device after continuous 1 sun illumination for 300 s (Fig. 6f), closely consistent with the J–V result. The unencapsulated TSC device exhibits decent operational stability by maintaining 90% of the initial PCE for 500 h after continuous operation in the MPP tracking regime under LED lamp illumination in a nitrogen environment (Fig. 6g). Here, it should be noted that the fabricated perovskite/c-Si TSCs exhibit good stability under ISOS-L-1 conditions, but there remains a gap compared to state-of-the-art tandem devices prepared by a one-step method. Although the all-solution two-step method successfully generated a uniform and pinhole-free perovskite film on the surface of the sub-micron pyramid structured c-Si cells, due to the limitation of the unreacted lead iodide at the pyramid bottom, the challenge of extracting photogenerated carriers from the perovskite layer has not been fully addressed.47 To address the challenge, achieving a balance between light management using submicron-scale pyramids and the existence of the unreacted lead iodide in the thick perovskite film prepared by all-solution two-step method is crucial for unifying the stability of tandem and single-junction photovoltaic cells.48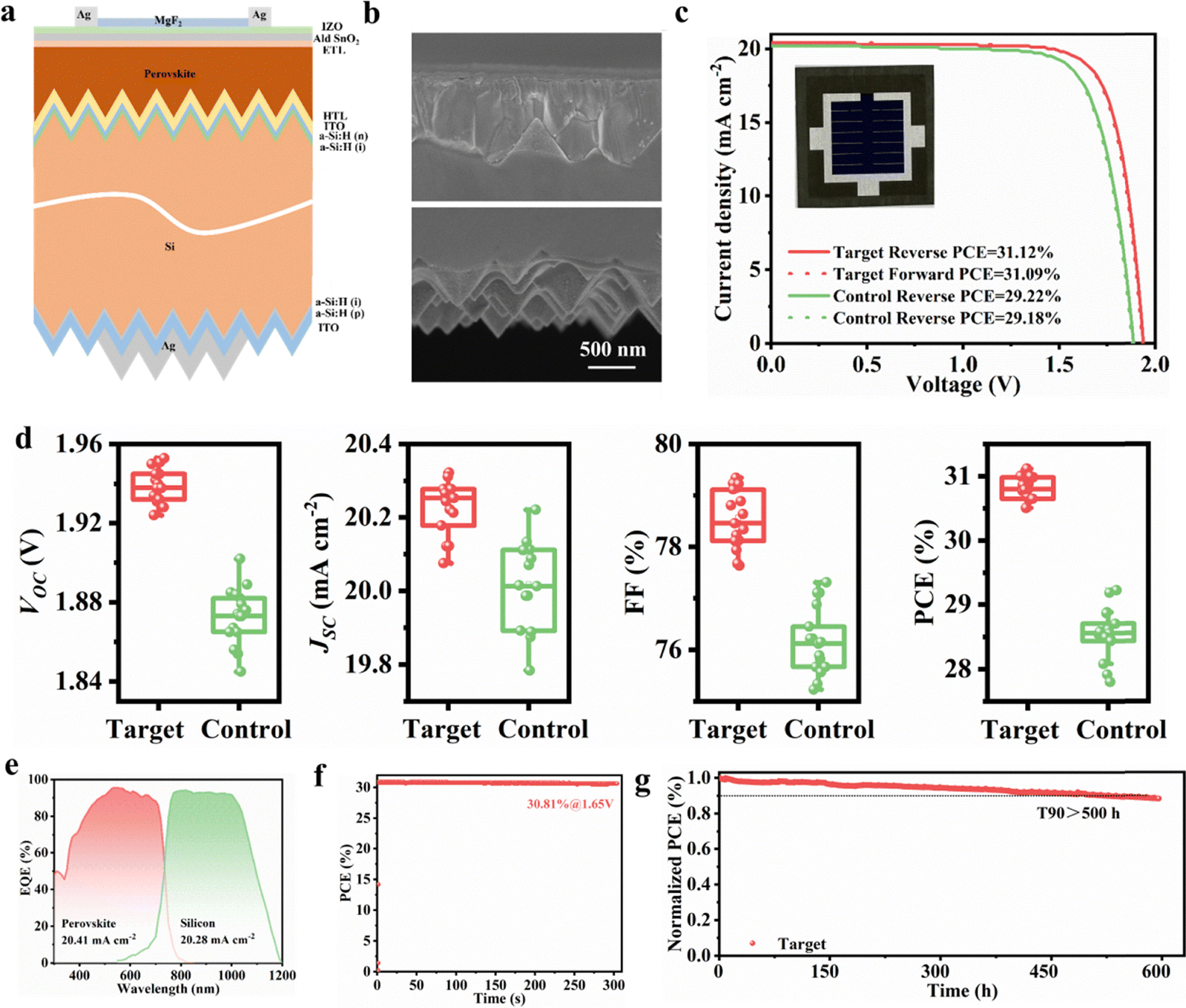 | ||
| Fig. 6 (a) Schematic structure of TSCs. (b) Cross-sectional SEM image of a perovskite/silicon tandem device. (c) J–V curve of the champion of TSCs. (d) The VOC, JSC, FF and PCE distributions for the different series of devices. (e) EQE spectra of the target tandem device and corresponding integrated JSC of the sub-cells. (f) A SPO at maximum power point as a function of time of TSCs. (g) MPP tracking of the unencapsulated devices in an N2 atmosphere under one-sun white-light LED illumination at a temperature of approximately 50 °C. | ||
3. Conclusion
The incorporation of BCF into the 1.68 eV wide-bandgap perovskite through the all-solution two-step method significantly enhanced both device efficiency and stability. By leveraging the synergistic effects of boron–halogen covalent bonding as well as hydrogen-bonded complexes, we successfully modulated crystallization of 1.68 eV wide-bandgap perovskite films, thus stabilizing uniform halide distribution and delaying the reaction between the organic ammonium salt and the inorganic precursor. BCF molecules are shown to localize on the surface of the perovskite films and at the gain boundaries, thereby inhibiting nonradiative recombination losses and suppressing phase segregation. As a result, we achieved single-junction wide-bandgap PSCs with a remarkable efficiency of 23.49% and a high open-circuit voltage (VOC) of 1.291 V, corresponding to a low VOC loss of only 0.39 V. This represents one of the highest reported PCE values for such devices to date. Furthermore, we pioneered an all-solution two-step sequential deposition method for preparing 1.68 eV perovskite/silicon tandem solar cells on textured silicon, exhibiting a PCE of 31.12% and a VOC output of 1.940 V, which are the highest among those of tandem cells prepared by a two-step method. Our rational modification strategy, utilizing the BCF molecule for perovskite crystallization regulation, offers a highly promising pathway toward achieving superior efficiency and stability in wide-bandgap PSCs. This study underscores the critical role of precise control over perovskite crystallization kinetics in advancing the performance of wide-bandgap PSCs and perovskite/silicon tandem solar cells, thus paving the way for their broader industrial application.Author contributions
S. Luo, X. Deng, P. Troshin, B. Yang and X. Xu conceived and supervised the research. S. Luo prepared the devices, executed the characterization, analyzed the data, and wrote the paper. L. G. Gutsev and G. L. Gutsev carried out computational simulations of the interactions between BCF and the key perovskite precursor components using the Vienna ab initio simulation package. A. F. Shestakov carried out computational simulations of the minimal-energy computed molecular structures between BCF with MABr and FAI and the PbI2–BCF system. D. Liu, S. Cao, T. Zheng, Z. Bi and L. Xiong assisted with the fabrication and analysis. H. Li and N. Li helped design the experiments and analyses of in situ ultraviolet-visible absorption spectroscopy. V. V. Ozerova and N. A. Slesarenko designed and implemented experiments related to NMR analysis. N. A. Emelianov performed IR s-SNOM imaging. X. Xu, P. A. Troshin, A. F. Shestakov, S. M. Aldoshin, G. L. Gutsev, Z. Zhao and X. Deng participated in the revision of the manuscript. All authors commented on the paper.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Data availability
The data that support the findings of this study are available from the corresponding author, Xueqing Xu, upon reasonable request.Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5ee03984c.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (W2412077), Guangdong Provincial Science and Technology Plan Project (Grant No. 2023A0505010003), CAS President's International Fellowship Initiative (Grant No. 2024VCC0012), Research and development projects in key areas of Dongguan City (Grant No. 22001200300143), Guangdong-Macao Science and Technology Project (Grant No. 2023A0505020010), 2024 Guangdong-Hong Kong-Macao Joint Innovation Area Plan Project (Grant No. 2024A0505040018), and Guangdong Basic and Applied Basic Research Foundation (Grant No. 2024B1515120023). Z. Bi acknowledges the Young Talent Support Project of Guangzhou Association for Science and Technology. V. V. Ozerova, N. A. Emelianov, S. M. Aldoshin, and P. A. Troshin acknowledge the funding from Russian Science Foundation (Grant No. 25-43-00162).References
- E. Aydin, E. Ugur, B. K. Yildirim, T. G. Allen, P. Dally, A. Razzaq, F. Cao, L. Xu, B. Vishal, A. Yazmaciyan, A. A. Said, S. Zhumagali, R. Azmi, M. Babics, A. Fell, C. Xiao and S. De Wolf, Enhanced optoelectronic coupling for perovskite/silicon tandem solar cells, Nature, 2023, 623(7988), 732–738 CrossRef CAS PubMed.
- A. Al-Ashouri, E. Köhnen, B. Li, A. Magomedov, H. Hempel, P. Caprioglio, J. A. Márquez, A. B. Morales Vilches, E. Kasparavicius, J. A. Smith, N. Phung, D. Menzel, M. Grischek, L. Kegelmann, D. Skroblin, C. Gollwitzer, T. Malinauskas, M. Jošt, G. Matič, B. Rech, R. Schlatmann, M. Topič, L. Korte, A. Abate, B. Stannowski, D. Neher, M. Stolterfoht, T. Unold, V. Getautis and S. Albrecht, Monolithic perovskite/silicon tandem solar cell with >29% efficiency by enhanced hole extraction, Science, 2020, 370(6522), 1300–1309 CrossRef CAS PubMed.
- Z. Liu, C. Zhu, H. Luo, W. Kong, X. Luo, J. Wu, C. Ding, Y. Chen, Y. Wang, J. Wen, Y. Gao and H. Tan, Grain Regrowth and Bifacial Passivation for High-Efficiency Wide-Bandgap Perovskite Solar Cells, Adv. Energy Mater., 2022, 13, 2203230 CrossRef.
- T. Duong, Y. Wu, H. Shen, J. Peng, X. Fu, D. Jacobs, E. C. Wang, T. C. Kho, K. C. Fong, M. Stocks, E. Franklin, A. Blakers, N. Zin, K. McIntosh, W. Li, Y. B. Cheng, T. P. White, K. Weber and K. Catchpole, Rubidium Multication Perovskite with Optimized Bandgap for Perovskite-Silicon Tandem with over 26% Efficiency, Adv. Energy Mater., 2017, 7, 1700228 CrossRef.
- X. Luo, H. Luo, H. Li, R. Xia, X. Zheng, Z. Huang, Z. Liu, H. Gao, X. Zhang, S. Li, Z. Feng, Y. Chen and H. Tan, Efficient Perovskite/Silicon Tandem Solar Cells on Industrially Compatible Textured Silicon, Adv. Mater., 2023, 35, 2207883 CrossRef CAS PubMed.
- Q. Xu, Y. Zhao and X. Zhang, Light Management in Monolithic Perovskite/Silicon Tandem Solar Cells, Sol. RRL, 2019, 4, 1900206 CrossRef.
- J. P. Mailoa, C. D. Bailie, E. C. Johlin, E. T. Hoke, A. J. Akey, W. H. Nguyen, M. D. McGehee and T. Buonassisi, A 2-terminal perovskite/silicon multijunction solar cell enabled by a silicon tunnel junction, Appl. Phys. Lett., 2015, 106, 121105 CrossRef.
- T. Xu, Y. Chen and Q. Chen, Improving intrinsic stability for perovskite/silicon tandem solar cells. Science China Physics, Mech. Astron., 2022, 66, 217305 CrossRef.
- D. P. McMeekin, G. Sadoughi, W. Rehman, G. E. Eperon, M. Saliba, M. T. Hörantner, A. Haghighirad, N. Sakai, L. Korte, B. Rech, M. B. Johnston, L. M. Herz and H. J. Snaith, A mixed-cation lead mixed-halide perovskite absorber for tandem solar cells, Science, 2016, 351, 151–155 CrossRef CAS PubMed.
- Z. Fang, Q. Zeng, C. Zuo, L. Zhang, H. Xiao, M. Cheng, F. Hao, Q. Bao, L. Zhang, Y. Yuan, W.-Q. Wu, D. Zhao, Y. Cheng, H. Tan, Z. Xiao, S. Yang, F. Liu, Z. Jin, J. Yan and L. Ding, Perovskite-based tandem solar cells, Sci. Bull., 2021, 66(6), 621–636 CrossRef CAS PubMed.
- F. Fu, J. Li, T. C. J. Yang, H. Liang, A. Faes, Q. Jeangros, C. Ballif and Y. Hou, Monolithic Perovskite-Silicon Tandem Solar Cells: From the Lab to Fab?, Adv. Mater., 2022, 34, 2106540 CrossRef CAS.
- C. Gao, D. Du, D. Ding, F. Qiao and W. Shen, A review on monolithic perovskite/c-Si tandem solar cells: progress, challenges, and opportunities, J. Mater. Chem. A, 2022, 10, 10811–10828 RSC.
- J. Lim, N.-G. Park, S. Il Seok and M. Saliba, All-perovskite tandem solar cells: from fundamentals to technological progress, Energy Environ. Sci., 2024, 17, 4390–4425 RSC.
- J. Werner, B. Niesen and C. Ballif, Perovskite/Silicon Tandem Solar Cells: Marriage of Convenience or True Love Story? – An Overview, Adv. Mater. Interfaces, 2018, 5, 1700731 CrossRef.
- Z. Zhang, Z. Li, L. Meng, S. Y. Lien and P. Gao, Perovskite-Based Tandem Solar Cells: Get the Most Out of the Sun, Adv. Funct. Mater., 2020, 30, 2001904 CrossRef CAS.
- R. Wang, X. Liu, S. Yan, N. Meng, X. Zhao, Y. Chen, H. Li, S. M. H. Qaid, S. Yang, M. Yuan and T. He, Efficient wide-bandgap perovskite photovoltaics with homogeneous halogen-phase distribution, Nat. Commun., 2024, 15(1), 8899 CrossRef CAS PubMed.
- L. Tang, L. Zeng, J. Luo, W. Wang, Z. Xue, Z. Luo, H. Yan, J. Gong, S. Wang, J. Li and X. Xiao, All-Round Passivation Strategy Yield Flexible Perovskite/CuInGaSe2 Tandem Solar Cells with Efficiency Exceeding 26.5%, Adv. Mater., 2024, 36, 2402480 CrossRef CAS PubMed.
- G. Y. Kim, A. Senocrate, T.-Y. Yang, G. Gregori, M. Grätzel and J. Maier, Large tunable photoeffect onion conduction in halide perovskites and implications for photodecomposition, Nat. Mater., 2018, 17, 445–449 CrossRef CAS.
- Y.-C. Zhao, W.-K. Zhou, X. Zhou, K.-H. Liu, D.-P. Yu and Q. Zhao, Quantification of light-enhanced ionic transport in lead iodide perovskite thin films and its solar cell applications, Light: Sci. Appl., 2016, 6(5), e16243 CrossRef PubMed.
- H. Guan, S. Zhou, S. Fu, D. Pu, X. Chen, Y. Ge, S. Wang, C. Wang, H. Cui, J. Liang, X. Hu, W. Meng, G. Fang and W. Ke, Regulating Crystal Orientation via Ligand Anchoring Enables Efficient Wide-Bandgap Perovskite Solar Cells and Tandems, Adv. Mater., 2024, 36, 2307987 CrossRef CAS.
- H. Rao, Y. Su, G. Liu, H. Zhou, J. Yang, W. Sheng, Y. Zhong, L. Tan and Y. Chen, Monodisperse Adducts-Induced Homogeneous Nucleation Towards High-Quality Tin-Based Perovskite Film, Angew. Chem., Int. Ed., 2023, 62, e202306712 CrossRef CAS PubMed.
- R. Wang, J. Zhu, J. You, H. Huang, Y. Yang, R. Chen, J. Wang, Y. Xu, Z. Gao, J. Chen, B. Xu, B. Wang, C. Chen, D. Zhao and W.-H. Zhang, Custom-tailored solvent engineering for efficient wide-bandgap perovskite solar cells with a wide processing window and low VOC losses, Energy Environ. Sci., 2024, 17, 2662–2669 RSC.
- L. G. Gutsev, S. Nations, B. R. Ramachandran, G. L. Gutsev, S. Wang, S. Aldoshin and Y. Duan, Redox Chemistry of the Subphases of α-CsPbI2Br and β-CsPbI2Br: Theory Reveals New Potential for Photostability, Nanomaterials, 2023, 13(2), 276 CrossRef CAS PubMed.
- L. Yang, Z. Fang, Y. Jin, H. Feng, B. Deng, L. Zheng, P. Xu, J. Chen, X. Chen, Y. Zhou, C. Shi, W. Gao, J. Yang, X. Xu, C. Tian, L. Xie and Z. Wei, Suppressing Halide Segregation via Pyridine-Derivative Isomers Enables Efficient 1.68 eV Bandgap Perovskite Solar Cells, Adv. Mater., 2024, 36, 2311923 CrossRef CAS PubMed.
- S. Li, Z. Zheng, J. Ju, S. Cheng, F. Chen, Z. Xue, L. Ma and Z. Wang, A Generic Strategy to Stabilize Wide Bandgap Perovskites for Efficient Tandem Solar Cells, Adv. Mater., 2024, 36, 2307701 CrossRef CAS PubMed.
- F. Pei, Y. Chen, Q. Wang, L. Li, Y. Ma, H. Liu, Y. Duan, T. Song, H. Xie, G. Liu, N. Yang, Y. Zhang, W. Zhou, J. Kang, X. Niu, K. Li, F. Wang, M. Xiao, G. Yuan, Y. Wu, C. Zhu, X. Wang, H. Zhou, Y. Wu and Q. Chen, A binary 2D perovskite passivation for efficient and stable perovskite/silicon tandem solar cells, Nat. Commun., 2024, 15, 7024 CrossRef CAS PubMed.
- Q. Jiang, J. Tong, R. A. Scheidt, X. Wang, A. E. Louks, Y. Xian, R. Tirawat, A. F. Palmstrom, M. P. Hautzinger, S. P. Harvey, S. Johnston, L. T. Schelhas, B. W. Larson, E. L. Warren, M. C. Beard, J. J. Berry, Y. Yan and K. Zhu, Compositional texture engineering for highly stable wide-bandgap perovskite solar cells, Science, 2022, 378, 1295–1300 CrossRef CAS PubMed.
- Z. Li, A. Sun, C. Tian, R. Zhuang, Y. Zheng, X. Wu, B. Ouyang, J. Du, J. Cai, J. Chen, T. Xue, R. Li, T. Cen, Y. Zhao, K. Zhao, Q. Chen and C.-C. Chen, Sustainable Molecular Passivation via Heat-Induced Disaggregation and Redox Reactions for Inverted Perovskite Solar Cells, ACS Energy Lett., 2024, 9(11), 5471–5482 CrossRef CAS.
- J. Liu, M. De Bastiani, E. Aydin, G. T. Harrison, Y. Gao, R. R. Pradhan, M. K. Eswaran, M. Mandal, W. Yan, A. Seitkhan, M. Babics, A. S. Subbiah, E. Ugur, F. Xu, L. Xu, M. Wang, Au Rehman, A. Razzaq, J. Kang, R. Azmi, A. A. Said, F. H. Isikgor, T. G. Allen, D. Andrienko, U. Schwingenschlögl, F. Laquai and S. De Wolf, Efficient and stable perovskite-silicon tandem solar cells through contact displacement by MgFx, Science, 2022, 377(6603), 302–306 CrossRef CAS PubMed.
- M. Chen, Y. Li, Z. Zeng, M. Liu, T. Du, X. Huang, L. Bi, J. Wang, W. Jiang, Y. An, S.-W. Tsang, J. Yin, S. Wu and A. K. Y. Jen, Regulating the crystallization of mixed-halide perovskites by cation alloying for perovskite–organic tandem solar cells, Energy Environ. Sci., 2024, 17, 9580–9589 RSC.
- Y. Chen, N. Yang, G. Zheng, F. Pei, W. Zhou, Y. Zhang, L. Li, Z. Huang, G. Liu, R. Yin, H. Zhou, C. Zhu, T. Song, C. Hu, D. Zheng, Y. Bai, Y. Duan, Y. Ye, Y. Wu and Q. Chen, Nuclei engineering for even halide distribution in stable perovskite/silicon tandem solar cells, Science, 2024, 385(6708), 554–560 CrossRef CAS PubMed.
- B. Chen, P. Wang, R. Li, N. Ren, W. Han, Z. Zhu, J. Wang, S. Wang, B. Shi, J. Liu, P. Liu, Q. Huang, S. Xu, Y. Zhao and X. Zhang, A Two-Step Solution-Processed Wide-Bandgap Perovskite for Monolithic Silicon-Based Tandem Solar Cells with >27% Efficiency, ACS Energy Lett., 2022, 7(8), 2771–2780 CrossRef CAS.
- J. L. Zhou, Y. F. Mu, M. Qiao, M. R. Zhang, S. X. Yuan, M. Zhang and T. B. Lu, Unlocking One-Step Two-Electron Oxygen Reduction via Metalloid Boron-Modified Zn3In2S6 for Efficient H2O2 Photosynthesis, Angew. Chem., Int. Ed., 2025, 64, e202506963 CrossRef CAS PubMed.
- S. Luo, S. Cao, Z. Bi, Y. Zheng, H. A. Tauqeer, Y. Zhuo, V. V. Ozerova, N. A. Emelianov, N. A. Slesarenko, L. A. Frolova, L. G. Gutsev, B. R. Ramachandran, G. L. Gutsev, P. A. Troshin and X. Xu, Interface contact optimization and defect passivation via tyramine hydrochloride for efficient and stable inverted perovskite solar cells, Nano Energy, 2025, 139, 110944 CrossRef CAS.
- S. Cao, S. Luo, T. Zheng, Z. Bi, J. Mo, L. G. Gutsev, N. A. Emelianov, V. V. Ozerova, N. A. Slesarenko, G. L. Gutsev, S. M. Aldoshin, F. Sun, Y. Tian, B. R. Ramachandran, P. A. Troshin and X. Xu, Hybrid Self-Assembled Molecular Interlayers for Efficient and Stable Inverted Perovskite Solar Cells, Adv. Energy Mater., 2025, 15, 2405367 CrossRef CAS.
- Z. Wang, T. Xu, N. Li, Z. Chang, J. Shan, Y. Wang, M. Wu, F. Xiao, S. Liu and W. Xiang, Comprehensive crystallization retardation of inorganic perovskites for high performance inverted solar cells, Energy Environ. Sci., 2025, 18(9), 4130–4141 RSC.
- H. Li, C. Liu, Y. Guo, H. Wu, X. Zhang, H. Tan, Z. Zhang, G. Zhou, X. Qiu, H. Wen, T. Yin and S. Huang, Optimizing Crystallization, Defects, and Toughness with an Amphiphilic Molecule for High-Performance Rigid and Flexible Perovskite Solar Cells, Adv. Funct. Mater., 2025, e14162 CrossRef.
- S. Luo, S. Cao, T. Zheng, Z. Bi, Y. Zheng, Y. Li, B. Z. Taye, V. V. Ozerova, L. A. Frolova, N. A. Emelianov, E. D. Tarasov, Z. Liang, L. G. Gutsev, S. M. Aldoshin, B. R. Ramachandran, P. A. Troshin and X. Xu, Melamine holding PbI2 with three “arms”: an effective chelation strategy to control the lead iodide to perovskite conversion for inverted perovskite solar cells, Energy Environ. Sci., 2025, 18, 2436–2451 RSC.
- W. Wu, H. Gao, L. Jia, Y. Li, D. Zhang, H. Zhan, J. Xu, B. Li, Z. Geng, Y. Cheng, H. Tong, Y. Pan, J. Liu, Y. He, X. Xu, Z. Li, B. He, M. Zhou, L. Wang and C. Qin, Stable and uniform self-assembled organic diradical molecules for perovskite photovoltaics, Science, 2025, 389(6756), 195–199 CrossRef CAS PubMed.
- X. Wei, Y. Sun, Y. Zhang, B. Yu and H. Yu, A X-bit optimized 2D solid solution Ti3CNTx MXene as the electron transport layer toward high-performance perovskite solar cells, Nano Energy, 2025, 133, 110513 CrossRef CAS.
- L. A. Frolova, S. Y. Luchkin, Y. Lekina, L. G. Gutsev, S. A. Tsarev, I. Zhidkov, E. Z. Kurmaev, Z. X. Shen, K. J. Stevenson, S. M. Aldoshin and P. A. Troshin, Reversible Pb2+/Pb0 and I−/I3− redox chemistry drives the light-induced phase segregation in all-inorganic mixed halide perovskites, Adv. Energy Mater., 2021, 11, 2002934 CrossRef CAS.
- A. F. Akbulatov, M. I. Ustinova, L. Gutsev, S. A. Tsarev, N. N. Dremova, I. Zhidkov, S. Yu. Luchkin, B. R. Ramachandran, L. A. Frolova, E. Z. Kurmaev, K. J. Stevenson, S. M. Aldoshin and P. A. Troshin, When iodide meets bromide: Halide mixing facilitates the light-induced decomposition of perovskite absorber films, Nano Energy, 2021, 86, 106082 CrossRef CAS.
- R. Guo, Q. Xiong, A. Ulatowski, S. Li, Z. Ding, T. Xiao, S. Liang, J. E. Heger, T. Guan, X. Jiang, K. Sun, L. K. Reb, M. A. Reus, A. Chumakov, M. Schwartzkopf, M. Yuan, Y. Hou, S. V. Roth, L. M. Herz, P. Gao and P. Müller-Buschbaum, Trace Water in Lead Iodide Affecting Perovskite Crystal Nucleation Limits the Performance of Perovskite Solar Cells, Adv. Mater., 2023, 36, 2310237 CrossRef PubMed.
- L. Tian, E. Bi, I. Yavuz, C. Deger, Y. Tian, J. Zhou, S. Zhang, Q. Liu, J. Shen, L. Yao, K. Zhao, J. Xu, Z. Chen, L. Xiao, Z. Yang, P. Shi, X. Zhang, S. Wang, S. Chu, M. Haider, J. Xue and R. Wang, Divalent cation replacement strategy stabilizes wide-bandgap perovskite for Cu(In,Ga)Se2 tandem solar cells, Nat. Photonics, 2025, 19(5), 479–485 CrossRef CAS.
- S. Qu, H. Huang, J. Wang, P. Cui, Y. Li, M. Wang, L. Li, F. Yang, C. Sun, Q. Zhang, P. Zhu, Y. Wang and M. Li, Revealing and Inhibiting the Facet-related Ion Migration for Efficient and Stable Perovskite Solar Cells, Angew. Chem., Int. Ed., 2024, 64, e202415949 CrossRef PubMed.
- J. Wei, D. Zhang, R. Li, H. Xin, D. Ding, X. Xu, S. Zhou, P. Hang, D. Yang and X. Yu, Operationally Stable Perovskite/Silicon Tandem Solar Cells via Suppression of Lead Iodide-Mediated Phase Segregation in Wide-Bandgap Perovskites, Adv. Energy Mater., 2025, 2502696 CrossRef CAS.
- J. He, W. Sheng, K. Liu, Q. Cai, Y. Zhong, L. Tan and Y. Chen, Unraveling the reaction dynamics mechanism and thermodynamic pathway of lead iodide and organic cations in the two-step sequential deposition process, Energy Environ. Sci., 2025, 18, 6983–7004 RSC.
- L. Yuan, Q. Xue, F. Wang, N. Li, G. I. N. Waterhouse, C. J. Brabec, F. Gao and K. Yan, Perovskite Solar Cells and Light Emitting Diodes: Materials Chemistry, Device Physics and Relationship, Chem. Rev., 2025, 125(11), 5057–5162 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |