
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuneable redox-responsive albumin-hitchhiking drug delivery to tumours for cancer treatment†
Shiwei
Fu
a,
Ajay
Zheng
 a,
Lukun
Wang
a,
Jiuyan
Chen
a,
Bowen
Zhao
a,
Xiao
Zhang
a,
Victoria A. A.
McKenzie
a,
Zixin
Yang
a,
Roger M.
Leblanc
a,
Rajeev
Prabhakar
a and
Fuwu
Zhang
*ab
a,
Lukun
Wang
a,
Jiuyan
Chen
a,
Bowen
Zhao
a,
Xiao
Zhang
a,
Victoria A. A.
McKenzie
a,
Zixin
Yang
a,
Roger M.
Leblanc
a,
Rajeev
Prabhakar
a and
Fuwu
Zhang
*ab
aDepartment of Chemistry, University of Miami, Coral Gables, FL 33146, USA. E-mail: fxz174@miami.edu
bThe Dr John T. Macdonald Foundation Biomedical Nanotechnology Institute, University of Miami, Miami, FL 33136, USA
First published on 20th June 2024
Abstract
This paper outlines a novel drug delivery system for highly cytotoxic mertansine (DM1) by conjugating to an albumin-binding Evans blue (EB) moiety through a tuneable responsive disulfide linker, providing valuable insights for the development of effective drug delivery systems toward cancer therapy.
 Fuwu Zhang | Fuwu Zhang is currently an Assistant Professor of Chemistry at the University of Miami. He received his PhD degree in chemistry from Texas A&M University in 2015 and was then a postdoctoral fellow at the National Institutes of Health and Texas A&M University. Research in his group broadly lies in organic chemistry, polymer chemistry, chemical biology, drug delivery, and nanomedicine, with an emphasis on the development of innovative delivery systems for small molecules, nucleic acids, peptides, and proteins. Dr Zhang received the University of Miami Provosts’ Research Awards multiple times and most notably, received the prestigious 2023 National Science Foundation CAREER Award for developing stimuli-responsive polypeptides for nucleic acid delivery. |
Significant advances in classical cancer treatment modalities, including chemotherapy, radiotherapy, and surgery, have contributed to a decline in cancer mortality rates in recent decades; however, cancer remains the second most common cause of death in the USA, with an estimated 609
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 820 deaths in 2023.1–3 Small molecular drugs play a pivotal role in treating cancer, but many of these small molecular drugs, such as camptothecin (CPT), monomethyl auristatin E (MMAE), and mertansine (DM1), suffer from limited solubility, severe side effects, and unsatisfactory efficacy due to unfavourable pharmacokinetics and inefficient drug delivery to diseased sites.4–6 Drug delivery systems, including polymeric micelles, liposomes, polymer–drug conjugates, antibody–drug conjugates, and nanoparticles, have been widely used to improve pharmacokinetics and biodistribution, ultimately leading to enhanced therapeutical efficacy and reduced adverse effects.7–14
820 deaths in 2023.1–3 Small molecular drugs play a pivotal role in treating cancer, but many of these small molecular drugs, such as camptothecin (CPT), monomethyl auristatin E (MMAE), and mertansine (DM1), suffer from limited solubility, severe side effects, and unsatisfactory efficacy due to unfavourable pharmacokinetics and inefficient drug delivery to diseased sites.4–6 Drug delivery systems, including polymeric micelles, liposomes, polymer–drug conjugates, antibody–drug conjugates, and nanoparticles, have been widely used to improve pharmacokinetics and biodistribution, ultimately leading to enhanced therapeutical efficacy and reduced adverse effects.7–14
Small molecular drug amphiphiles (SMDAs) have drawn great interest in drug delivery, which enhance drug solubility, stability, and bioavailability by conjugating hydrophilic molecules to hydrophobic drugs resulting in self-assembly into nanoparticles in aqueous environments.15–19 SMDAs have demonstrated drug delivery compared to unmodified, small molecular drugs with poor water solubility, ensuring efficient and effective accumulation in targeted areas.20–23 For instance, hydrophilic oligo ethylene glycol (OEG) was conjugated to CPT to form SMDA that self-assembled into nanoparticles with high in vitro and in vivo antitumor activity.24 However, self-assembled SMDA nanoparticles are subject to disintegration at concentrations below their critical aggregation centration (CAC), and cannot take advantage of nanomedicine, including increased drug stability and high accumulation in the tumour.
As a natural carrier for various nutrients and metabolites, albumin is abundant (35–50 g L−1) and stable (t1/2:19 days) in humans.25 The tumour microenvironment is characterized by an overexpression of specific proteins, such as the gp60 receptor and SPARC (secreted protein, acidic and rich in cysteine), which exhibit a high affinity for albumin.26 Therefore, employing albumin as a carrier for anticancer drugs not only prolongs the half-life of these drugs, but also optimizes their delivery and retention within the tumour. Indeed, albumin has been utilized to deliver various therapeutic agents, for example, the FDA-approved albumin-bound paclitaxel (Abraxane).26–30 The binding of paclitaxel to albumin leads to an improvement in solubility, a decrease in toxicity, and an enhancement of the drug pharmacokinetics.31 However, many drugs are not suitable for albumin-based delivery, where their binding to albumin is either too weak or irreversible to release drugs in tumour cells. To achieve optimized binding with endogenous albumin and controlled release drugs in tumour cells, we have conjugated the drug molecule to a clinically used albumin-binding small molecule Evans blue (EB). The transformative SMDAs were converted to nanosized prodrug-albumin complexes that had long blood circulation time and remarkable anticancer activities in vivo.32–34 The therapeutic efficacy of the transformative SMDA in cancer therapy is inherently limited due to relatively moderate toxicity of the therapeutic drug CPT. Novel and potent drug molecules are expected to further enhance SMDA's potential in cancer therapy.
DM1 is renowned for its potent anticancer activity at extremely high cytotoxicities, being 20–500 times more toxic than CPT, vinblastine, and paclitaxel.35,36 Due to its superior antitumor efficacy, DM1 has shown promise in effectively treating a variety of malignancies, including breast cancer, melanoma, multiple myeloma, liver cancer, and lung cancer.35,37 Nevertheless, its clinical application has been constrained by significant limitations, including severe side effects, a narrow therapeutic window, and poor water solubility.38 Despite these challenges, DM1's high antitumor activity positions it as a potential great candidate for SMDAs. In this communication, we report a novel DM1-based SMDA where DM1 was conjugated to EB via a responsive disulfide bond leveraging an albumin-hitchhiking strategy. Linkers with different length and hydrophobicity were used for optimized drug delivery with tuneable albumin binding, drug release, and biological performances towards cancer therapy.
Redox-responsive albumin-binding DM1 analogues were synthesized in four steps using EB-NH2 and commercially available DM1 (Fig. 1A).32 Briefly, by reacting with a bifunctional linker 2, the amino group of EB-NH2 was first converted to compound 3 with an activated disulfide. The final product EB-ss-DM1 prodrugs (EB-ss-DM1-3, EB-ss-DM1-16) with varying chain lengths were obtained via thiol exchange reactions between the thiol group from DM1 and compound 3. EB-ss-DM1 and other compounds were thoroughly characterized by 1H NMR and high-resolution mass spectrometry (HRMS) (Experimental details and Fig. S8–S18, ESI†).
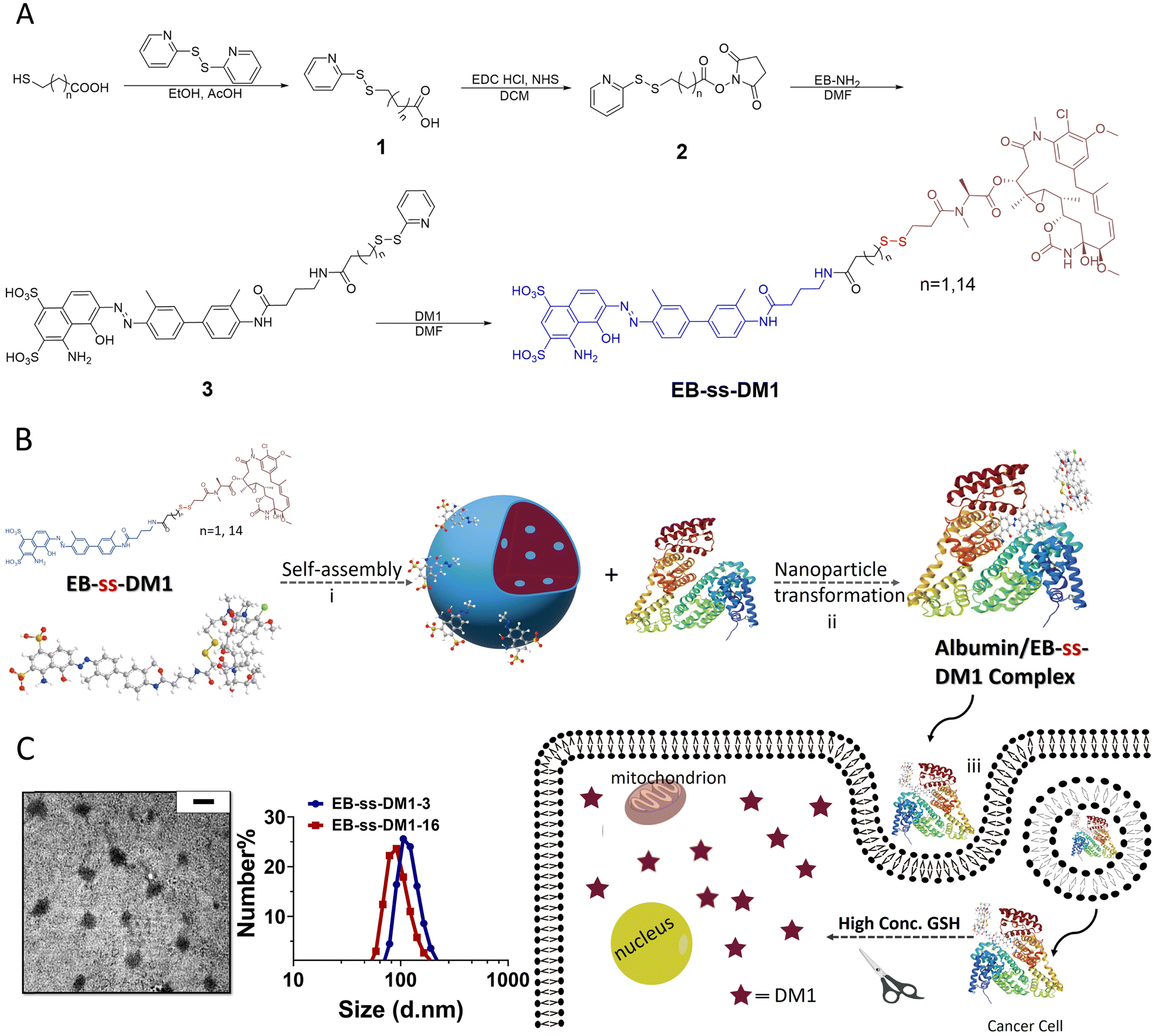 | ||
| Fig. 1 (A) Synthesis of EB-ss-DM1 (EB-ss-DM1-3, n = 1; EB-ss-DM1-16, n = 14). (B) (i) Formation of the nanoparticle from the self-assembly of amphiphilic DM1 prodrug. (ii) Nanoparticle transformation through binding with albumin. (iii) The responsive DM1 release in cancer cells. (C) Characterization of self-assembled EB-ss-DM1 nanoparticles (Left: A TEM image of EB-ss-DM1-3. The scale bar represents 200 nm; Right: Size distribution as measured by DLS). | ||
Then, we studied the self-assembly of a DM1 amphiphilic prodrug by directly resuspending into an aqueous solution, followed by 2 min sonication. It is spontaneously self-assembled into well-defined nanoparticles as a result of its inherent amphiphilic nature. Dynamic light scattering (DLS) analyses revealed that the number-averaged hydrodynamic diameter of the EB-ss-DM1-3 nanoparticles was 121 ± 29 nm (Fig. 1C), with a zeta potential of −31.9 mV (Fig. S2A, ESI†). The EB-ss-DM1-16 nanoparticles were slightly smaller with a number-averaged hydrodynamic diameter of 97 ± 26 nm (Fig. 1C), and a zeta potential of −43.4 mV (Fig. S2B, ESI†). As shown in Fig. 1C left panel, the EB-ss-DM1 nanoparticles, as observed through transmission electron microscopy (TEM), appeared to be spherical and exhibited a relatively uniform size, with each particle's diameter of roughly 105 ± 22 nm for EB-ss-DM1-3 (Fig. 1C) and 56 ± 11 nm for EB-ss-DM1-16 (Fig. S1A, ESI†). The smaller size of EB-ss-DM1-16 compared to EB-ss-DM1-3 might be attributed to the increase in the chain length of the hydrophobic part, which led to stronger hydrophobic interactions and more compact molecular packing.
One of the key features of EB-ss-DM1 prodrugs is their ability to bind with albumin, resulting in the formation of albumin/EB-ss-DM1 nanocomplexes (Fig. 1B(ii)). Owing to albumin's prolonged circulation in the bloodstream and its ability to accumulate in cancer cells via the so-called enhanced permeability and retention (EPR) effect, EB-ss-DM1 prodrugs could potentially enhance their in vivo blood circulation via binding with albumin, leading to much higher accumulation in the tumour. To confirm the prodrugs’ ability to bind with albumin and further elucidate their binding mechanism, a series of in vitro experiments were conducted (Fig. 2). First, we monitored the fluorescence intensity of EB-ss-DM1 in the presence of different concentrations of bovine serum albumin (BSA). The prodrugs themselves displayed really weak fluorescence (Fig. 2A & B and Fig. S3, ESI†). However, a significant increase in fluorescence intensity was observed when incubated with BSA with an approximate 30-fold fluorescence intensity when 6.0 equivalent of BSA was used, indicative of a strong binding interaction between EB-ss-DM1 and albumin (Fig. 2A and B and Fig. S3, ESI†). This phenomenon was consistent across both EB-ss-DM1-3 and EB-ss-DM1-16 (Fig. S3, ESI†). Next, we explored the size changes in EB-ss-DM1 prodrugs upon adding varying amounts of BSA, as determined by DLS. Both EB-ss-DM1-3 (Fig. 2C) and EB-ss-DM1-16 (Fig. S1B, ESI†) nanoparticles displayed an initial size increase at very low BSA equivalences (0.1 and 0.2 equivalence) and remarkable reductions in their intensity-averaged hydrodynamic diameters. The diameter of EB-SS-DM1-3 decreased from 121 nm to 9 nm, while that of EB-ss-DM-16, changed from 97 nm to 9 nm when higher equivalences of BSA were used. These observations demonstrated that the majority of the EB-ss-DM1 prodrugs were converted from larger self-assembled nanoparticles into much smaller albumin/EB-ss-DM1 nanocomplexes. Furthermore, we determined the binding kinetics of BSA and EB-ss-DM1 prodrugs in PBS using isothermal titration calorimetry (ITC). As shown in Fig. 2D, the dissociation constant (Kd) of EB-ss-DM1-3/BSA was determined to be 0.066 mM (Fig. 2D) and that of EB-ss-DM1-16/BSA was determined to be 0.104 mM (Fig. S4, ESI†), suggesting a similar binding affinity of our prodrugs to albumin. The interactions between the EB-ss-DM-3 and EB-ss-DM1-16 prodrugs and HSA were investigated by combining molecular docking and molecular dynamics (MD) simulations. The poses with the lowest binding energies provided by the docking procedure were equilibrated utilizing MD simulations. The structures derived from MD simulations showed that both prodrugs interacted in the same region of HSA, i.e. EB-ss-DM-3 with the E277, R160, K281, and H288 residues, while EB-ss-DM1-16 with H440, R160, K281, and H288 (Fig. 2E and Fig. S5, ESI†). The binding energies of −37 kJ mol−1 and −39 kJ mol−1 for EB-ss-DM-3 and EB-ss-DM1-16, respectively computed using the molecular mechanics Poisson–Boltzmann surface area (MMPBSA) approach39 were also quite similar. They showed that a difference in linker length doesn’t change their interaction sites and binding energies. Collectively, these experimental and computational findings underscore the remarkable binding affinity of EB-ss-DM1 prodrugs to albumin, facilitating the formation of albumin/EB-ss-DM1 nanocomplexes. Furthermore, our study reveals that variations in linker length and hydrophobicity minimally affect the prodrug's binding capacity to albumin.
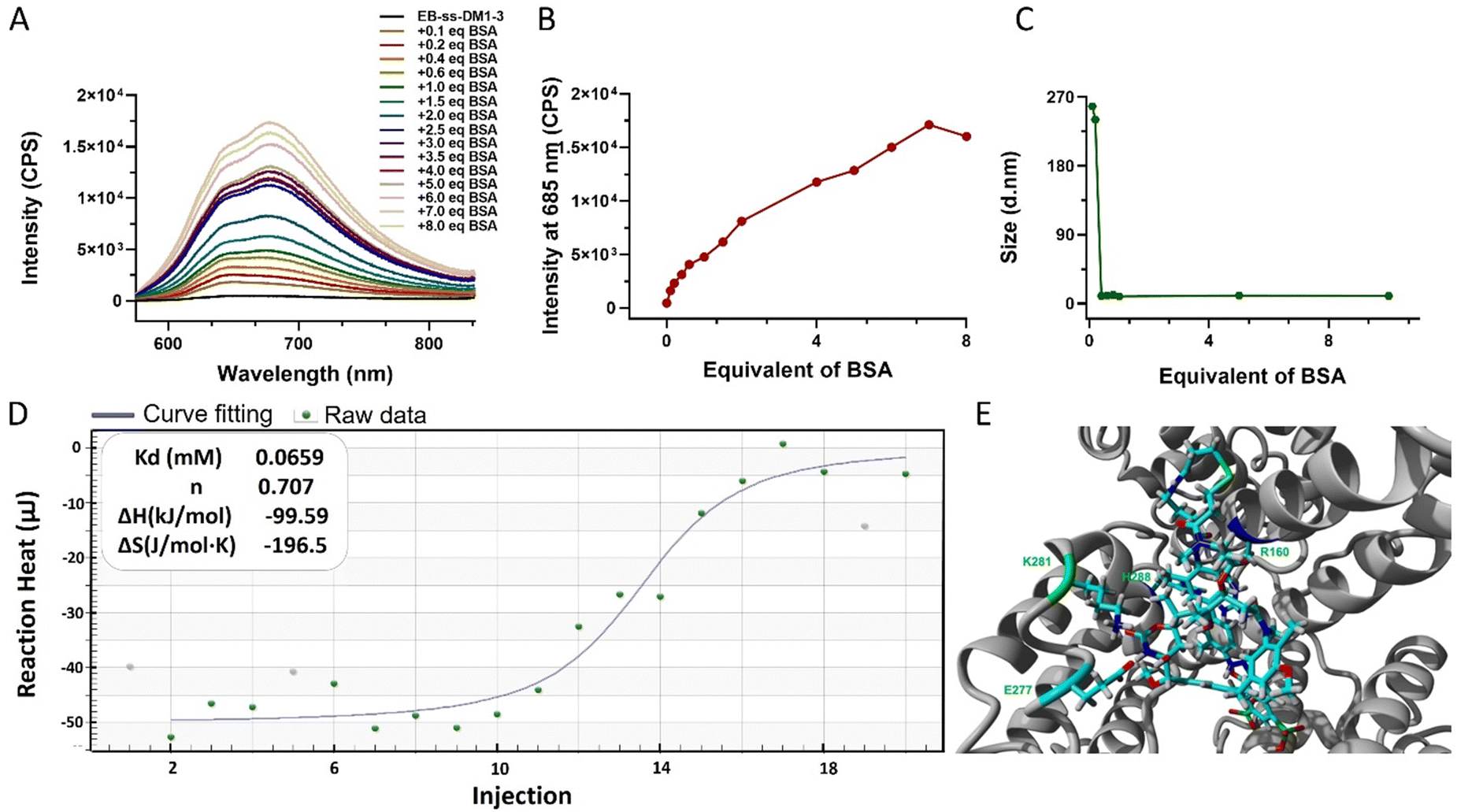 | ||
| Fig. 2 (A) The emission spectra of EB-ss-DM1-3 and (B) fluorescence intensity at 685 nm and (C) hydrodynamic diameter in the presence of different equivalents of BSA (EB-ss-DM1-3, 5 μM in PBS; BSA, 0.5–40 μM in PBS. Excitation: 560 nm). (D) ITC determination of the binding thermodynamics of EB-ss-DM1-3 and BSA binding affinity. (E) Predicted structure of the EB-ss-DM1-3/HSA nanocomplex. HSA (gray) is represented in solid ribbon. EB-ss-DM1 prodrugs and the residues in the binding site of HSA are represented in stick form. | ||
The responsive release of DM1 from EB-ss-DM1 nanoparticles at the target site is a crucial factor for their therapeutic effectiveness. To achieve this, our prodrugs incorporate a redox-responsive disulfide bond, designed to be cleaved in the presence of elevated GSH levels inside cancer cells, thereby enabling accelerated drug release at the tumour site. EB-ss-DM1 remained very stable in PBS without GSH (Fig. 3B) as monitored by high-performance liquid chromatography (HPLC), while 61% DM1 degraded in 18 h in PBS, demonstrating a much higher aqueous stability in PBS. To avoid the complications with DM1 degradation in aqueous solutions, we then used the concentration decrease of the EB-ss-DM1 prodrug over time as an indicator of DM1. In the presence of 5 mM GSH, the concentration of EB-ss-DM1-3 decreased by approximately 60% in 2 h and 90% in 6 h as a result of disulfide bond cleavage by GSH, which led to the release of DM1 (Fig. 3A). As a comparison, the EB-ss-DM1-16 concentration decreased by only 33% in 24 h in 5 mM GSH and 68% in 40 mM GSH (Fig. 3A). The responsive yet much slower release of DM1 from EB-ss-DM1-16 over EB-ss-DM1-3 was indeed as expected because the higher hydrophobicity of the long alky chain led to lower accessibility by the hydrophilic GSH. The release rate substantially influences the therapeutic efficacy of the drug. A slower release kinetics may be beneficial for sustained delivery of DM1 over a longer period, avoiding premature drug release during blood circulation and enhancing prodrug accumulation in the tumour via EPR effects.
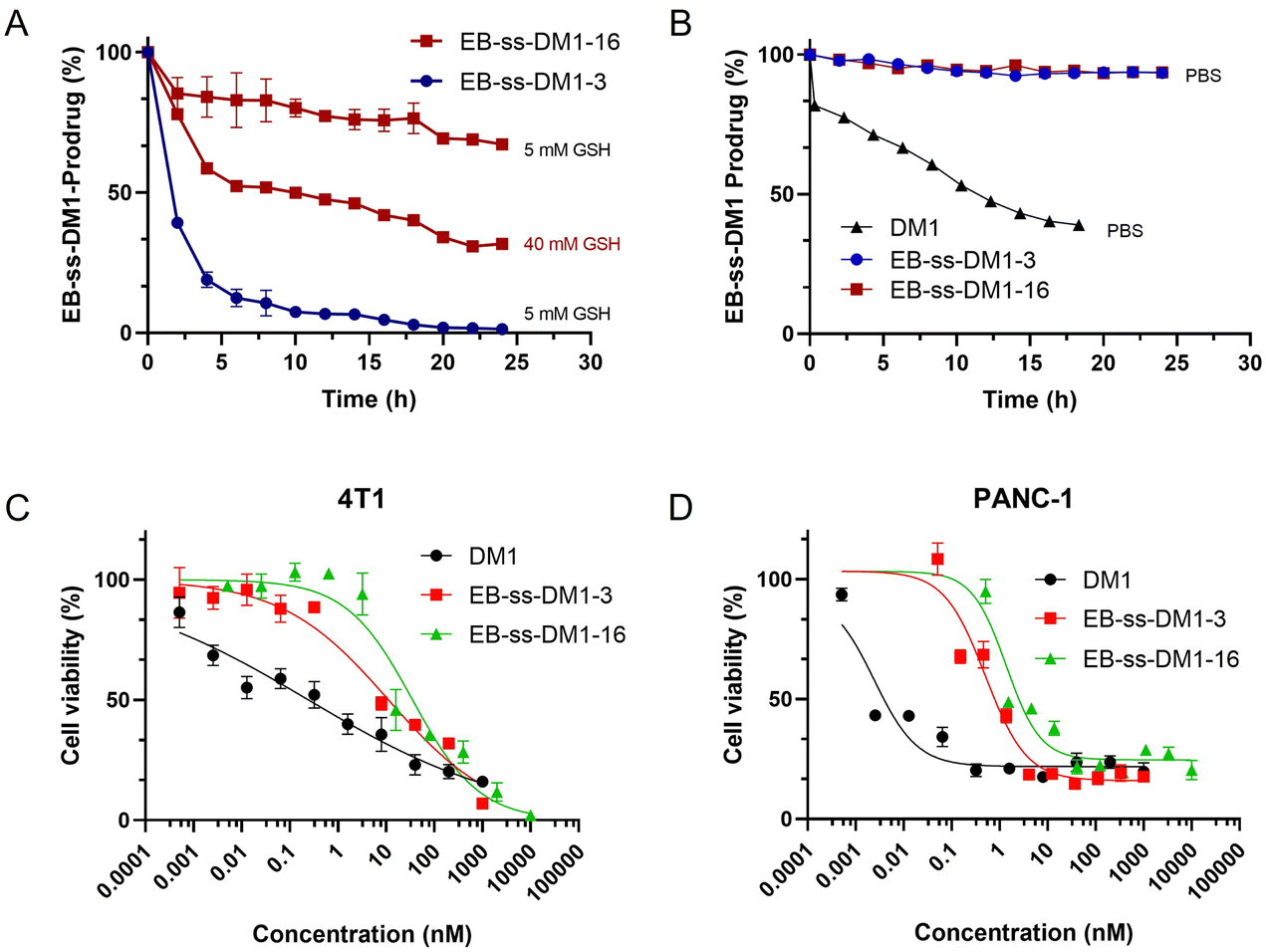 | ||
| Fig. 3 In vitro drug release (A) in the presence of 5 mM or 40 mM GSH and (B) without GSH over 24 h at 25 °C in PBS, as monitored by the decrease of EB-ss-DM1 prodrugs by HPLC, and in vitro cytotoxicities of EB-ss-DM1-3, EB-ss-DM1-16, and DM1 against (C) 4T1 and (D) PANC-1 cancer cells. | ||
Furthermore, we investigated the cellular uptake of EB-ss-DM1 prodrugs using confocal microscopy. Benefiting from the inherent fluorescence of EB, these prodrugs can be directly imaged without additional dye labelling. Cell nuclei were stained with a Hoechst dye. In 4T1 cells, both EB-ss-DM1-3 and EB-ss-DM1-16 successfully enter the cell as indicated by red fluorescence in the cytoplasm around the nuclei. EB-ss-DM1-16 with a longer hydrophobic linker exhibited slightly higher cellular uptake compared to that of EB-ss-DM1-3 with a shorter chain linker (Fig. 4B). A similar trend was observed in PANC-1 pancreatic cancer cells and hTERT-HPNE immortalized pancreatic epithelial cells (Fig. S7, ESI†). These observations demonstrated effective intracellular uptake of EB-ss-DM1 prodrugs and that the longer alkyl hydrophobic linker may lead to high cellular uptake, highlighting their potential for cancer therapy with tuneable physiochemical and biological properties.
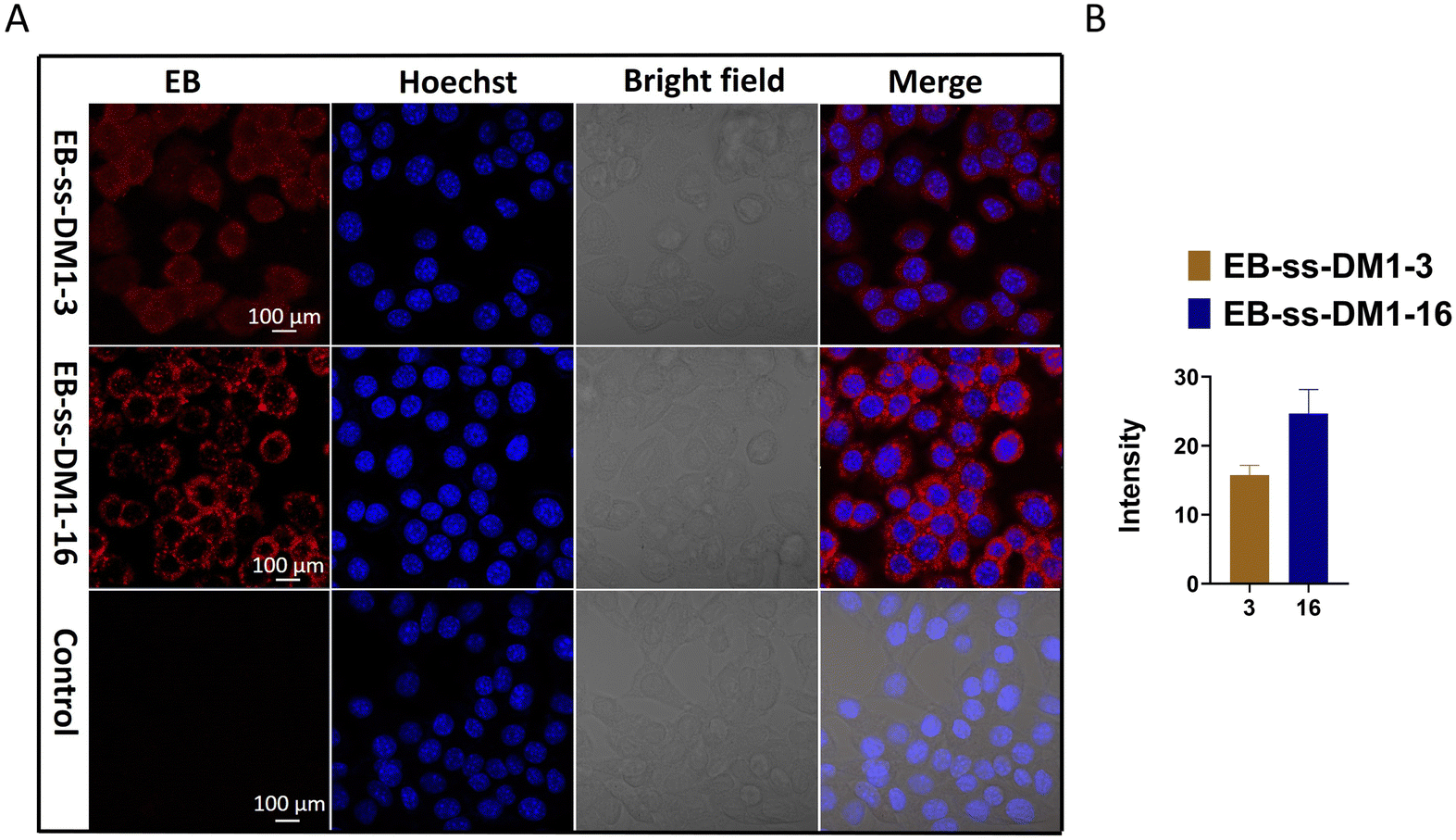 | ||
| Fig. 4 (A) Confocal microscopic images of 4T1 cells treated with 10 μM of EB-ss-DM1-3, EB-ss-DM1-16, and RPMI-1640 media. Red: EB fluorescence from EB-ss-DM1 prodrugs; Blue: Hoechst 33342 (nuclear stain). (B) The average fluorescence intensity of each cell treated with either EB-ss-DM1-3 or EB-ss-DM1-16. (The fluorescence intensity was analysed by ZEISS ZEN 3.8.). | ||
Building on the understanding of the release profiles and cellular uptake of EB-ss-DM1 prodrugs, we further investigated DM1 prodrugs’ in vitro cytotoxicity against multiple cancer cell lines. In 4T1 murine mammary cancer cells, the IC50 value of EB-ss-DM1-3 was 17.5 ± 0.5 nM, while the IC50 value of EB-ss-DM1-16 was 37.2 ± 7.4 nM. Similar results were observed in PANC-1 pancreatic cancer cells, where EB-ss-DM1-3 also displayed lower cytotoxicity with an IC50 value of 0.51 ± 0.02 nM vs. 1.3 ± 0.1 nM for EB-ss-DM1-16. The difference in cytotoxicity was more pronounced in AsPC-1 pancreatic cancer cells, respectively (Fig. S6 and Table S1, ESI†). Furthermore, prodrugs’ cytotoxicities were also evaluated in a normal pancreatic cell line hTERT-HPNE. DM1 was found to be extremely cytotoxic with an IC50 value of ca. 0.000029 nM, whereas EB-ss-DM1-3 and EB-ss-DM1-16 were much less cytotoxic to the healthy cell lines with IC50 values of ca. 0.61 nM and ca. 1.15 nM, respectively (Fig. S6 and Table S1, ESI†). The relatively much lower cytotoxicity in the healthy cell line of these prodrugs vs. DM1 suggests the great potential of our strategy in drug delivery. It is worth noting that EB-ss-DM1 prodrugs displayed much lower cytotoxicity in all cell lines, partly due to slower drug release. These results confirmed the potent cytotoxicity of the EB-ss-DM1 prodrugs compared to many chemotherapeutic agents such as CPT,33,40 cisplatin,41 and doxorubicin.42 Though higher cellular uptake was observed, the longer hydrophobic chain linker of EB-ss-DM1-16 exhibited reduced cytotoxicity compared to the shorter chain in EB-ss-DM1-3, which could be explained by the significantly slower drug release (Fig. 3). The faster DM1 release of EB-ss-DM1-3 endowed it with higher cytotoxicity with a rapid onset of action. These insights are pivotal for prodrug optimization, highlighting the importance of linker design in drug development.
In summary, we have developed an innovative approach for DM1 delivery towards cancer therapy, hijacking albumin as a drug carrier through the conjugation of an albumin-binding EB via a responsive linker. We demonstrated that these EB-based prodrugs maintain their ability to bind with albumin and enter cancer cells. The incorporation of disulfide bonds in these prodrugs allows for responsive and controlled release of DM1 within tumour cells. Importantly, our research highlights the importance of the linker design, which significantly affects prodrug's self-assembly, drug release, cellular uptake, and cytotoxicity against multiple cancer cells, thereby impacting their therapeutic potential. The straightforward synthesis and tuneable physicochemical and biological performances render our approach extremely promising for drug optimization towards specific diseases. These findings offer valuable insights for the development of effective drug delivery systems for cancer therapy.
Author contributions
F. Z. directed the project and oversaw the work. S. F., L. W., F. Z., R. M. L., and R. P. contributed to conceptualization of the projects and data analyses. S. F., A. Z., L. W., J. C., B. Z., X. Z. V. A. A. M., and Z. Y. conducted experiments and data collection. All authors contributed to writing, review and editing of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Fuwu Zhang thanks the financial support from the National Science Foundation (Grant Number DMR-2238812). The computational results are based upon work supported by a grant from the National Science Foundation (Grant Number CHE-2102563) to R.P. We acknowledge Vania Almeida and the University of Miami Transmission Electron Microscopy Core for EM sample preparation and assistance with the generation of EM images.References
- R. L. Siegel, K. D. Miller, N. S. Wagle and A. Jemal, Ca-Cancer J. Clin., 2023, 73, 17–48 CrossRef PubMed.
- B. K. Kashyap, V. V. Singh, M. K. Solanki, A. Kumar, J. Ruokolainen and K. K. Kesari, ACS Omega, 2023, 8, 14290–14320 CrossRef CAS PubMed.
- B. Zhao, X. Zhang, M. S. Bickle, S. Fu, Q. Li and F. Zhang, Nanoscale, 2024, 16, 2250–2264 RSC.
- L. Zhong, Y. Li, L. Xiong, W. Wang, M. Wu, T. Yuan, W. Yang, C. Tian, Z. Miao, T. Wang and S. Yang, Signal Transduction Targeted Ther., 2021, 6, 201 CrossRef.
- V. J. Venditto and E. E. Simanek, Mol. Pharm., 2010, 7, 307–349 CrossRef CAS.
- M. Wu, W. Huang, N. Yang and Y. Liu, Exp. Hematol. Oncol., 2022, 11, 93 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. D. P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H. S. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef PubMed.
- A. M. Vargason, A. C. Anselmo and S. Mitragotri, Nat. Biomed. Eng., 2021, 5, 951–967 CrossRef PubMed.
- K. Elumalai, S. Srinivasan and A. Shanmugam, Biomed. Technol., 2024, 5, 109–122 CrossRef CAS.
- S. F. A. Rizvi, L. Zhang, H. Zhang and Q. Fang, ACS Pharmacol. Transl. Sci., 2024, 7, 309–334 CrossRef CAS PubMed.
- Z. Fu, S. Li, S. Han, C. Shi and Y. Zhang, Signal Transduction Targeted Ther., 2022, 7, 93 CrossRef CAS PubMed.
- K. Tsuchikama, Y. Anami, S. Y. Y. Ha and C. M. Yamazaki, Nat. Rev. Clin. Oncol., 2024, 21, 203–223 CrossRef CAS PubMed.
- X. Zhang, B. Zhao, S. Fu, R. S. Seruya, J. F. Madey III, E. Bukhryakova and F. Zhang, Macromolecules, 2024, 57, 2858–2867 CrossRef CAS.
- J. Xi and H. Liu, Adv. Ther., 2020, 3, 1900107 CrossRef.
- H. Su, F. Wang, W. Ran, W. Zhang, W. Dai, H. Wang, C. F. Anderson, Z. Wang, C. Zheng, P. Zhang, Y. Li and H. Cui, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 4518–4526 CrossRef CAS PubMed.
- N. Amirmahani, N. O. Mahmoodi, M. Mohammadi Galangash and A. Ghavidast, J. Ind. Eng. Chem., 2017, 55, 21–34 CrossRef CAS.
- P. Liu, Y. Huang, C. Zhan, F. Zhang, C. Deng, Y. Jia, T. Wan, S. Wang and B. Li, Mater. Today Bio., 2023, 21, 100722 CrossRef CAS PubMed.
- L. Han, S. Liang, W. Mu, Z. Zhang, L. Wang, S. Ouyang, B. Yao, Y. Liu and N. Zhang, Asian J. Pharm. Sci., 2022, 17, 129–138 CrossRef.
- S. Dragojevic, J. S. Ryu and D. Raucher, Molecules, 2015, 20, 21750–21769 CrossRef CAS PubMed.
- P. Huang, D. Wang, Y. Su, W. Huang, Y. Zhou, D. Cui, X. Zhu and D. Yan, J. Am. Chem. Soc., 2014, 136, 11748–11756 CrossRef CAS PubMed.
- L. Zhu, Y. Guo, Q. Qian, D. Yan, Y. Li, X. Zhu and C. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17944–17950 CrossRef CAS PubMed.
- T. C. Ezike, U. S. Okpala, U. L. Onoja, C. P. Nwike, E. C. Ezeako, O. J. Okpara, C. C. Okoroafor, S. C. Eze, O. L. Kalu, E. C. Odoh, U. G. Nwadike, J. O. Ogbodo, B. U. Umeh, E. C. Ossai and B. C. Nwanguma, Heliyon, 2023, 9, e17488 CrossRef CAS PubMed.
- H. Su, W. Zhang, H. Wang, F. Wang and H. Cui, J. Am. Chem. Soc., 2019, 141, 11997–12004 CrossRef CAS PubMed.
- C. Li, X. Wang, H. Song, S. Deng, W. Li, J. Li and J. Sun, Asian J. Pharm. Sci., 2020, 15, 1–12 CrossRef PubMed.
- E. N. Hoogenboezem and C. L. Duvall, Adv. Drug Delivery Rev., 2018, 130, 73–89 CrossRef CAS PubMed.
- H. Cho, S. I. Jeon, C. H. Ahn, M. K. Shim and K. Kim, Pharmaceutics, 2022, 14, 728 CrossRef CAS PubMed.
- I. Cucinotto, L. Fiorillo, S. Gualtieri, M. Arbitrio, D. Ciliberto, N. Staropoli, A. Grimaldi, A. Luce, P. Tassone, M. Caraglia and P. Tagliaferri, J. Drug Delivery, 2013, 2013, 905091 Search PubMed.
- A. Spada, J. Emami, J. A. Tuszynski and A. Lavasanifar, Mol. Pharmaceutics, 2021, 18, 1862–1894 CrossRef CAS PubMed.
- S. Teixeira, M. A. Carvalho and E. M. S. Castanheira, Biomedicines, 2022, 10, 486 CrossRef CAS PubMed.
- J. Cortes and C. Saura, Eur. J. Cancer Suppl., 2010, 8, 1–10 CrossRef CAS.
- Z. Zhou, C. Du, Q. Zhang, G. Yu, F. Zhang and X. Chen, Angew. Chem., Int. Ed., 2021, 60, 21033–21039 CrossRef CAS PubMed.
- F. Zhang, G. Zhu, O. Jacobson, Y. Liu, K. Chen, G. Yu, Q. Ni, J. Fan, Z. Yang, F. Xu, X. Fu, Z. Wang, Y. Ma, G. Niu, X. Zhao and X. Chen, ACS Nano, 2017, 11, 8838–8848 CrossRef CAS PubMed.
- D. Zhao, H. Zhang, W. Tao, W. Wei, J. Sun and Z. He, Biomater. Sci., 2017, 5, 502–510 RSC.
- J. Huang, S. Song, M. Wang and H. Wang, Nanoscale, 2023, 15, 10110–10124 RSC.
- J. M. Baron, B. L. Boster and C. M. Barnett, J. Oncol. Pharm. Pract., 2015, 21, 132–142 CrossRef CAS PubMed.
- M. Lopus, Cancer Lett., 2011, 307, 113–118 CrossRef CAS PubMed.
- X. Yu, H. Wu, H. Hu, Z. Dong, Y. Dang, Q. Qi, Y. Wang, S. Du and Y. Lu, Drug Delivery, 2020, 27, 100–109 CrossRef CAS PubMed.
- C. Wang, D. Greene, L. Xiao, R. Qi and R. Luo, Front. Mol. Biosci., 2017, 4, 87 CrossRef PubMed.
- F. Zhang, Q. Ni, O. Jacobson, S. Cheng, A. Liao, Z. Wang, Z. He, G. Yu, J. Song, Y. Ma, G. Niu, L. Zhang, G. Zhu and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 7066–7070 CrossRef CAS PubMed.
- F. Zhang, M. Elsabahy, S. Zhang, L. Y. Lin, J. Zou and K. L. Wooley, Nanoscale, 2013, 5, 3220–3225 RSC.
- Q. Ni, F. Zhang, Y. Zhang, G. Zhu, Z. Wang, Z. Teng, C. Wang, B. C. Yung, G. Niu, G. Lu, L. Zhang and X. Chen, Adv. Mater., 2018, 30, 1870064 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00751d |
| This journal is © The Royal Society of Chemistry 2024 |