
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Setting up the HyDRA blind challenge for the microhydration of organic molecules†
Taija L.
Fischer
 a,
Margarethe
Bödecker
a,
Anne
Zehnacker-Rentien
b,
Ricardo A.
Mata
a and
Martin A.
Suhm
*a
a,
Margarethe
Bödecker
a,
Anne
Zehnacker-Rentien
b,
Ricardo A.
Mata
a and
Martin A.
Suhm
*a
aInstitut für Physikalische Chemie, Universität Göttingen, Tammannstr. 6, 37077 Göttingen, Germany. E-mail: msuhm@gwdg.de
bInstitut des Sciences Moléculaires d'Orsay, Université Paris-Saclay, CNRS, 91405, Orsay, France
First published on 29th April 2022
Abstract
The procedure leading to the first HyDRA blind challenge for the prediction of water donor stretching vibrations in monohydrates of organic molecules is described. A training set of 10 monohydrates with experimentally known and published water donor vibrations is presented and a test set of 10 monohydrates with unknown or unpublished water donor vibrational wavenumbers is described together with relevant background literature. The rules for data submissions from computational chemistry groups are outlined and the planned publication procedure after the end of the blind challenge is discussed.
1 Introduction
The microhydration of organic matter1–3 deserves a systematic, unbiased comparison between theory and experiment.4 The primary and most fundamental quantities to be benchmarked are probably structure and energy of cold, neutral 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hydrate complexes prepared in supersonic jet expansions.5 Any isomerism in the docking of individual water molecules to multifunctional organic molecules6 is of particular interest in this context, because theoretical input is most valuable and reliable for trends and comparisons.7 This is where systematic model errors often tend to cancel,8 paving the way for helpful approximations. The properties of monoisomeric systems are valuable for a first calibration of absolute quantities, which are typically more challenging because they require highly accurate treatments of electronic structure and nuclear dynamics effects. It is rewarding that there are numerous rotational spectroscopy studies of monohydrates with more or less direct structural and sometimes relative energy information available in the literature.5,9–11 These provide a lot of detail on the shallow potential energy landscapes of solvation phenomena.
:1 hydrate complexes prepared in supersonic jet expansions.5 Any isomerism in the docking of individual water molecules to multifunctional organic molecules6 is of particular interest in this context, because theoretical input is most valuable and reliable for trends and comparisons.7 This is where systematic model errors often tend to cancel,8 paving the way for helpful approximations. The properties of monoisomeric systems are valuable for a first calibration of absolute quantities, which are typically more challenging because they require highly accurate treatments of electronic structure and nuclear dynamics effects. It is rewarding that there are numerous rotational spectroscopy studies of monohydrates with more or less direct structural and sometimes relative energy information available in the literature.5,9–11 These provide a lot of detail on the shallow potential energy landscapes of solvation phenomena.
A currently underexplored proxy to the hydrogen bond structure and energy of microhydrates is the wavenumber of the hydrogen-bonded OH stretching vibration ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) OHb of any docking water serving as a hydrogen bond donor.12 As a single number for a given monohydrate complex which can be derived from vibrational spectroscopy in a straightforward way, it offers a set of advantages over more demanding structural studies. For multifunctional substrates, linear IR and Raman spectroscopy can provide an immediate survey over competing hydrogen bond docking sites due to the simplicity and intensity of the vibrational fingerprint of the OH group.13,14 To compensate for the low spectral resolution, double resonance techniques allow for rigorous size and conformational distinction,15 relaxation experiments can identify the most stable conformation,16 and 18O labelling of water can unambiguously discriminate from other hydride stretching modes in the vicinity.17 The spectral position of the OH stretching vibration is very sensitive to the strength of the hydrogen bond, spanning about two orders of magnitude in relation to the typical spectral width of an isolated vibrational transition.
OHb of any docking water serving as a hydrogen bond donor.12 As a single number for a given monohydrate complex which can be derived from vibrational spectroscopy in a straightforward way, it offers a set of advantages over more demanding structural studies. For multifunctional substrates, linear IR and Raman spectroscopy can provide an immediate survey over competing hydrogen bond docking sites due to the simplicity and intensity of the vibrational fingerprint of the OH group.13,14 To compensate for the low spectral resolution, double resonance techniques allow for rigorous size and conformational distinction,15 relaxation experiments can identify the most stable conformation,16 and 18O labelling of water can unambiguously discriminate from other hydride stretching modes in the vicinity.17 The spectral position of the OH stretching vibration is very sensitive to the strength of the hydrogen bond, spanning about two orders of magnitude in relation to the typical spectral width of an isolated vibrational transition.
A theoretical model which successfully predicts only a single observable quantity such as OHb is likely to initially get the right answer for the wrong reason, as multidimensional anharmonicity is not easy to model rigorously. This is particularly true if the experimental answer is known beforehand, due to a multitude of available and conceivable model variants and parameters. Therefore, large training sets, blind test components and iterative refinements are essential ingredients on the path towards systematic success for the modeling of OH stretching wavenumbers of monohydrates. Even a largely empirical model with a high success rate for a selected observable can be useful for the experimental gas phase cluster community. This is still close to the current status, where each observable and sometimes even each research group has its favourite recipes (functionals, basis sets, scaling factors) to assist experiment. There may be more or less systematic correlations between the observed wavenumber shifts and experimental or computed proton affinities and gas phase basicities18–20 or calculated hydrogen bond lengths,21 to name just a few possible links to be exploited. Ultimately, one must hope for a number of systematically successful models which connect theory and experiment on multiple observables, not only the vibrational wavenumber. Meaningful approximations such as elaborate scaling techniques,22,23 local mode models,24,25 parameterised harmonic DFT predictions,26ab initio molecular dynamics,27 polarisable force fields28 or hierarchical models building on high level treatments of the smallest systems or subgroups29 may be identified or we may even witness the victory of machine-learning approaches30 towards such challenges.
While blind challenges have become popular in different chemical subcommunities such as protein31 and crystal structure prediction32 or physicochemical data for drug molecules in solution,33–36 we are not aware of a blind challenge explicitly addressing vibrational spectroscopy data as a rigorous connecting point between theory and experiment. This may also be due to the fact that this connection relies on challenging electronic structure and vibrational anharmonicity issues at the same time. However, this is also subtly true for observables like molecular structure37 and energy differences,38 and therefore it appears timely to investigate a bolder case.
The HyDRA challenge is meant to kick off such a systematic approach to microhydration, with numerous conceivable follow-up options. HyDRA stands for Hydrate Donor Redshift Anticipation, where redshift refers to the wavenumber downshift of the donor OH vibration relative to the symmetric stretching fundamental of the water molecule39,40 at 3657 cm−1. The switch between absolute values and relative trends is thus trivial in this case, because it only involves adding or subtracting this experimental monomer value. In the present work, we use the sign convention that a positive downshift describes a lower wavenumber in the complex than in monomeric water. Here, we outline the construction of an initial training and test set for neutral organic monohydrates which was recently offered to theory groups,41 connected with the invitation to participate in a blind challenge.
2 Molecular systems
2.1 Experimental training set
The first goal in this challenge was to identify a number of vibrationally well-characterised, diverse monohydrate complexes of small organic molecules in the existing experimental literature, for which the lowest energy structure involves water as the hydrogen bond donor and thus generates a characteristic donor OH stretching vibrational fundamental νOHb. For this purpose, a call among vibrational spectroscopists was initiated early in 2021 and the suggestions were collected on a website,41 summarising the observed wavenumberOHb, the organic species (with CAS number), the DOI of the relevant publication and the contact email of the submitter.
Further mandatory entries of every submission included the proposing researcher and institution (not necessarily from the group publishing the experimental data) as well as the spectroscopic detection, wavenumber calibration, and cooling techniques used. Concerning the observed wavenumber, the submitter had to estimate its accuracy, to predict the relevant docking site for the observed water molecule and to assess whether the monohydrate was the global minimum arrangement of the two complex partners.
Optional entries for further assessment and use of the submissions beyond the initial training included supporting researchers like coworkers and endorsing colleagues as well as spectroscopic data on the free (or otherwise engaged) OH stretching vibration of the hydrate water (OHf), the wavenumber and relative intensity of a potential resonance partner to νOHb (possibly stealing intensity and shifting its band centre), and any isotope substitution information or information on observed docking isomers.
Because it is essential for the challenge that the training set data are well secured, it was possible to list additional assignment measures concerning the organic molecule conformation, the docking site, and the size assignment to a monohydrate, as well as to comment on any other relevant issue.
Based on more than a dozen submissions from half a dozen countries over half a year, a small committee consisting of a PhD student, an undergraduate research student and two experimental spectroscopy group leaders made a selection of 10 systems to be recommended to the theory community for the training of their models. Among others, methanol14 was not included into the core training set because of its very large amplitude internal motion and a capped phenylalanine42 was not included because the global minimum hydrate structure presumably does not involve water as a hydrogen bond donor, although a higher-energy conformer does. Several ketones (cycloheptanone, cyclohexanone, 4,4-dimethylcyclohexanone, (−)-fenchone, 2-fluoroacetophenone, pinacolone, 3,3,5,5-tetramethylcyclohexanone)43,44 were not included to keep the balance between represented functional groups. The growing set of non-selected submissions is still made available, with the goal of a comprehensive curated database on OH stretching wavenumbers of hydrate complexes in mind. Numerous interesting monohydrates are still missing in the database, such as systems where the water induces a strong restructuring of the solute42 or floppy ring systems.45
The 10 systems selected for the HyDRA training set are shown in Fig. 1 together with the three-letter acronym used in this work and with their CAS registry number. Each of them fulfills a number of desirable properties for this initial training process (Table 1). All are believed to have water acting as a hydrogen bond donor in the global minimum structure, all contain less than 100 electrons to facilitate accurate quantum-chemical modeling. All except APH (where IR spectroscopy was required to detect the second isomer43,46) have only one experimentally observed monohydrate conformation in the jet-cooled vibrational spectrum. For all except IMZ, the free OH stretching wavenumber of the solvating water is also known experimentally. All except for the methyl ketones ACE and APH with their low barrier methyl torsions are fairly stiff, with no monomer fundamental wavenumber significantly below 30 cm−1. For most monohydrates of the training set, there is more than one spectroscopic study in the literature, and in a few cases (ACE, IMZ) there are also isotope substitution experiments. In particular, many monohydrates (APH, ANL, CBU, DBF, IMZ) have been characterised structurally by rotational spectroscopy.46–50 In two cases (ACE, APH), there is a well-established vibrational b2lib resonance43 (a resonance of the water OH stretch with the bending overtone (b2) and a hydrogen bond librational (lib) mode) which makes these systems particularly interesting for advanced anharmonic models, but models not including such a resonance can also be applied, because we provide the deperturbed OH stretching wavenumber as well. The rationale behind offering this choice shall be explained briefly for the case of acetone, where it is particularly pronounced. Experimentally, one finds a dominant transition at 3538 cm−1 and a secondary transition with about half the intensity at 3516 cm−1. The latter is believed to be a three-quantum state involving the water bending overtone and a librational motion of the water molecule against the acetone. It would normally be too weak to be observed, but its vicinity to the OHb stretching state leads to intensity borrowing from the OHb state. A perfect vibrational model would thus predict both states at 3538 and 3516 cm−1 and it would predict the 3538 cm−1 signal to carry more intensity, because it has more OHb character. However, such a vibrational model would have to include higher order anharmonic couplings which shift around the levels and redistribute their IR transition strength. More approximate theoretical models and in particular (scaled) harmonic models are unable to describe this resonance and therefore they are better compared to the hypothetical OHb stretching band centre which would be observed if this resonance were absent (e.g. due to a softening of the water bending vibration). In a two-state analysis, this hypothetical or deperturbed band centre can be estimated from the experimental band positions and intensities.43 It is 3531 cm−1 and corresponds to the intensity centre of the experimental doublet. To properly train a harmonic or simplified anharmonic model, one should therefore try to match 3531 cm−1 instead of 3538 or 3516 cm−1 for ACE, and 3530 cm−1 instead of 3536 or 3514 cm−1 for APH. In two other cases (ANL, DBN), there may be an analogous resonance, but this remains experimentally open and therefore the deperturbed value for OHb spans the experimentally observed main transition. To analyse such resonances, it is essential to avoid embedding of the monohydrates in matrices, because matrices may also induce site splittings which are difficult to distinguish from a resonance.51
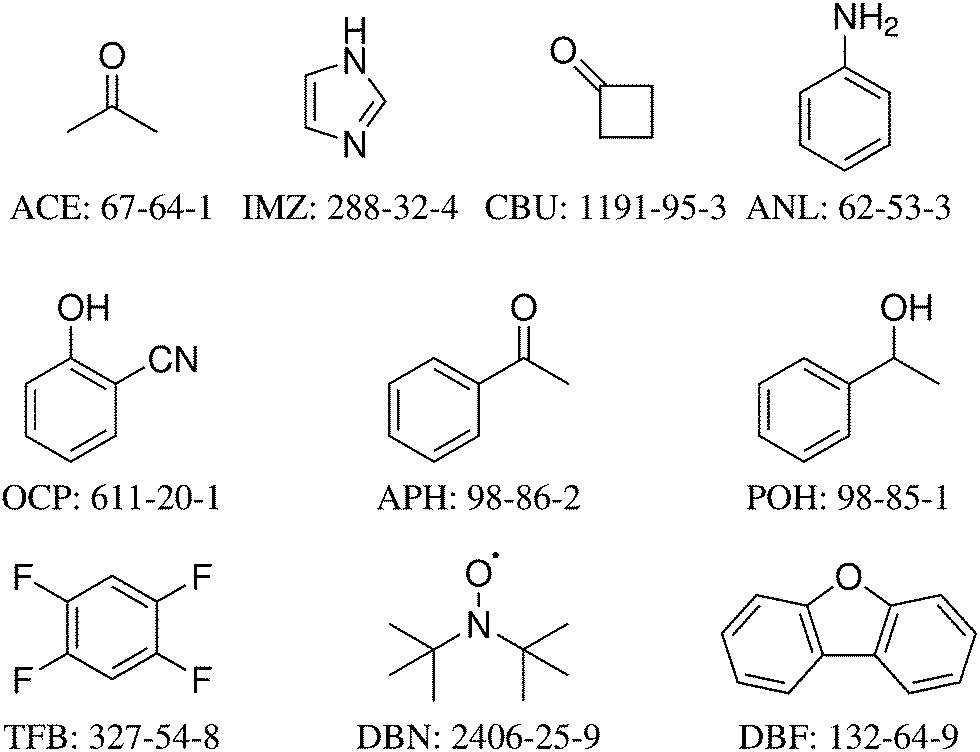 | ||
| Fig. 1 The structural formulas, abbreviations, and CAS registry numbers of the 10 systems selected for the HyDRA training set, sorted according to their electron number. | ||
| ACE | APH | ANL | CBU | DBF | DBN | IMZ | OCP | POH | TFB | |
|---|---|---|---|---|---|---|---|---|---|---|
| Unique observed 1:1 conformer? |
+ | − | + | + | + | + | + | + | + | + |
| Global minimum? | + | + | + | + | + | + | + | + | + | + |
| Isotope data? | + | − | − | − | − | − | + | − | − | − |
| Further experimental references? | + | + | + | + | + | + | + | − | + | − |
| OHf known? | + | + | + | + | + | + | − | + | + | + |
| Potential b2lib-resonance? | + | + | + | − | − | + | − | − | − | − |
| Less than 100 e−? | + | + | + | + | + | + | + | + | + | + |
| Lowest monomer-vibration >30 cm−1? | − | − | + | + | + | + | + | + | + | + |
In terms of experimental techniques, 5 monohydrates were characterised by UV/IR double resonance techniques (in different laboratories in France, Germany, Japan and Spain) and 6 by FTIR spectroscopy (in Germany), all in supersonic jet expansions. 6 compounds are aromatic, 1 is a radical, 3 are ketones, 2 are alcohols/phenols. The hydrogen bond acceptor atoms include O, F, N and π-systems.
The target benchmark observables are summarised in Table 2. Either the absolute OHb-wavenumbers exp(OHb) of the hydrogen-bonding water or equivalently their downshifts ΔOH from the isolated water symmetric stretching fundamental can be targeted. For models including full anharmonicity and thus the postulated Darling–Dennison-like fourth-rank 1–3 resonance for four systems, the most intense OHb transition can be addressed directly. For models which do not include such resonances, it may be better to address the values in square brackets, which attempt to correct for this anharmonic perturbation with a coupling matrix element on the order of 10 cm−1 and involve a larger error bar due to the correction uncertainty. For imidazole it is worth mentioning that most techniques detect both the NH- and OH-bonded isomers,50,57 whereas theory and a slit jet expansion study17 indicate a clear energetical preference for the OH-bonded isomer which is of interest in the context of this challenge.
exp(OHb) (with estimated uncertainty in parentheses) of the acceptor molecules in the monohydrates selected for the training set. In square brackets, estimated deperturbed OHb-wavenumbers after removal of a three-quantum resonance (see text) are provided. These wavenumbers in brackets should be targeted by vibrational models which do not include such an anharmonic resonance. Where the resonance remains hypothetical, the deperturbed wavenumber uncertainty (in parentheses) spans the experimentally observed main transition wavenumber. Also provided are downshifts ΔOH/cm−1 (positive, if the complex has a lower wavenumber) from the water monomer fundamental at 3657 cm−1
| Acceptor molecule | Abbreviation | CAS RN |
exp (OHb)/cm−1 |
Δ OH/cm−1 |
|---|---|---|---|---|
| Acetone43 | ACE | 67-64-1 | 3538(1) [3531(2)] | 119(1) [126(2)] |
| Acetophenone43 | APH | 98-86-2 | 3536(1) [3530(2)] | 121(1) [127(2)] |
| Aniline52 | ANL | 62-53-3 | 3524(1) [3526(3)] | 133(1) [131(3)] |
| Cyclobutanone43 | CBU | 1191-95-3 | 3548(1) | 109(1) |
| Dibenzofuran49 | DBF | 132-64-9 | 3623(1) | 34(1) |
| Di-tert-butyl nitroxide53 | DBN | 2406-25-9 | 3484(2) [3487(4)] | 173(2) [170(4)] |
| Imidazole17 | IMZ | 288-32-4 | 3458(2) | 199(2) |
| o-Cyanophenol54 | OCP | 611-20-1 | 3595(2) | 62(2) |
| 1-Phenylethanol55 | POH | 98-85-1 | 3620(2) | 37(2) |
| 1,2,4,5-Tetrafluorobenzene56 | TFB | 327-54-8 | 3647(1) | 10(1) |
2.2 Experimental test set
For the experimental test set of vibrationally uncharacterised monohydrates, a multifaceted approach was chosen. Because their experimental data had to be secured and kept secret until the end of the blind challenge, some of the considered systems were already experimentally characterised in the research groups forming the selection committee, but not yet published. Others were at least pre-explored to increase the likelihood of a successful assignment during the runtime of the blind challenge. Furthermore, an informal call to microwave spectroscopy groups was made to collect suggestions for systems which had already been characterised structurally, to minimise the risk that a subsequent vibrational characterisation meets unexpected difficulties. This approach led to a preselection of 15 monohydrate proposals, for which there is no gas phase vibrational spectroscopy record in the literature. From those, the final selection of 10 official test systems was made one month after the publication of the training set, by the same selection committee. These 10 target systems for the blind challenge are summarised in Fig. 2 (ordered by number of electrons) and Table 3 (in alphabetical sequence). Note that if the model to be submitted by a participant involves a three-quantum b2lib resonance (vide supra), a short discussion of the consequences for the deperturbed water OHb wavenumber is recommended. | ||
| Fig. 2 The structural formulas, abbreviations, and CAS registry numbers of the 10 systems selected for the HyDRA test set. | ||
exp(OHb) (or its downshift ΔOH/cm−1 from the water monomer fundamental at 3657 cm−1) should be predicted. If a three-quantum resonance is predicted (see text), estimated deperturbed water OHb wavenumbers after removal of this resonance43 may also be provided. For n(OH) > 2 (hydroxy compounds), we recommend that the theory groups report all OH stretching fundamentals, because there may be mode mixing between the hydrogen-bonded alcoholic and water OH bonds. Alternatively, we recommend to report the mode which shows the strongest downshift when H216O is replaced by H218O, because it carries the strongest water stretching character
| Acceptor molecule | Abbreviation | CAS RN | n(OH) |
|---|---|---|---|
| Cyclooctanone58,59 | CON | 502-49-8 | 2 |
| 1,3-Dimethyl-2-imidazolidinone60,61 | DMI | 80-73-9 | 2 |
| Formaldehyde62,63 | FAH | 50-00-0 | 2 |
| Methyl lactate64,65 | MLA | 547-64-8 | 3 |
| 1-Phenylcyclohexane-cis-1,2-diol66 | PCD | 125132-75-4 | 4 |
| Pyridine67,68 | PYR | 110-86-1 | 2 |
| Tetrahydrofuran69,70 | THF | 109-99-9 | 2 |
| Tetrahydrothiophene71 | THT | 110-01-0 | 2 |
| 2,2,2-Trifluoroacetophenone72 | TPH | 434-45-7 | 2 |
| 2,2,2-Trifluoroethan-1-ol73,74 | TFE | 75-89-8 | 3 |
Each target system, ordered by increasing number of electrons and thus computational complexity, will be briefly discussed in the following to introduce the most relevant literature sources for structural and other information.
For FAH as the simplest molecule in the test set, it is long established by theory75–77 and experiment63 that water coordinates asymmetrically. The OH stretching vibration which is of interest in this challenge has been determined in matrix isolation,62,78 but to the best of our knowledge not in the gas phase without environmental influence. In cryogenic matrices, split signals were found (3580, 3585 cm−1 in neon and argon, 3573, 3578 cm−1 in a nitrogen matrix). These can give a first orientation, but the vacuum-isolated transition may be located above, below or in this range and the splitting may be lifted if it is caused by matrix interaction. Whether the complex is planar or quasiplanar is difficult to determine with the available experimental data63, but we expected to provide a single experimental vacuum-isolated water OHb stretching wavenumber to be predicted by the different models.
Because of its relevance for clathrate formation, the OH stretching spectrum of the THF monohydrate has been obtained in CCl4 solution79 at 268 K. A broad (about 80 cm−1 FWHM) hydrogen-bonded OH stretching fundamental was located near 3450 cm−1 at high dilution. Due to thermal and solvent effects, this absolute value is not suitable for the direct comparison to environment-free ground state calculations. In a theoretical study from 2005, it was concluded that the complex has never been observed in spectroscopic experiments.70 With respect to gas phase investigations, we found no evidence to revise this statement. In particular, there appears to be no rotational spectroscopy characterisation of the monohydrate, but one can expect one or two isomers, due to the complex pseudorotational dynamics of the monomer.69,80
The PYR monohydrate is a model system for photoinduced homolytic water splitting.81 Its structure is quite flexible, as evidenced by rotational spectroscopy.68 It includes a tunneling motion with an inverted H/D isotope effect, but this is not directly relevant for the vibrational transition at low resolution. There is a room temperature gas phase value for the hydrogen-bonded OH stretching mode in PYR monohydrate,82 at 3480 cm−1. However, for such a soft and flexible hydrogen bond, it is likely that the cold transition obtainable in a jet expansion occurs at significantly lower wavenumber. While ultracold helium droplets should exhibit much smaller, but still significant17,83 environmental shifts than a room temperature gas phase measurement, a corresponding recent study67 does not address the relevant spectral range and isotope composition.
In contrast to the THF situation, the monohydrate of THT has been structurally characterised in much detail,71 also with respect to the water orientation relative to the ring. We are not aware of a complementary vibrational gas phase study and thus considered this to be a valuable diversification of the test set, also in preparation for more challenging thio compounds.84
In TFE, as in many fluorinated alcohols,73 the first solvating water acts primarily as a hydrogen bond acceptor, but the symmetric stretching mode is only subtly shifted from the water monomer value by this interaction and by an additional donor contact to the CF3 group. This provides valuable benchmark information for small complexation shifts, but the IR activity of such weakly perturbed symmetric OH stretching motions in water molecules is low. Therefore, Raman spectroscopy14 was expected to provide key vibrational information in this case. Structurally, the preference of the monohydrate for an insertion complex is well established74 and the related, structurally more diverse case of hexafluoroisopropanol is also well studied.85,86 By including the simplest model of a peptide co-solvent87 monohydrate into the vibrational target systems, the characterisation of their unusual biochemical properties can be supported.88
For MLA with its weak internal hydrogen bond between the hydroxy group and (preferentially) the carbonyl group, a solvent water has different options to coordinate. It can insert into the internal hydrogen bond, or add to either end, or even to the ester oxygen. Due to the activation barrier for the first process, jet experiments have to make sure that insertion is not kinetically hindered.89 This is important, because theory predicts insertion to be most stable. In this context, it is essential that there is experimental microwave evidence for the inserted structure,64 in which the solute also directs the dangling water hydrogen to one side. The competition from lactate conformations with the carbonyl group pointing away from the hydroxy group is intense in aqueous solution,65 but not yet in the monohydrate. In contrast to jet studies of the methanol complex89 and matrix studies of the ammonia complex,90 we are not aware of matrix isolation or jet studies of the OH stretching vibration of the monohydrate.
For the urea derivative DMI, a solution study60 has revealed broad absorption maxima of the monohydrate in 1,2-dichloroethane (3431 cm−1) and CCl4 (3460 cm−1) solution, downshifted by 162 and 155 cm−1, respectively, from the monomer symmetric OH stretch of water in the same solution. We are not aware of structural data for the monohydrate in the gas phase, but there is evidence for involvement of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group in the hydrogen bond60 and the DMI monomer has been characterised by rotational spectroscopy.61
O group in the hydrogen bond60 and the DMI monomer has been characterised by rotational spectroscopy.61
Monomeric CON has been structurally characterised by microwave jet spectroscopy and shown to exist in a dominant boat-chair conformation with non-equivalent lone pairs of the oxygen.58 The energy difference to the next conformation is so large that monohydration is unlikely to switch the energy sequence. Therefore up to two isomers of the monohydrate are expected59 and the more stable one is of interest in the present challenge. As the energy balance between the two docking isomers could be subtle, we recommend to calculate both and to provide at least the donor OH stretching wavenumber of the more stable one. Note that there is isotopic exchange evidence for transient formation of the geminal diol from the ketone monohydrate,59 but such a competing structural isomer should be easily distinguishable from the monohydrate complex also in the vibrational spectrum and is not the target for the present investigation. We are not aware of any vibrational study of the monohydrate of CON.
TPH is an unsymmetric ketone, in which the water molecule is strongly directed to one of the CO lone electron pairs. Its monohydrate was recently accurately characterised by rotational spectroscopy and theory72 but we are not aware of a published gas phase vibrational study. Comparison to the well-studied non-fluorinated parent compound APH from the training set will reveal the influence of fluorination through space and through several bonds.
PCD as the most complex member of the test set is of stereochemical and hydrogen bond topological interest. Note that the two OH groups are cis-configured, whereas the absolute chirality is not relevant for the monohydrate. The diol forms two weak, but cooperative hydrogen bonds (OH–OH and OH–π) which give rise to separate OH stretching transitions in CCl4 solution.66 The water in the monohydrate has several options to attach or insert into this hydrogen bond chain and the most stable conformation is sought. In this particular case, we recommend an extensive search among the possible conformations to identify the global minimum conformation. It is going to be characterised by IR/UV spectroscopy and isotope labelling. We are not aware of previous gas phase work on this system.
Fig. 3 correlates the number of vibrational degrees of freedom 3Nn − 6 for Nn nuclei with the number of electrons ne of the solutes for the training and test sets. The sets span an order of magnitude in each quantity and deviate significantly from a linear relationship, with some systems being more challenging for the nuclear dynamics treatment and others for the electronic structure treatment. Two of the test set members (FAH, PCD) represent an extrapolation relative to the training set, whereas the others tend to fill gaps in terms of electron and nuclear degrees of freedom.
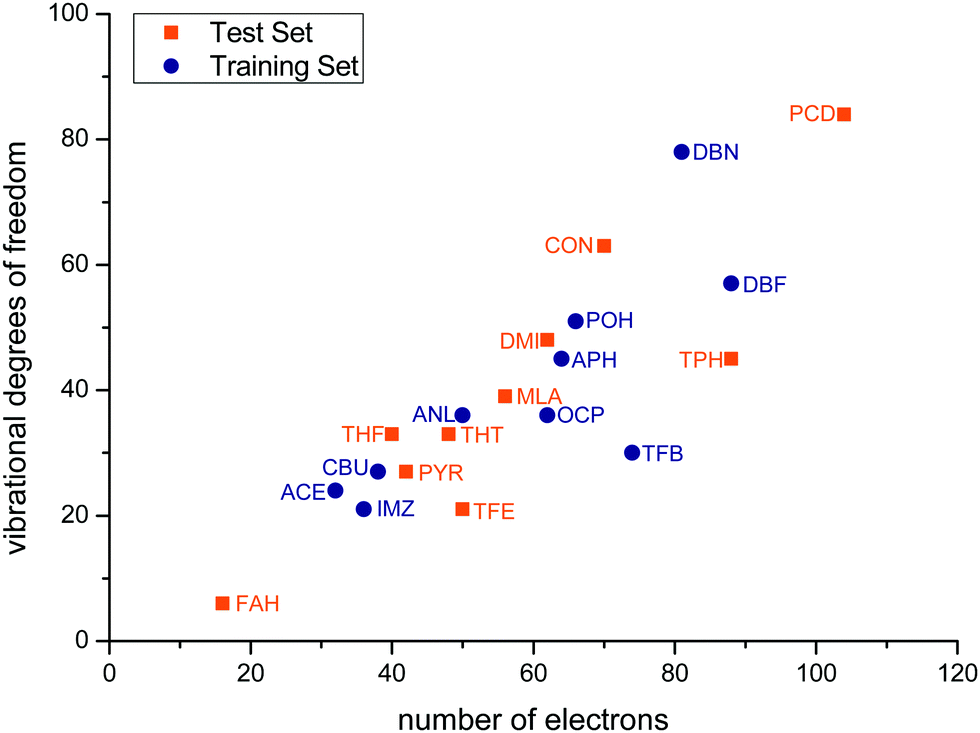 | ||
| Fig. 3 Vibrational degrees of freedom 3Nn − 6 of the training (blue) and test (orange) solute molecules with Nn nuclei for which monohydrates are investigated as a function of their number of electrons ne. | ||
Acceptors which were considered as suitable candidates for the test set, but are postponed for later investigation to maximise its diversity and to reduce its conformational complexity, include acetaldehyde (AAH: 75-07-0), 4′-fluoroacetophenone (FPH: 403-42-9), hexafluoroisopropanol (HFP: 920-66-1),85,86N,N-dimethylformamide (DMF: 68-12-2) and 2,2,4,4-tetramethylpentan-3-one (TPO: 815-24-7). Other candidates which require further testing or turned out to be less suitable at this stage include benzaldehyde, 4-fluorobenzaldehyde, furan, thiophene,91 propylene oxide, and 4-methoxy-2,2,6,6-tetramethyl-1-piperidinyloxy. Further interesting candidates which were suggested by microwave groups and will have to be further analysed in terms of suitability include propargyl alcohol,92 furfuryl alcohol, and furfuryl mercaptane93 as well as thenyl alcohol and thenyl mercaptane.84
3 Experimental methods
3.1 Description of experimental procedures
All experimental data of this blind challenge refer to translationally and rotationally cold molecules (T < 20 K) without contact to other molecules or carrier gas atoms, but with some residual vibrational excitation in soft modes from the initial thermal population. There is the possibility of conformational trapping behind barriers, which may partially avoid relaxation to the global minimum structure. This is the typical situation in adiabatic carrier gas expansion experiments into vacuum, either free94 or skimmed to form a molecular beam,95 with a starting temperature of 300 K or somewhat higher for low-volatility compounds. The specific monohydrates were chosen to minimise the risk of significant conformational trapping in most cases. The trapping can be intermolecular, if two or more hydrogen bond formation sites are available in the solute molecule. In such a case, cold monohydrates with water docking on the less favourable site cannot quantitatively interconvert into the most favourable site on the timescale of the experiment in all cases. This is particularly relevant for the three alcohols in the test set, where water can attach to any lone pair or insert into a pre-existing hydrogen bond of the solute.89 The insertion is typically an activated process and if the activation energy significantly exceeds 5 kJ mol−1, it may not happen sufficiently fast under the low collision rates of the supersonic expansion. The trapping can also be of intramolecular nature, if the solute exists in different conformations with higher barriers. For ring puckering modes, the barriers are not always sufficiently low for efficient funneling into the vibrational ground state. For intramolecular hydrogen bonds, the solutes are chosen such that relaxation into the global minimum structure was typically proven experimentally, or is at least likely. Constitutional relaxation is evidently not considered. Thus, the decomposition of formaldehyde into CO and H2 is disregarded for the global minimum criterion, although it is exothermic by about 9 kJ mol−1 at low temperature.96 Also, the exothermic diol formation from the reaction of a formaldehyde molecule with a water molecule97,98 is not considered. The solutes always remain in the constitution given in Fig. 2.The spectroscopic methods used to probe the monohydrates may be distinguished into action and linear techniques. In action techniques, the effect of the photons on the monohydrates is probed, e.g. ionisation, dissociation or fluorescence. In linear techniques, the effect of the monohydrates on the photons is probed, e.g. direct infrared absorption13 or inelastic (Raman) scattering.87 Action techniques often offer much higher sensitivity, they can be size-selective and conformationally selective.15,99–101
3.2 Challenges and experimental difficulties
Linear techniques probe all the molecular systems present in the expansion, based on their photon absorption or scattering cross sections. They can usually identify size (e.g. hydration number) by variation of the expansion conditions and by following the signal evolution, at least for 1:1 complexes if those do not spectrally overlap with other cluster compositions. Conformations can only be distinguished if they differ in their spectral fingerprint, sometimes with the help of isotope substitution. There is a less liberal molecular size limit for linear techniques due to the decreasing vapour pressure and the limited sensitivity. Action techniques may in turn suffer from unwanted competing processes such as fragmentation and may still be problematic for many non-aromatic systems and even some aromatic systems with fast processes in the electronically excited state.102,103 The best is always a combination of both techniques, such as in the case of POH from the training set. This is, however, unrealistic on the time scale of this challenge and for the choice of molecules. It was anticipated that one member of the test set (PCD) would only be characterised by an action technique (IR/UV double resonance), whereas the others would be mostly accessed by direct infrared absorption. For the latter, a recent instrumental development104 is very helpful for monohydrates, because its gas recycling allows for the use of expensive isotopologues, solutes and carrier gases. In some cases, where the water vibration of interest has a low infrared intensity or may be distributed over several OH stretching modes in the complex, linear Raman spectroscopy14,87 could provide the required information.
Aggravated by the partial double-blind character of the challenge, it could not be strictly ruled out that a training or test set member has to be removed completely from the challenge, because experiments or their interpretations reveal a major problem. Such problems may include the discovery of new monohydrate or even monomer conformations which complicate an unambiguous assignment,38 issues with the cluster size assignment, experimental downtimes due to instrument failure, commercial availability bottlenecks, etc. However, we expected that such problems would not exceed a 10% threshold and thus do not significantly narrow the significance of the challenge. Indeed, at the time of writing, only one of the test systems contains a non-critical degree of experimental uncertainty, whereas the others appear to be unambiguous in their interpretation.
4 Challenge procedure
4.1 Theory group involvement
Theory groups and other researchers interested in the systematic prediction of OH stretching wavenumbers for monohydrates were alerted about this blind challenge via news groups, conferences, and direct contact. The training set list of 10 organic solvates was provided at this time. Interested groups were given the possibility to register for the challenge before the test set was published. They could then be alerted immediately upon the start of the actual competition. Registrations up to the submission deadline 6 months later were accepted. A unique entry code was provided to each participant in order to allow them later to review their own submitted data.Although it did not stand as a strict rule, the calculations for all molecules in both sets (test and training) had to be carried out as consistently as possible. This meant that the level of theory at all different stages (conformer search, optimisation, calculation of frequencies, etc.) were expected to be the same. The same also applied to methods not based on molecular structure. For example, if a regression was used, the same model was expected to be applied to all systems, while some parameters of the protocol were allowed to change. For example, in some systems conformational searches might be redundant, or carry different parameters due to the range of molecular sizes. It is also possible that some geometry optimisation criteria call for adaptation. These computational details were expected to be provided at the end of the challenge. Assistance to all the participating groups was provided throughout the challenge duration, strictly avoiding any direct contact between the theory and the experimental groups, in the true spirit of a double-blind challenge.
One cannot rule out that we have overlooked published cold gas phase vibrational work on a system in the test set, or that such work appeared on the scientific record before the experimental HyDRA data are released after the end of the challenge. This could have been due to activities of other experimental groups not involved in the challenge, or due to data leakage from our experimental groups despite substantial effort to keep the results confidential among the contributing experimentalists. In such a case, the affected test set member would be shifted to training set status, because one cannot rule out that some model-proposing participants had a knowledge advantage over others. However, at the time of writing, we are not aware of such an incidence. The goal was clearly to keep the blind character as unquestionable as possible.33
4.2 Mandatory data submission
The mandatory part of the submissions for this blind challenge was compact. For each of the training and test set members, a single predicted wavenumber for the hydrogen-bonded water OH stretching mode in the most stable monohydrate in cm−1 had to be provided in a standardised online form. We strongly recommended to provide together with this number an error estimate. This could be based on an actual statistical analysis (e.g., from the errors observed in the training systems) or even from personal convictions. Each participant group had the freedom to select which systems to compute (and ultimately submit data). In other words, all submissions were eligible to acceptance even if incomplete.This compact table of up to 20 numbers (plus uncertainties) had to be accompanied by a detailed description of the rational prediction method employed, to be included as a separate, autonomous file in the supplementary information of the planned publication. The description had to contain all the details required for the reproduction of the results, such as the employed methods and program packages, with suitable references. Scaling parameters, employed atomic masses and any other parameters and computational keywords required for reproduction had to be provided. Any additional computed data deemed of interest for the monohydrates (see next section) could be included into the supplement. This document will remain in the full responsibility of the submitting authors and should include all the names and affiliations of the contributing co-authors as well as the role of each co-author in the submission. The use of ORCID IDs is strongly recommended.
4.3 Optional data submission
Optional parts of the submission could be, in the sequence of decreasing importance for the goals of this blind challenge, and in free format to be included in the supplementary information:(a) A compact table of possible resonance partners of the hydrogen-bonded OH stretching mode of water, where predicted. This should include the wavenumber of the perturber and its fractional share of the intensity (square of the zero order OH stretching wavefunction), and for control purposes also the fractional share of the OH intensity and the stretching wavenumber of the perturbed OH stretching state. From this and the mandatory submission of the predicted OH stretching wavenumber, a coupling matrix element can be obtained in the 2-level dark perturber approximation, see ref. 43. Such information will only be possible if anharmonic calculations or coupling models are included. For other submissions, the submitted OH stretching wavenumber will be taken as the deperturbed zero order OH stretch which is obtained from experiment in such a 2-level deperturbation, in the few cases where such a resonance is observed.
(b) A table of any other OH stretching wavenumbers of the monohydrate, in particular the free OH stretch of the water and any other OH stretching mode in the acceptor molecule. If strong mixing of the OH oscillators is predicted, the mode with the strongest hydrogen-bonded water character should be submitted in the mandatory part and all the others in this part. One way to identify the mode with dominant water vibration character is to calculate the spectrum for H218O. The OH stretching mode with the strongest isotope downshift is the one of interest.
(c) IR intensities and Raman scattering activities for all the OH stretching modes of the monohydrate, where available, preferably in km mol−1 and in Å4 u−1, including the depolarisation ratio for polarised light.
(d) Any spectral information on isotopologues which may be useful for the experimental analysis and for follow-up work (D, 18O), always specifying the exact employed atomic masses. We recommend to always use the explicit isotopic masses for best method comparison.105 Some programs use masses averaged over natural isotopic composition by default. This may have relatively small consequences for the current low resolution vibrational spectra and systems (still up to 5 cm−1 for vibrations involving sulfur due to a ratio of 1.003 between the natural atomic mass and the main isotope mass), but is clearly detrimental when comparing to high resolution (rotational) spectroscopy in the future.
(e) Relative energy (in kJ mol−1, excluding and including zero-point vibrational energy corrections) and spectral properties of any higher-lying conformations of the monohydrates which were investigated, within a range of 3–5 kJ mol−1 of the global minimum structure. Here, global is defined under the restriction that the chemical bond connectivity remains the same, so rearranged bonding patterns like geminal diols59,97,98 as structural isomers of noncovalent ketone monohydrates96 are not considered. Geometries, when available, may be provided.
After the official submission deadline for model contributions, there was a limited time window for the correction of submissions. After this window was closed, the raw experimental data were made publicly available on a data repository, such that the challenge was closed on both ends.
4.4 Evaluation of the blind challenge
The submitted OH stretching data will be carefully evaluated and compared with the experimental values in different ways. Correlation plots for training, test and combined sets will be produced and analysed for all complete submissions, with a possible focus on which set is predicted better. Where several incomplete submissions focus on the most simple systems, which is to be expected for high-level theoretical treatments, analysis of such subsets with benchmark character will be attempted.4.5 Publication of the results
The results will be published in a relatively compact many-author manuscript with extensive supplementary information in the responsibility of separate experimental and computational author subgroups, planned to be part of the present PCCP themed collection on benchmarking in 2022. The final selection of the methods and results included will be subject to different criteria including diversity of methods, sophistication of the approach and total number of ‘coincident’ submissions. Our expectation is, however, that all submissions will be considered. The submission of the data does not change the ownership of the data, the latter remains exclusive to the participating group. Any participants whose results are not included are still welcome to submit their work independently. Participants selected to participate in the joint paper are expected to respond to two rounds of manuscript reviews and to remove inconsistencies and gaps. Failure to reply within a two-week period effectively confirming co-authoring could result in exclusion from the joint publication. The publication should be viewed as the starting point for numerous follow-up activities by the individual groups, such as completing or improving their predictions in a non-blind fashion. Groups which did not meet the submission deadline are invited to present their results independently as well. Some experimental assignments may have to be revisited in the light of systematically conflicting predictions. Optional data provided by the computational approaches may help to better understand the spectra. The double-sided theory-experiment interplay will continue, as usual in the field. All these activities profit from a fast publication of the primary blind challenge results.4.6 Post-publication activities
After publication of the results of the HyDRA blind challenge, the training and test sets can be extended to more strongly and even more weakly bound monohydrates, to charged systems (where more powerful experimental techniques are available106–108), to dihydrates, to isotope effects, to other vibrations, or to further observables such as structures and relative energies. These extensions may be targeted by different groups and will hopefully lead to a continuous refinement of the theoretical modelling of the microhydration of organic matter, identifying the most powerful and universal models in the field.There are indeed many interesting monohydrates which were not considered for the present challenge for various reasons. This includes systems where the global minimum structure is hidden behind a large monomer isomerisation barrier,109 and some systems which are closely related to members of the training or test set.43,110 It also includes the numerous systems where the global minimum structure involves water as a pure acceptor. In terms of acceptor elements beyond O and N, there are many extension possibilities towards S84,111,112 which remain to be explored. For the structurally well characterised acid hydrates,113,114 one may expect complex vibrational dynamics whenever the OH stretching excitation comes close to degenerate proton transfer barriers.
5 Conclusions
We have described a first systematic blind challenge initiative in the field of hydrogen bond-induced shifts in solvating water molecules. It consists of a training set of 10 literature-known 1:1 hydrate complexes spanning a downshift window of nearly 200 cm−1. 10 vibrationally unassigned monohydrates were proposed for the actual blind test and promised to be experimentally characterised during the runtime of the challenge. The combined experimental results and predictions will be jointly published to identify the most promising empirical, semiempirical and ab initio models in the field. Several options for the extension of this initial challenge in chemical and spectral space are proposed, in the spirit of continuous benchmarking.115
We see this direct match between electronic structure theory and vibrational spectroscopy as an important complement to more traditional and well-established blind challenges in the field of protein116 or crystal structure117 prediction, where subtle energy differences also relate to zero point or thermal vibrational energy and thus anharmonicity treatments, besides getting the potential energy hypersurfaces right. This is even more obvious for the field of room temperature hydration,118 where an atomistic method which systematically predicts correct free energy differences for a good reason is likely to predict reliable vibrational shifts of monohydrates as well. However, there is a long path of convergence between rigorously atomistic and other, more empirical or approximate or machine-learning models which are more easily extendable to macroscopic samples. Clearly, there is plenty of room in between. Blind challenges may be among the most objective procedures to find out where we stand.
Author contributions
T. L. F.: data curation, formal analysis, investigation, visualization, writing – review and editing; M. B.: data curation, investigation, visualization, writing – review and editing; A. Z.: investigation, methodology, validation, writing – review and editing; R. A. M.: conceptualization, funding acquisition, methodology, project administration, resources, software, writing – review and editing; M. A. S.: conceptualization, formal analysis, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, writing – original draft, writing – review and editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank colleagues who have made helpful suggestions for the training and test sets, namely E. Arunan, D. Bernhard, J. Fernandez, A. Fujii, E. Gloaguen, A. Lesarri, M. Mons, D. Obenchain, M. Sanz, K. Schwing, R. Wugt Larsen, Y. Xu, J. Zischang. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 389479699/GRK2455.References
- P. M. Felker and A. H. Zewail, Stepwise solvation of molecules as studies by picosecond-jet spectroscopy: dynamics and spectra, Chem. Phys. Lett., 1983, 94, 454–460 CrossRef CAS.
- R. D. Knochenmuss and D. E. Smith, Time and internal energy dependent fluorescence spectra of naphthol_water clusters, J. Chem. Phys., 1994, 101, 7327–7336 CrossRef CAS.
- R. Bombach, E. Honegger and S. Leutwyler, Solute-solvent interactions in microhydrate clusters: Carbazole-(H2O)n, Chem. Phys. Lett., 1985, 118, 449–454 CrossRef CAS.
- C. Puzzarini, J. Bloino, N. Tasinato and V. Barone, Accuracy and interpretability: the devil and the holy grail. New routes across old boundaries in computational spectroscopy, Chem. Rev., 2019, 119, 8131–8191 CrossRef CAS PubMed.
- M. Becucci and S. Melandri, High-resolution spectroscopic studies of complexes formed by medium-size organic molecules, Chem. Rev., 2016, 116, 5014–5037 CrossRef CAS PubMed.
- M. Mons, I. Dimicoli, B. Tardivel, F. Piuzzi, E. G. Robertson and J. P. Simons, Energetics of the gas phase hydrates of trans-formanilide: A microscopic approach to the hydration sites of the peptide bond, J. Phys. Chem. A, 2001, 105, 969–973 CrossRef CAS.
- A. Poblotzki, H. C. Gottschalk and M. A. Suhm, Tipping the scales: Spectroscopic tools for intermolecular energy balances, J. Phys. Chem. Lett., 2017, 8, 5656–5665 CrossRef CAS PubMed.
- C. Zimmermann, H. Gottschalk and M. Suhm, Three-dimensional docking of alcohols to ketones: an experimental benchmark based on acetophenone solvation energy balances, Phys. Chem. Chem. Phys., 2020, 22, 2870–2877 RSC.
- S. E. Novick, Bibliography of rotational spectra of weakly bound complexes. 2019, https://wesfiles.wesleyan.edu/home/snovick/SN_webpage/vdw.pdf.
- W. Caminati and J.-U. Grabow, Microwave Spectroscopy - Molecular Systems, in Frontiers of molecular spectroscopy, Section 4.2, ed. J. Laane, Elsevier, 2011, ch. 15 Search PubMed.
- P. Pinacho, D. A. Obenchain and M. Schnell, New findings from old data: A semiexperimental value for the eQq of the nitrogen atom, J. Chem. Phys., 2020, 153, 234307 CrossRef CAS PubMed.
- A. Potapov and P. Asselin, High-resolution jet spectroscopy of weakly bound binary complexes involving water, Int. Rev. Phys. Chem., 2014, 33, 275–300 Search PubMed.
- M. Suhm and F. Kollipost, Femtisecond single-mole infrared spectroscopy of molecular clusters, Phys. Chem. Chem. Phys., 2013, 15, 10702–10721 RSC.
- M. Nedić, T. N. Wassermann, R. W. Larsen and M. A. Suhm, A combined Raman- and infrared jet study of mixed methanol-water and ethanol-water clusters, Phys. Chem. Chem. Phys., 2011, 13, 14050–14063 RSC.
- E. Gloaguen, M. Mons, K. Schwing and M. Gerhards, Neutral peptides in the gas phase: conformation and aggregation issues, Chem. Rev., 2020, 120, 12490–12562 CrossRef CAS PubMed.
- T. S. Zwier, The spectroscopy of solvation in hydrogen-bonded aromatic clusters, Annu. Rev. Phys. Chem., 1996, 47, 205–241 CrossRef CAS.
- J. Zischang, J. J. Lee and M. A. Suhm, Communication: Where does the first water molecule go in imidazole?, J. Chem. Phys., 2011, 135, 061102 CrossRef PubMed.
- G. Maes, J. Smets, L. Adamowicz, W. McCarthy, M. Van Bael, L. Houben and K. Schoone, Correlations between ab initio and experimental data for isolated H-bonded complexes of water with nitrogen bases, J. Mol. Struct., 1997, 410, 315–322 Search PubMed.
- J. Graton, M. Berthelot, F. Besseau and C. Laurence, An enthalpic scale of hydrogen-bond basicity. 3. Ammonia, primary, secondary, and tertiary amines, J. Org. Chem., 2005, 70, 7892–7901 CrossRef CAS PubMed.
- B. Chan, J. E. Del Bene, J. Elguero and L. Radom, On the relationship between the preferred site of hydrogen bonding and protonation, J. Phys. Chem. A, 2005, 109, 5509–5517 CrossRef CAS PubMed.
- M. Rozenberg, A. Loewenschuss and Y. Marcus, An empirical correlation between stretching vibration redshift and hydrogen bond length, Phys. Chem. Chem. Phys., 2000, 2, 2699–2702 RSC.
- R. J. Plowright, E. Gloaguen and M. Mons, Compact folding of isolated four-residue neutral peptide chains: H-bonding patterns and entropy effects, ChemPhysChem, 2011, 12, 1889–1899 CrossRef CAS PubMed.
- F. Ben Nasr, I. Alata, D. Scuderi, V. Lepère, V. Brenner, N.-E. Jadane and A. Zehnacker, Effects of complexation with sulfuric acid on the photodissociation of protonated Cinchona alkaloids in the gas phase, Phys. Chem. Chem. Phys., 2019, 21, 15439–15451 RSC.
- E. Vogt, P. Bertran Valls and H. G. Kjaergaard, Accurate calculations of OH-stretching intensities with a reduced-dimensional local mode model including Eckart Axis embedding, J. Phys. Chem. A, 2020, 124, 932–942 CrossRef CAS PubMed.
- K. N. Blodgett, J. L. Fischer, T. S. Zwier and E. L. Sibert, The missing NH stretch fundamental in S1 methyl anthranilate: IR-UV double resonance experiments and local mode theory, Phys. Chem. Chem. Phys., 2020, 22, 14077–14087 RSC.
- R. Medel and M. A. Suhm, Predicting OH stretching fundamental wavenumbers of alcohols for conformational assignment: Different correction patterns for density functional and wave-function-based methods, Phys. Chem. Chem. Phys., 2021, 23, 5629–5643 RSC.
- M. Thomas, M. Brehm, R. Fligg, P. Vöhringer and B. Kirchner, Computing vibrational spectra from ab initio molecular dynamics, Phys. Chem. Chem. Phys., 2013, 15, 6608–6622 RSC.
- D. Semrouni, A. Sharma, J.-P. Dognon, G. Ohanessian and C. Clavaguera, Finite temperature infrared spectra from polarizable molecular dynamics simulations, J. Chem. Theory Comput., 2014, 10, 3190–3199 CrossRef CAS PubMed.
- B. J. Miller, J. R. Lane and H. G. Kjaergaard, Intramolecular OH…π interactions in alkenols and alkynols, Phys. Chem. Chem. Phys., 2011, 13, 14183–14193 RSC.
- W. Fu and W. S. Hopkins, Applying machine learning to vibrational spectroscopy, J. Phys. Chem. A, 2018, 122, 167–171 CrossRef CAS PubMed.
- J. Moult, K. Fidelis, A. Kryshtafovych, T. Schwede and A. Tramontano, Critical assessment of methods of protein structure prediction (CASP)|Round XII, Proteins: Struct., Funct., Bioinform., 2018, 86, 7–15 CrossRef CAS PubMed.
- A. M. Reilly, R. I. Cooper, C. S. Adjiman, S. Bhattacharya, A. D. Boese, J. G. Brandenburg, P. J. Bygrave, R. Bylsma, J. E. Campbell and R. Car, et al., Report on the sixth blind test of organic crystal structure prediction methods, Acta Crystallograph. Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 439–459 CrossRef CAS PubMed.
- J. P. Guthrie, A blind challenge for computational solvation free energies: introduction and overview, J. Phys. Chem. B, 2009, 113, 4501–4507 CrossRef CAS PubMed.
- D. L. Mobley, K. L. Wymer, N. M. Lim and J. P. Guthrie, Blind prediction of solvation free energies from the SAMPL4 challenge, J. Comput.-Aided Mol. Des., 2014, 28, 135–150 CrossRef CAS PubMed.
- C. C. Bannan, K. H. Burley, M. Chiu, M. R. Shirts, M. K. Gilson and D. L. Mobley, Blind prediction of cyclohexane-water distribution coefficients from the SAMPL5 challenge, J. Comput.-Aided Mol. Des., 2016, 30, 927–944 CrossRef CAS PubMed.
- M. Amezcua, L. El Khoury and D. L. Mobley, SAMPL7 Host-guest challenge overview: Assessing the reliability of polarizable and non-polarizable methods for binding free energy calculations, J. Comput.-Aided Mol. Des., 2021, 35, 1–35 CrossRef CAS PubMed.
- H. C. Gottschalk, A. Poblotzki, M. Fatima, D. A. Obenchain, C. Pérez, J. Antony, A. A. Auer, L. Baptista, D. M. Benoit and G. Bistoni, et al., The first microsolvation step for furans: New experiments and benchmarking strategies, J. Chem. Phys., 2020, 152, 164303 CrossRef CAS PubMed.
- H. C. Gottschalk, A. Poblotzki, M. A. Suhm, M. M. Al-Mogren, J. Antony, A. A. Auer, L. Baptista, D. M. Benoit, G. Bistoni and F. Bohle, et al., The furan microsolvation blind challenge for quantum chemical methods: First steps, J. Chem. Phys., 2018, 148, 014301 CrossRef PubMed.
- W. Benedict and E. K. Plyler, Absorption spectra of water vapor and carbon dioxide in the region of 2.7 microns, J. Res. Natl. Bur. Stand., 1951, 46, 246–265 CrossRef CAS.
- D. Bender, The Raman effect of water vapor, Phys. Rev., 1935, 47, 252 CrossRef CAS.
- QMbench – challenges for numerical quantum chemistry, 2021, https://qmbench.net/.
- H. S. Biswal, Y. Loquais, B. Tardivel, E. Gloaguen and M. Mons, Isolated monohydrates of a model peptide chain: Effect of a first water molecule on the secondary structure of a capped phenylalanine, J. Am. Chem. Soc., 2011, 133, 3931–3942 CrossRef CAS PubMed.
- T. L. Fischer, T. Wagner, H. C. Gottschalk, A. Nejad and M. A. Suhm, A rather universal vibrational resonance in 1:1 hydrates of carbonyl compounds, J. Phys. Chem. Lett., 2021, 12, 138–144 CrossRef CAS PubMed.
- M. Chrayteh, E. Burevschi, D. Loru, T. R. Huet, P. Dréan and M. E. Sanz, Disentangling the complex network of non-covalent interactions in fenchone hydrates via rotational spectroscopy and quantum chemistry, Phys. Chem. Chem. Phys., 2021, 20686–20694 RSC.
- A. Bouchet, J. Altnöder, M. Broquier and A. Zehnacker, IR-UV spectroscopy of jet-cooled 1-indanol: Restriction of the conformational space by hydration, J. Mol. Struct., 2014, 1076, 344–351 CrossRef CAS.
- J. Lei, J. Zhang, G. Feng, J.-U. Grabow and Q. Gou, Conformational preference determined by inequivalent n-pairs: Rotational studies on acetophenone and its monohydrate, Phys. Chem. Chem. Phys., 2019, 21, 22888–22894 RSC.
- U. Spoerel and W. Stahl, The aniline-water complex, J. Mol. Spectrosc., 1998, 190, 278–289 CrossRef CAS PubMed.
- S. Melandri, A. Maris, B. M. Giuliano and W. Caminati, Water-ketones hydrogen bonding: The rotational spectrum of cyclobutanone-water, J. Chem. Phys., 2005, 123, 164304 CrossRef PubMed.
- D. Bernhard, M. Fatima, A. Poblotzki, A. Steber, C. Pérez, M. Suhm, M. Schnell and M. Gerhards, Dispersion-controlled docking preference: Multi-spectroscopic study on complexes of dibenzofuran with alcohols and water, Phys. Chem. Chem. Phys., 2019, 21, 16032–16046 RSC.
- E. Gougoula, D. J. Cole and N. R. Walker, Bifunctional hydrogen bonding of imidazole with water explored by rotational spectroscopy and DFT calculations, J. Phys. Chem. A, 2020, 124, 2649–2659 CrossRef CAS PubMed.
- X. K. Zhang, E. G. Lewars, R. E. March and J. M. Parnis, Vibrational spectrum of the acetone-water complex: A matrix isolation FTIR and theoretical study, J. Phys. Chem., 1993, 97, 4320–4325 CrossRef CAS.
- I. León, P. Arnáiz, I. Usabiaga and J. Fernández, Mass resolved IR spectroscopy of aniline- water aggregates, Phys. Chem. Chem. Phys., 2016, 18, 27336–27341 RSC.
- E. M. Brás, T. L. Fischer and M. A. Suhm, The hydrates of TEMPO: Water vibrations reveal radical microsolvation, Angew. Chem., Int. Ed., 2021, 60, 19013–19017 CrossRef PubMed.
- M. Broquier, F. Lahmani, A. Zehnacker-Rentien, V. Brenner, P. Millié and A. Peremans, Hydrogen-bonded bridges in complexes of o-cyanophenol: Laser-induced fluorescence and IR/UV double-resonance studies, J. Phys. Chem. A, 2001, 105, 6841–6850 CrossRef CAS.
- K. Le Barbu, F. Lahmani, M. Mons, M. Broquier and A. Zehnacker, IR-UV investigation of the structure of the 1-phenylethanol chromophore and its hydrated complexes, Phys. Chem. Chem. Phys., 2001, 3, 4684–4688 RSC.
- V. Venkatesan, A. Fujii, T. Ebata and N. Mikami, A direct experimental evidence for an aromatic C–H…O hydrogen bond by fluorescence-detected infrared spectroscopy, Chem. Phys. Lett., 2004, 394, 45–48 CrossRef CAS.
- M. K. Van Bael, J. Smets, K. Schoone, L. Houben, W. McCarthy, L. Adamowicz, M. J. Nowak and G. Maes, Matrix-isolation FTIR studies and theoretical calculations of hydrogen-bonded complexes of imidazole. A comparison between experimental results and different calculation methods, J. Phys. Chem. A, 1997, 101, 2397–2413 CrossRef CAS.
- E. Burevschi, I. Peña and M. E. Sanz, Medium-sized rings: Conformational preferences in cyclooctanone driven by transannular repulsive interactions, Phys. Chem. Chem. Phys., 2019, 21, 4331–4338 RSC.
- E. Burevschi, I. Peña and M. E. Sanz, Geminal diol formation from the interaction of a ketone with water in the gas phase: Structure and reactivity of cyclooctanone-(H2O)1,2 clusters, J. Phys. Chem. Lett., 2021, 12, 12419–12425 CrossRef CAS PubMed.
- E. Vrolix, M. Goethals and T. Zeegers-Huyskens, Infrared study of hydrogen bond complexes involving 1, 3-dimethyl, 2-imidazolidinone and hydroxylic derivatives, Spectrosc. Lett., 1993, 26, 497–507 CrossRef CAS.
- A. Vigorito, L. Paoloni, C. Calabrese, L. Evangelisti, L. B. Favero, S. Melandri and A. Maris, Structure and dynamics of cyclic amides: The rotational spectrum of 1, 3-dimethyl-2-imidazolidinone, J. Mol. Spectrosc., 2017, 342, 38–44 CrossRef CAS.
- B. Nelander, A matrix isolation study of the water-formaldehyde complex. The far-infrared region, Chem. Phys., 1992, 159, 281–287 CrossRef CAS.
- F. J. Lovas and C. Lugez, The microwave spectrum and structure of CH2O-H2O, J. Mol. Spectrosc., 1996, 179, 320–323 CrossRef CAS.
- J. Thomas, O. Sukhorukov, W. Jäger and Y. Xu, Direct spectroscopic detection of the orientation of free OH groups in methyl lactate-(water)1,2 clusters: Hydration of a chiral hydroxy ester, Angew. Chem., Int. Ed., 2014, 53, 1156–1159 CrossRef CAS PubMed.
- S. A. Katsyuba, S. Spicher, T. P. Gerasimova and S. Grimme, Revisiting conformations of methyl lactate in water and methanol, J. Chem. Phys., 2021, 155, 024507 CrossRef CAS PubMed.
- E. Galantay, 1-phenylcyclohexane-1,2-diols and their geometry, Tetrahedron, 1963, 19, 319–321 CrossRef CAS.
- P. Nieto, M. Letzner, T. Endres, G. Schwaab and M. Havenith, IR spectroscopy of pyridine- water structures in helium nanodroplets, Phys. Chem. Chem. Phys., 2014, 16, 8384–8391 RSC.
- R. B. Mackenzie, C. T. Dewberry, R. D. Cornelius, C. Smith and K. R. Leopold, Multidimensional large amplitude dynamics in the pyridine-water complex, J. Phys. Chem. A, 2017, 121, 855–860 CrossRef CAS PubMed.
- D. G. Melnik, S. Gopalakrishnan, T. A. Miller and F. C. De Lucia, The absorption spectroscopy of the lowest pseudorotational states of tetrahydrofuran, J. Chem. Phys., 2003, 118, 3589–3599 CrossRef CAS.
- P. K. Sahu and S.-L. Lee, Hydrogen-bond interaction in 1:1 complexes of tetrahydrofuran with water, hydrogen fluoride, and ammonia: A theoretical study, J. Chem. Phys., 2005, 123, 044308 CrossRef PubMed.
- M. E. Sanz, J. C. López, J. L. Alonso, A. Maris, P. G. Favero and W. Caminati, Conformation and stability of adducts of sulfurated cyclic compounds with water: Rotational spectrum of tetrahydrothiophene-water, J. Phys. Chem. A, 1999, 103, 5285–5290 CrossRef CAS.
- J. Lei, S. Alessandrini, J. Chen, Y. Zheng, L. Spada, Q. Gou, C. Puzzarini and V. Barone, Rotational spectroscopy meets quantum chemistry for analyzing substituent effects on non-covalent interactions: The case of the trifluoroacetophenone-water complex, Molecules, 2020, 25, 4899 CrossRef CAS PubMed.
- M. Heger, T. Scharge and M. A. Suhm, From hydrogen bond donor to acceptor: The effect of ethanol fluorination on the first solvating water molecule, Phys. Chem. Chem. Phys., 2013, 15, 16065–16073 RSC.
- J. Thomas and Y. Xu, Structure and tunneling dynamics in a model system of peptide co-solvents: Rotational spectroscopy of the 2,2,2-trifluoroethanol…water complex, J. Chem. Phys., 2014, 140, 06B616_1 Search PubMed.
- T. K. Ha, J. Makarewicz and A. Bauder, Ab initio study of the water-formaldehyde complex, J. Phys. Chem., 1993, 97, 11415–11419 CrossRef CAS.
- Y. Dimitrova and S. D. Peyerimhoff, Theoretical study of hydrogen-bonded formaldehyde-water complexes, J. Phys. Chem., 1993, 97, 12731–12736 CrossRef CAS.
- T. A. Ramelot, C.-H. Hu, J. E. Fowler, B. J. DeLeeuw and H. F. Schaefer, Theoretical study of hydrogen-bonded formaldehyde-water complexes, J. Chem. Phys., 1994, 100, 4347–4354 CrossRef CAS.
- B. Nelander, Infrared spectrum of the water formaldehyde complex in solid argon and solid nitrogen, J. Chem. Phys., 1980, 72, 77–84 CrossRef CAS.
- M. J. Shultz and T. H. Vu, Hydrogen bonding between water and tetrahydrofuran relevant to clathrate formation, J. Phys. Chem. B, 2015, 119, 9167–9172 CrossRef CAS PubMed.
- R. Meyer, J. C. López, J. L. Alonso, S. Melandri, P. G. Favero and W. Caminati, Pseudorotation pathway and equilibrium structure from the rotational spectrum of jet-cooled tetrahydrofuran, J. Chem. Phys., 1999, 111, 7871–7880 CrossRef CAS.
- X. Liu, A. L. Sobolewski, R. Borrelli and W. Domcke, Computational investigation of the photoinduced homolytic dissociation of water in the pyridine-water complex, Phys. Chem. Chem. Phys., 2013, 15, 5957–5966 RSC.
- D. J. Millen and G. W. Mines, Hydrogen bonding in the gas phase. Part 5.|Infrared spectroscopic investigation of O–H…N complexes formed by water: ammonia monohydrate and amine and pyridine monohydrates, J. Chem. Soc., Faraday Trans. 2, 1977, 73, 369–377 RSC.
- M. Y. Choi and R. E. Miller, Infrared laser spectroscopy of imidazole complexes in helium nanodroplets: Monomer, dimer, and binary water complexes, J. Phys. Chem. A, 2006, 110, 9344–9351 CrossRef CAS PubMed.
- M. Juanes, R. T. Saragi, R. Pinacho, J. E. Rubio and A. Lesarri, Sulfur hydrogen bonding and internal dynamics in the monohydrates of thenyl mercaptan and thenyl alcohol, Phys. Chem. Chem. Phys., 2020, 22, 12412–12421 RSC.
- A. Shahi and E. Arunan, Microwave spectroscopic and theoretical investigations of the strongly hydrogen bonded hexafluoroisopropanol…water complex, Phys. Chem. Chem. Phys., 2015, 17, 24774–24782 RSC.
- B. Wu, A. S. Hazra, N. A. Seifert, S. Oswald, W. Jäger and Y. Xu, Higher-energy hexafluoroisopropanol…water isomer and its large amplitude motions: Rotational spectra and DFT calculations, J. Phys. Chem. A, 2021, 125, 10401–10409 CrossRef CAS PubMed.
- T. Scharge, C. Cézard, P. Zielke, A. Schütz, C. Emmeluth and M. A. Suhm, A peptide co-solvent under scrutiny: Self-aggregation of 2,2,2-trifluoroethanol, Phys. Chem. Chem. Phys., 2007, 9, 4472–4490 RSC.
- D. Roccatano, G. Colombo, M. Fioroni and A. E. Mark, Mechanism by which 2,2,2- triuoroethanol/water mixtures stabilize secondary-structure formation in peptides: A molecular dynamics study, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12179–12184 CrossRef CAS PubMed.
- N. Borho, M. A. Suhm, K. Le Barbu-Debus and A. Zehnacker, Intra- vs. intermolecular hydrogen bonding: dimers of alpha-hydroxyesters with methanol, Phys. Chem. Chem. Phys., 2006, 8, 4449–4460 RSC.
- C. Merten and Y. Xu, Chirality transfer in a methyl lactate-ammonia complex observed by matrix-isolation vibrational circular dichroism spectroscopy, Angew. Chem., 2013, 125, 2127–2130 CrossRef.
- W. G.-D. P. Silva and J. van Wijngaarden, Characterization of large-amplitude motions and hydrogen bonding interactions in the thiophene-water complex by rotational spectroscopy, J. Phys. Chem. A, 2021, 125, 3425–3431 CrossRef CAS PubMed.
- S. P. Gnanasekar and E. Arunan, Structure and internal motions of a multifunctional alcohol-water complex: rotational spectroscopy of the propargyl alcohol…H2O Dimer, J. Phys. Chem. A, 2021, 7138–7150 CrossRef CAS PubMed.
- M. Juanes, A. Lesarri, R. Pinacho, E. Charro, J. E. Rubio, L. Enrquez and M. Jaraz, Sulfur hydrogen bonding in isolated monohydrates: Furfuryl mercaptan versus furfuryl alcohol, Chem. – Eur. J., 2018, 24, 6564–6571 CrossRef CAS PubMed.
- C. M. Lovejoy and D. J. Nesbitt, Slit pulsed valve for generation of long-path-length supersonic expansions, Rev. Sci. Instrum., 1987, 58, 807–811 CrossRef CAS.
- R. Campargue, Progress in overexpanded supersonic jets and skimmed molecular beams in free-jet zones of silence, J. Phys. Chem., 1984, 88, 4466–4474 CrossRef CAS.
- J. Altnoeder, K. Krueger, D. Borodin, L. Reuter, D. Rohleder, F. Hecker, R. A. Schulz, X. T. Nguyen, H. Preiss and M. Eckhoff, et al., The Guinness molecules for the carbohydrate formula, Chem. Rec., 2014, 14, 1116–1133 CrossRef CAS PubMed.
- C. Zhu, N. F. Kleimeier, A. M. Turner, S. K. Singh, R. C. Fortenberry and R. I. Kaiser, Synthesis of methanediol [CH2(OH)2]: The simplest geminal diol, Proc. Natl. Acad. Sci. U. S. A., 2021, 119, e2111938119 CrossRef PubMed.
- H.-Y. Jian, C.-T. Yang and L.-K. Chu, Gaseous infrared spectra of the simplest germinal diol CH2(OH)2 and the isotopic analogues in the hydration of formaldehyde, Phys. Chem. Chem. Phys., 2021, 23, 14699–14705 RSC.
- J. Simons, Good vibrations: probing biomolecular structure and interactions through spectroscopy in the gas phase, Mol. Phys., 2009, 107, 2435–2458 CrossRef CAS.
- S. Bakels, M.-P. Gaigeot and A. M. Rijs, Gas-phase infrared spectroscopy of neutral peptides: Insights from the far-IR and THz domain, Chem. Rev., 2020, 120, 3233–3260 CrossRef CAS PubMed.
- T. Nakamura, M. Schmies, A. Patzer, M. Miyazaki, S.-I. Ishiuchi, M. Weiler, O. Dopfer and M. Fujii, Solvent migration in microhydrated aromatic aggregates: Ionizationinduced site switching in the 4-aminobenzonitrile-water cluster, Chem. – Eur. J., 2014, 20, 2031–2039 CrossRef CAS PubMed.
- A. R. Auty, A. C. Jones and D. Phillips, Fluorescence excitation spectra and decay times of jet-cooled dibenzofuran and the dibenzofuran-water complex, Chem. Phys. Lett., 1984, 112, 529–533 CrossRef CAS.
- G. Piani, M. Pasquini, I. López-Tocón, G. Pietraperzia, M. Becucci and E. Castellucci, The aniline-water and aniline-methanol complexes in the S1 excited state, Chem. Phys., 2006, 330, 138–145 CrossRef CAS.
- H. C. Gottschalk, T. L. Fischer, V. Meyer, R. Hildebrandt, U. Schmitt and M. A. Suhm, A sustainable slit jet FTIR spectrometer for hydrate complexes and beyond, Instruments, 2021, 5, 12 CrossRef CAS.
- Atomic weights and isotopic compositions for all elements, https://physics.nist.gov/cgi-bin/Compositions/stand_alone.pl.
- A. Ben Faleh, S. Warnke and T. R. Rizzo, Combining ultrahigh-resolution ion-mobility spectrometry with cryogenic infrared spectroscopy for the analysis of glycan mixtures, Anal. Chem., 2019, 91, 4876–4882 CrossRef CAS PubMed.
- S. Spieler, C. H. Duong, A. Kaiser, F. Duensing, K. Geistlinger, M. Fischer, N. Yang, S. S. Kumar, M. A. Johnson and R. Wester, Vibrational predissociation spectroscopy of cold protonated tryptophan with different messenger tags, J. Phys. Chem. A, 2018, 122, 8037–8046 CrossRef CAS PubMed.
- K. C. Fischer, S. L. Sherman and E. Garand, Competition between solvation and intramolecular hydrogen-bonding in microsolvated protonated glycine and β-alanine, J. Phys. Chem. A, 2020, 124, 1593–1602 CrossRef CAS PubMed.
- W. Caminati, J. C. López, S. Blanco, S. Mata and J. L. Alonso, How water links to cis and trans peptidic groups: the rotational spectrum of N-methylformamide-water, Phys. Chem. Chem. Phys., 2010, 12, 10230–10234 RSC.
- W. Caminati, L. B. Favero, P. G. Favero, A. Maris and S. Melandri, Intermolecular hydrogen bonding between water and pyrazine, Angew. Chem., Int. Ed., 1998, 37, 792–795 CrossRef CAS PubMed.
- X. Li, T. Lu, D. A. Obenchain, J. Zhang, S. Herbers, J.-U. Grabow and G. Feng, The characteristics of disulfide-centered hydrogen bonds, Angew. Chem., Int. Ed., 2021, 60, 5838–5842 CrossRef CAS PubMed.
- L. Du, S. Tang, A. S. Hansen, B. N. Frandsen, Z. Maroun and H. G. Kjaergaard, Subtle differences in the hydrogen bonding of alcohol to divalent oxygen and sulfur, Chem. Phys. Lett., 2017, 667, 146–153 CrossRef CAS.
- K. R. Leopold, Hydrated acid clusters, Annu. Rev. Phys. Chem., 2011, 62, 327–349 CrossRef CAS PubMed.
- E. G. Schnitzler, N. A. Seifert, S. Ghosh, J. Thomas, Y. Xu and W. Jäger, Hydration of the simplest α-keto acid: A rotational spectroscopic and ab initio study of the pyruvic acid-water complex, Phys. Chem. Chem. Phys., 2017, 19, 4440–4446 RSC.
- M. Reiher, Molecule-specific uncertainty quantification in quantum chemical studies, Israel J. Chem., 2021, 62, e2021001 Search PubMed.
- A. Kryshtafovych, T. Schwede, M. Topf, K. Fidelis and J. Moult, Critical assessment of methods of protein structure prediction (CASP)|Round XIII, Proteins: Struct., Funct., Bioinform., 2019, 87, 1011–1020 CrossRef CAS PubMed.
- C. R. Groom and A. M. Reilly, Sixth blind test of organic crystal-structure prediction methods, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 776–777 CrossRef CAS PubMed.
- A. Kjaersgaard, E. Vogt, A. S. Hansen and H. G. Kjaergaard, Room temperature gas-phase detection and Gibbs energies of water amine bimolecular complex formation, J. Phys. Chem. A, 2020, 124, 7113–7122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Short profiles for the 10 training set members to help contributors in the computational treatment and in the judgement of its feasibility and correctness. It also describes details on the online submission of theoretical data. See DOI: https://doi.org/10.1039/d2cp01119k |
| This journal is © the Owner Societies 2022 |