
Method selection for liquid chromatography
Analytical Methods Committee AMCTB No. 107
First published on 9th July 2021
Abstract
The accurate measurement of trace level substances in complex mixtures requires advanced instrumental techniques that generally use some form of separation prior to analysis, such as chromatography. There is an increasing trend for users that do not specialise in separation science to undertake this work. With the plethora of available liquid chromatographic techniques this can lead to the inappropriate selection of separation methods, resulting in data that is incoherent or misleading. Hence, despite the existence of comprehensive guidance for users, there remains a need to provide quick and easy to use educational tools that are applicable across the natural and physical sciences.
Societies and organisations associated with measurement (e.g., IUPAC, Eurachem, Royal Society of Chemistry Analytical Methods Committee) have published a number of comprehensive guides1–6 and there are also online commercial resources such as ChromAcademy.7 However, with the broad range of applications utilizing liquid separations, and specifically liquid chromatography (LC) with its many modes of separation, there remains a clear need for straightforward guidance. This should enable readers to understand when and, importantly, why specific modes are chosen for the required analyte and/or matrix types. This technical brief provides an overview of the decision making process for selecting LC techniques to separate known analyte(s) from a complex liquid matrix. A subsequent brief will provide guidance to assist in the optimisation of the chosen LC technique(s) during method development.

Modalities for liquid separations
Prior to analysis, separations may be undertaken as a crude preparative process or at a higher resolution using more sophisticated equipment, such as column chromatography. However, regardless of the level of equipment required, the ‘mode of action’ of the separation will abide by fundamental physicochemical principles that can be loosely described by the following categories:(1) Partition (depending on solubility e.g. hydrophilic interaction liquid chromatography (HILIC))
(2) Adsorption (depending on surface interaction e.g. reversed-phase liquid chromatography (RPLC))
(3) Porosity (depending on size/time e.g. size exclusion chromatography (SEC)).
The first practical applications of separation methods based on these categories were very simple. For example, compounds in solution could be partitioned between two immiscible liquid phases by shaking them together in a separating funnel. However, this method was highly dependent on the dissolution of the solute and the immiscibility of the selected phases to ensure separation was achieved. By replacing one phase as a solid stationary material, the immiscibility of the two phases was ensured, regardless of solvent choice, providing greater flexibility in method development of the separation. This form of liquid separation (e.g. liquid chromatography) began with simple adsorbent stationary phases such as alumina or silica but a variety of materials and separation modes have been introduced since the 1950s. Similarly to liquid–liquid separations, those using a stationary phase can also operate with common interactions such as those based on polarity (e.g. like is attracted to like) and ion exchange (e.g. opposites attract). The variety of separation modes stimulated a steady growth in the number of applications but most were still based on a traditional chromatographic approach. These typically used thin layer chromatography (TLC) silica-coated plates, or short large diameter glass columns held vertically with solvent flow relying on gravity, resulting in relatively slow and low-resolution separations. However, this growth accelerated with the development of high-performance liquid chromatography (HPLC) using a pumped mobile phase flow through relatively long, narrow bore columns to achieve enhanced separation capability. These more current techniques are abbreviated to liquid chromatography (LC) and the more common separation modes are described briefly below with their applicability summarised in Fig. 1.
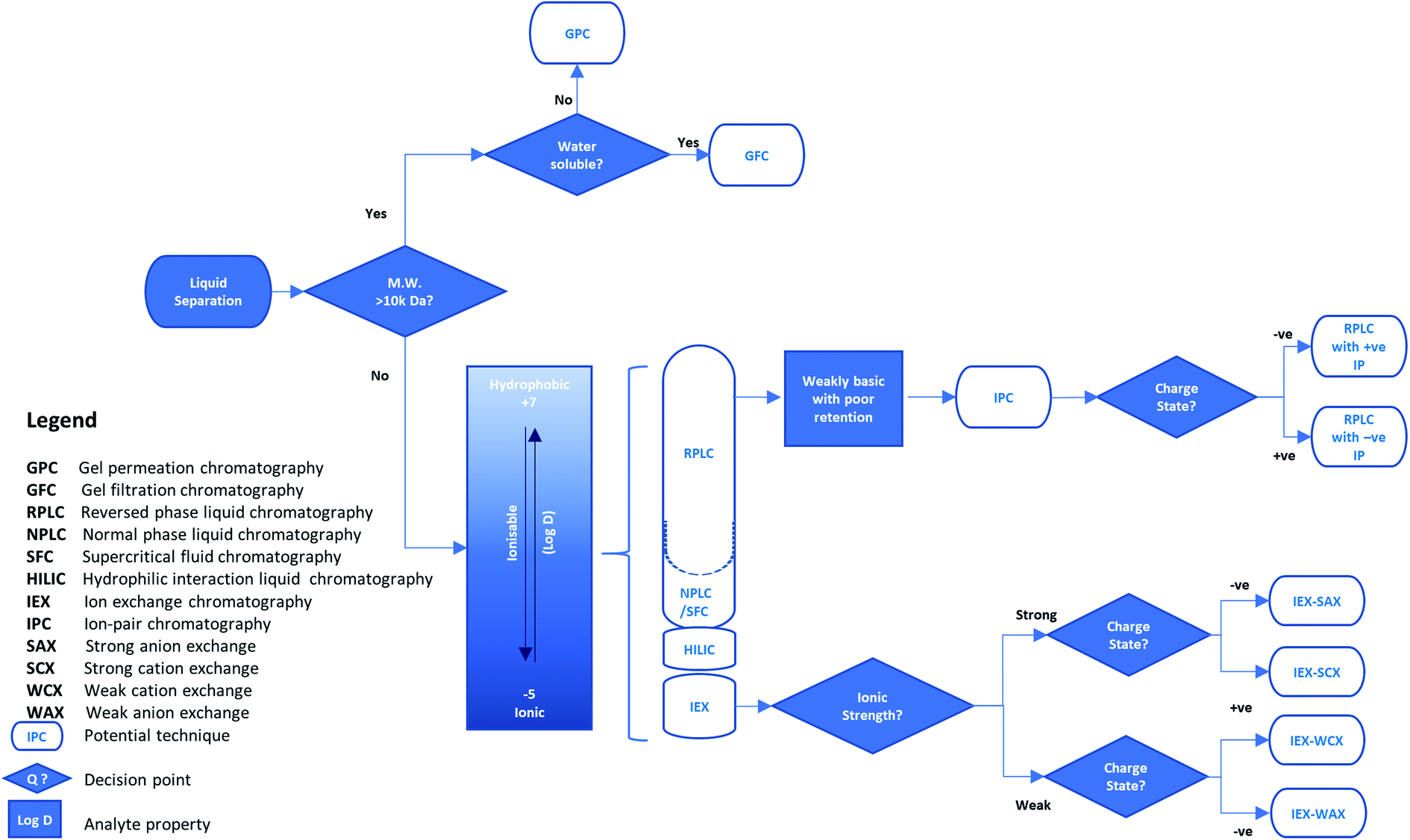 | ||
| Fig. 1 An example decision tree for selecting the liquid phase separation approach for reducing sample matrix complexity in isolating a target analyte. | ||
Normal-phase liquid chromatography (NPLC) is the classical mode of chromatography based on polarity. It uses a polar stationary phase with a non-polar (hydrophobic) mobile phase for sample loading, such as silica with hexane. The strength of interaction, and therefore retention time on column, increases with polarity. With this type of separation, retention is underpinned by a hydrophilic interaction. Hence, normal-phase liquid chromatography on silica typically requires low amounts of water from both the organic mobile phase and sample solutions loaded on the column. Therefore, this mode is unsuitable for analytes with poor solubility in organic solvents but is preferred over reversed-phase liquid chromatography for those with limited water solubility.
Reversed-phase liquid chromatography (RPLC) is also based on polarity but is the opposite of normal phase, using a polar mobile phase and a hydrophobic stationary phase. A variety of the latter are available but one of the most popular is C18-bonded silica (sometimes referred to as octadecyl-silica, ODS). Here, retention is governed by non-polar interactions, and as such, the mobile phase during sample loading is commonly an aqueous blend of water or buffer and a miscible, polar organic solvent, such as acetonitrile or methanol. This allows interaction of a wide range of analytes with non-polar functionalities and is one of the most widely used HPLC methods.
Hydrophilic interaction liquid chromatography (HILIC) is a derivative of normal-phase chromatography and uses a polar stationary phase such as silica. A small amount of water (usually <20%) is added to an organic mobile phase such as acetonitrile. As water is a very polar solvent it competes with polar analytes for adsorption on the polar stationary phase. This allows separation and elution of polar analytes which are very strongly retained using normal-phase chromatography and would be only weakly retained with a reversed-phase interaction. Therefore, analytes are eluted in order of increasing hydrophilicity relative to water.
Ion exchange chromatography (IEX) is based on electrical charge which acts using interactions where opposite charges attract and like charges repel each other. This mode of separation is split into four categories (see Fig. 1) which are distinguished by the type and strength of the acidic (anion) or basic (cation) functional groups on the sorbent. Strong ion exchangers have functional groups such as sulfonic acids or quaternary amines that are always ionized. However, they may retain weakly ionized species which can be eluted by disrupting the interaction through neutralization of the weak ion by changing the pH of the mobile phase. Conversely, stationary phases may also contain weak ion exchangers such as secondary-amine or carboxylic-acid functions, which may be neutralized above or below a certain pH value and lose their ability to retain ions by charge. Hence, the pH of the mobile phase is an important factor in developing ion exchange separations.
Ion-pair chromatography (IPC) is a version of RPLC that uses an ion-pair reagent added to the mobile phase. The ion-pair reagent is typically an alkylsulfonate, alkylsulfate or alkylammonium salt which changes the retention time of charged analytes, i.e. those which are acidic or basic, by adding an ion-exchange character to the hydrophobic column. For example, increasing the concentration of the ion-pair reagent increases the retention of an oppositely charged analyte but reduces the retention of one with a similar charge.
Separations based on size have developed in several forms since the 1950s. All depend on separating analytes according to their molecular size (effectively their molecular weight) rather than their charge or polarity. The original applications separated large biomolecules, typically water soluble analytes in an aqueous mobile phase, by passing, or filtering, them through a controlled-porosity, hydrophilic dextran polymer, and were termed gel-filtration chromatography (GFC). Subsequently, synthetic organic oligomers and polymers, typically soluble in an organic mobile phase, were separated using organic-polymer packings and referred to as gel-permeation chromatography (GPC). Finally, similar separations were done using controlled-porosity silica packings and the term size-exclusion chromatography (SEC) was introduced. However, any type of separation based on molecular size is now frequently referred to as SEC. In each case the mobile phase must be chosen to prevent any polarity or charge interactions between the analytes and the stationary phase surface as well as demonstrating good solubility for the analytes.
Supercritical fluid chromatography (SFC) is a form of chromatography that uses a supercritical fluid such as carbon dioxide under controlled pressure and temperature conditions, as the mobile phase. Various stationary phases are used including both bare and C18-modified silica. CO2 on its own is highly non-polar but co-solvents such as simple alcohols may be added to modify the mobile phase polarity. Advantages over HPLC using conventional solvents include rapid equilibration and excellent reproducibility together with the ability to easily recover and reuse the solvent. SFC is widely used in a variety of fields including bioanalysis and pharmaceutical analysis, but is particularly important for chiral separations.
Method selection
The separation mode for instrumental separation techniques is typically chosen according to two criteria: compatibility with the sample and detection system; and the ability to discriminate the target analyte(s) from each other and the remaining matrix constituents. For example, where the target analyte is known, a ‘specific’ analyte characteristic can be selected as the basis of method development. Higher resolution liquid separations, such as LC, can be applied when the isolation of a solubilised molecule in a complex liquid sample is required. In its simplest terms, if the analyte can be differentiated from other sample matrix components by orders of magnitude in size or molecular weight,† separations can be achieved using size exclusion. However, where greater selectivity is required for liquid samples, analyte solubility into, and/or adsorption onto, another phase using lipophilicity or charge etc., can be employed. The likelihood of method success may be estimated from the physicochemical properties of the analyte and sample matrix. For method development, these physicochemical properties can enable manipulation of the separation conditions to enhance the process by changing the degree of analyte interaction with the separation phase. Therefore, knowing the analyte hydrophobicity or ionisability (represented by the distribution coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D)) enables the user to select a more appropriate separation approach from five potential chromatographic modes (see Fig. 1). The chosen separation can be optimised through adjustment of the mobile phase pH or counter ion concentration (polarity). This optimisation is underpinned by established principles of acid–base chemistry,8 where acidic and basic species are considered as proton donors and acceptors, respectively. Using these principles, adjustments in pH can be used to neutralise or ionise weakly acidic or weakly basic analytes, and increase their interaction with organic (hydrophobic) or ionic separation phases, respectively.
D)) enables the user to select a more appropriate separation approach from five potential chromatographic modes (see Fig. 1). The chosen separation can be optimised through adjustment of the mobile phase pH or counter ion concentration (polarity). This optimisation is underpinned by established principles of acid–base chemistry,8 where acidic and basic species are considered as proton donors and acceptors, respectively. Using these principles, adjustments in pH can be used to neutralise or ionise weakly acidic or weakly basic analytes, and increase their interaction with organic (hydrophobic) or ionic separation phases, respectively.
However, to achieve this outcome, the user must select an appropriate pH, and this requires an understanding of the acid dissociation (strength) constant of the analyte, Ka, as defined by eqn (1).
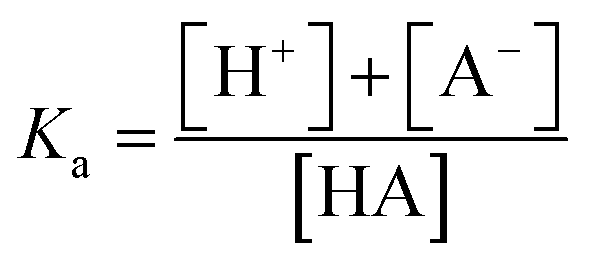 | (1) |
| pKa = −log10Ka | (2) |
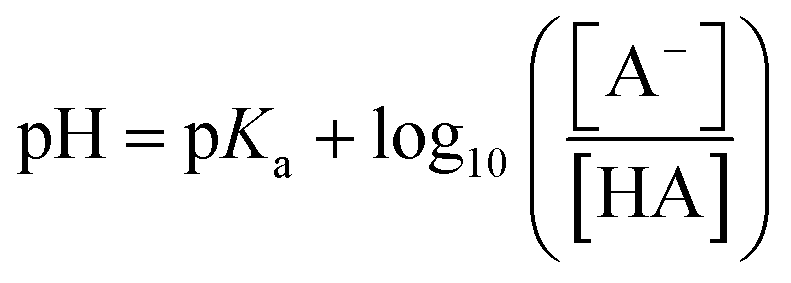 | (3) |
To ensure 99% of the analyte is converted to this charge state, a pH that exceeds the pKa by 2 units is required. Therefore, to facilitate hydrophobic (non-polar) interactions by neutralising an acidic or basic analyte, a pH that is 2 units below or above the analyte pKa, respectively, is used. Whilst, for ionic interactions where the acidic or basic analyte is charged during the separation, a pH of 2 units above or below the analyte pKa, respectively, is applied. Furthermore, by combining these physiochemical properties as complementary chromatographic interactions (e.g. ion-pair or multi-dimensional chromatography), the selectivity of the separation can be enhanced enabling the separation and analysis of a greater number of components in one attempt. Therefore, knowing as much as possible about the target analyte(s) and sample matrices is critical to the success of the separation and the value of the analysis.
Concluding remarks
The expanded range of modalities now available for LC separations has enabled a more tailored approach to method development, and facilitates an ‘untangling’ of complex mixtures to measure the analyte(s) in question without significant interference. However, with the wide choice of approaches there is also a considerable challenge in ensuring methods are applied appropriately to provide meaningful data. This requires, for example, avoiding unnecessarily convoluted or expensive protocols/techniques whilst maintaining the robustness of the approach. With increasing accessibility of LC techniques, and the number of users with limited knowledge of the topic, there remains a need for more informed use of these approaches particularly when, and why, they are used in combination. This Technical Brief is aimed at providing some assistance in selecting these approaches to develop methods for known target analytes. A subsequent brief will provide guidance to assist in the optimisation of the chosen LC technique(s) during method development.Ruth Godfrey (Swansea University), Scott Fletcher and Robert Boughtflower (RSC Separation Sciences Group (SSG)), and Mike Sargent (Royal Society of Chemistry Analytical Methods Committee, AMC).
This Technical Brief was prepared for the AMC, with contributions from members of the AMC Instrumental Analysis Expert Working Group and the SSG, and approved by the AMC on 5 May 2021.

Further reading
- International Union of Pure and Applied Chemistry (IUPAC), Terminology of separation methods (IUPAC Recommendations 2017), Pure Appl. Chem., 2018, 90(1), 181–231 CrossRef
.
-
World Health Organization, Good chromatography practices, Working document QAS/19.791/Rev.1, July 2019 Search PubMed
-
Guide to achieving reliable quantitative LC-MS measurements, ed. M. Sargent, RSC Analytical Methods Committee, 2013 Search PubMed
-
Eurachem, The fitness for purpose of analytical methods: A laboratory guide to method validation and related topics, 2nd edn, 2014 Search PubMed
-
European Medicines Agency, ICH guideline M10 on bioanalytical method validation: Draft version, March 2019 Search PubMed
-
Food and Drug Administration, Bioanalytical Method Validation: Guidance for industry, May 2018 Search PubMed
- www.chromacademy.com/index.html .
-
J. McMurry, Organic Chemistry, Student Edition, Cengage Learning, 2015 Search PubMed
Footnote |
| † Strictly speaking the analyte hydrodynamic volume, which is the sum of the time-average of the molecular volume and the volume of the solvent molecules associated with it. |
| This journal is © The Royal Society of Chemistry 2021 |