
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA LiPO2F2/LiPF6 dual-salt electrolyte enabled stable cycling performance of nickel-rich lithium ion batteries
Lili Liu†
ab,
Shijie Gu†c,
Shili Wangab,
Xiuyun Zhang d and
Shimou Chen*ef
d and
Shimou Chen*ef
aDepartment of Chemistry, School of Science, Beijing Technology and Business University, Beijing, 100048, China
bBeijing Key Laboratory of Plant Resource Research and Development, Beijing Technology and Business University, Beijing, 100048, China
cSchool of Materials Science and Engineering, Hebei University of Technology, Tianjin, 300401, China
dCollege of Physics Science and Technology, Yangzhou University, Yangzhou, Jiangsu 225002, China
eBeijing Key Laboratory of Ionic Liquids Clean Process, CAS Key Laboratory of Green Process and Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing, 100190, China. E-mail: chenshimou@ipe.ac.cn
fMaterials Center, Beijing Institute of Collaborative Innovation, Beijing 100081, China
First published on 9th January 2020
Abstract
In this work, a dual-lithium salt was proposed for constructing an electrolyte for high energy density lithium ion batteries. LiPO2F2 was composed with traditional LiPF6 to enhance the high voltage performance of the electrolyte. The electrochemical performance of the NCM811/Li cells with LiPO2F2/LiPF6 dual-lithium salt at 2.8–4.5 V was investigated. It was found that the dual-lithium salt can inhibit the oxidative decomposition of the electrolyte, suppress the dissolution of the transition metal ions in the electrode material, and reduce the side reaction between the transition metal ions and the electrolyte. We believe that the strategy of a dual-lithium salt electrolyte may provide a new idea to stabilize the electrode/electrolyte interface at high voltage, which is very important for developing high energy density batteries.
1. Introduction
In the past decades, lithium-ion batteries (LIBS) have made remarkable progress in the fields of 3C portable devices, aerospace, UAV and electric vehicles.1–3 With the environmental pollution caused by fossil energy becoming more and more serious, electric vehicles are more appealing and are being developed more and more rapidly.4,5 However, poor battery life limits their development.It is well known that the energy density of cathode materials is the main factor affecting the performance of batteries. In recent years, new ternary nickel–cobalt–manganese cathode materials have attracted wide attention of researchers.6 A material with a nickel content of more than 60% is called a nickel-rich material. With the increase of nickel content, the thermal stability and high voltage stability of the cathode will decrease. The dissolution of transition metals will have side effects with the electrolyte, consume the electrolyte and reduce the cycling stability of the cell.7 In order to solve these problems, surface coating and electrolyte modification are the most commonly used methods. But the latter is more convenient and effective. The simplest way to modify the electrolyte is to add other components, which are mainly divided into two kinds: dual-lithium salts and electrolyte additives.8–12
On the one hand, researchers are devoted to the study of electrolyte additives. Wang et al. reported that trimethyl borate was used as an additive in electrolytes to improve the cycling stability of NCM333/Li batteries.13 It was found that the capacity retention rate increased from 48% to 89% after 200 cycles by adding trimethyl borate into the base electrolyte, and the discharge specific capacity increased from 78 mA h g−1 to 102 mA h g−1 at high rate of 6C. Some researchers have proposed that the interface between the cathode and electrolyte has a similar structure to that between the anode and electrolyte. They named this interface the solid electrolyte interface (SEI). This interface can transport lithium ions but prevent electrons from passing through. Dong et al. studied the problem of poor stability of the interface between an NCM523 cathode and electrolyte by using p-toluenesulfonyl isocyanate as a film-forming additive.14 The improvement in cycling performance of the cell is attributed to the decomposition of –S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in PTSI, which inhibits the contact between lithium salt and water in the electrolyte, reduces the generation of HF and the formation of LiF. On the other hand, researchers are also working on the research of dual-lithium salts. Zhao et al. combined LiBOB with LiPF6 to improve the long-term cycling performance, rate performance and voltage stability of lithium (Li) metal batteries with a LiNi0.76Mn0.14Co0.10O2 cathode.15 The electrolyte not only helps to form a denser SEI layer on the surface of lithium metal, but also forms an enhanced cathode electrolyte interface layer, which prevents the corrosion of the cathode interface and reduces the formation of a disordered phase after cycling. Li et al. studied the effects of imine lithium and dual-lithium salt electrolytes with different salt chemistries in carbonate solvents on the cycling stability of lithium metal batteries.16 The stability order of common lithium salts is as follows: LiTFSI–LiBOB > LiTFSI–LiDFOB > LiFSI–LiDFOB > LiPF6 > LiFSI–LiBOB. This provides a new way to improve the stability of electrolyte by using dual-lithium salts.
O in PTSI, which inhibits the contact between lithium salt and water in the electrolyte, reduces the generation of HF and the formation of LiF. On the other hand, researchers are also working on the research of dual-lithium salts. Zhao et al. combined LiBOB with LiPF6 to improve the long-term cycling performance, rate performance and voltage stability of lithium (Li) metal batteries with a LiNi0.76Mn0.14Co0.10O2 cathode.15 The electrolyte not only helps to form a denser SEI layer on the surface of lithium metal, but also forms an enhanced cathode electrolyte interface layer, which prevents the corrosion of the cathode interface and reduces the formation of a disordered phase after cycling. Li et al. studied the effects of imine lithium and dual-lithium salt electrolytes with different salt chemistries in carbonate solvents on the cycling stability of lithium metal batteries.16 The stability order of common lithium salts is as follows: LiTFSI–LiBOB > LiTFSI–LiDFOB > LiFSI–LiDFOB > LiPF6 > LiFSI–LiBOB. This provides a new way to improve the stability of electrolyte by using dual-lithium salts.
In our work, LiPO2F2 and LiPF6 with different molar ratios were combined to improve the high voltage stability of nickel-rich NCM811/Li cells. The stability of cells is evaluated by constant current charging and discharging. The electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV) were used to study the redox stability of electrolyte. Scanning electron microscopy (SEM) was used to study their electrochemical behaviour, morphology of electrodes and stability of electrolytes. It was found that the decomposition products of dual-lithium salts electrolyte were less and the structure of electrodes remained intact.
2. Experimental
2.1 Preparation of electrode and electrolyte
A mixture of ethylene carbonate (EC)/ethyl methyl carbonate (EMC)/diethyl carbonate (DEC) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 in wt ratio) containing 1 M LiPF6 was used as the base electrolyte (base). The LiPO2F2 (LiDFP) dissolves in dimethoxymethane (DME). The base electrolyte was then diluted to 0.95 M, 0.9 M, 0.85 M, 0.8 M, 0.7 M, and add LiDFP (in DME) in the base solvent. Finally, they are mixed to control the concentration of Li+ to 1 M named dual-lithium salts.
:1:1 in wt ratio) containing 1 M LiPF6 was used as the base electrolyte (base). The LiPO2F2 (LiDFP) dissolves in dimethoxymethane (DME). The base electrolyte was then diluted to 0.95 M, 0.9 M, 0.85 M, 0.8 M, 0.7 M, and add LiDFP (in DME) in the base solvent. Finally, they are mixed to control the concentration of Li+ to 1 M named dual-lithium salts.
The LiNi0.8Co0.1Mn0.1O2 powder and Super P (conductive agent) were evenly mixed in an agate mortar. The NCM811 cathodes were then mixed 80 wt% NCM811, 10 wt% polyvinylidene fluoride (PVDF), and 10 wt% Super P binder in n-methyl-2-pyrrolidone to make slurry. The slurry was coated on an Al foil using doctor blading technique. Finally, the coated electrode was vacuum-dried at 90 °C for 10 h to remove the residual water, and then followed by roll-pressing by using a pressing machine to enhance the adhesion and electrical contact between LiNi0.8Co0.1Mn0.1O2, Super P (conductive carbon), and PVDF (binder) on the aluminum current collector. The composite electrode was assembled into CR2025 half-cells in an Ar-filled glove box (Ar, 99.999% purity). Polypropylene (Celgard 2400) was utilized as a separator. Li metal foil (99.9% purity, Alfa Aesar.) was used both as a counter and a reference electrode.
2.2 Electrochemical measurements
The charging and discharging tests of cells at constant current of 0.3C (1C = 200 mA h g−1) were carried out using LAND (CT2001A, China) testing system. Linear sweep voltammetry test (LSV, 0–6 V, 0.1 mV s−1), the cyclic voltammetry (CV, 2.8–4.6 V, 0.1 mV s−1), and the electrochemical impedance spectroscopy (EIS) measurements via Metrohm Auto lab M204.2.3 Physical characterization
Viscosity and conductivity were measured at room temperature by viscometer (DMA5000M-Lovis2000ME) and conductivity meter (FE38), respectively. The morphology and interface composition of the electrodes fresh and after cycling were characterized by XPS and SEM (Hitachi SU8020, Japan). Samples were rinsed 3–5 times with anhydrous DMC to remove excess components. The Escalab 250xi system of high voltage Al-Kα radiation = 1486.6 eV is used as X-ray source. The C1s (284.8 eV) peak of graphite (C–C) is used for final correction.3. Results and discussion
In lithium-ion batteries, the conductivity of electrolyte is reflected in Li+ transport. It is generally believed that when the conductivity of the electrolyte reaches about 10 mS cm−1 at room temperature, the batteries can maintain good cycling stability and rate performance.17Fig. 1 shows the effect of different concentrations of LiDFP on the conductivity and viscosity of the electrolyte. When the concentration of LiDFP is lower than 0.2 mol L−1, the conductivity increases with the increase of the concentration of LiDFP, but decreases when the concentration of LiDFP is higher than 0.2 mol L−1 (Fig. 1a). However, the conductivity of the electrolyte with LiDFP is higher than that of the base electrolyte in the experimental concentration range. In particular, when the concentration of LiDFP is 0.2 mol L−1, the conductivity reaches 11.61 mS cm−1. Similarly, the electrolyte viscosity decreases with the increase of the concentration of LiDFP (Fig. 1b), which provides a good interfacial wettability between the electrolyte and the separator.18
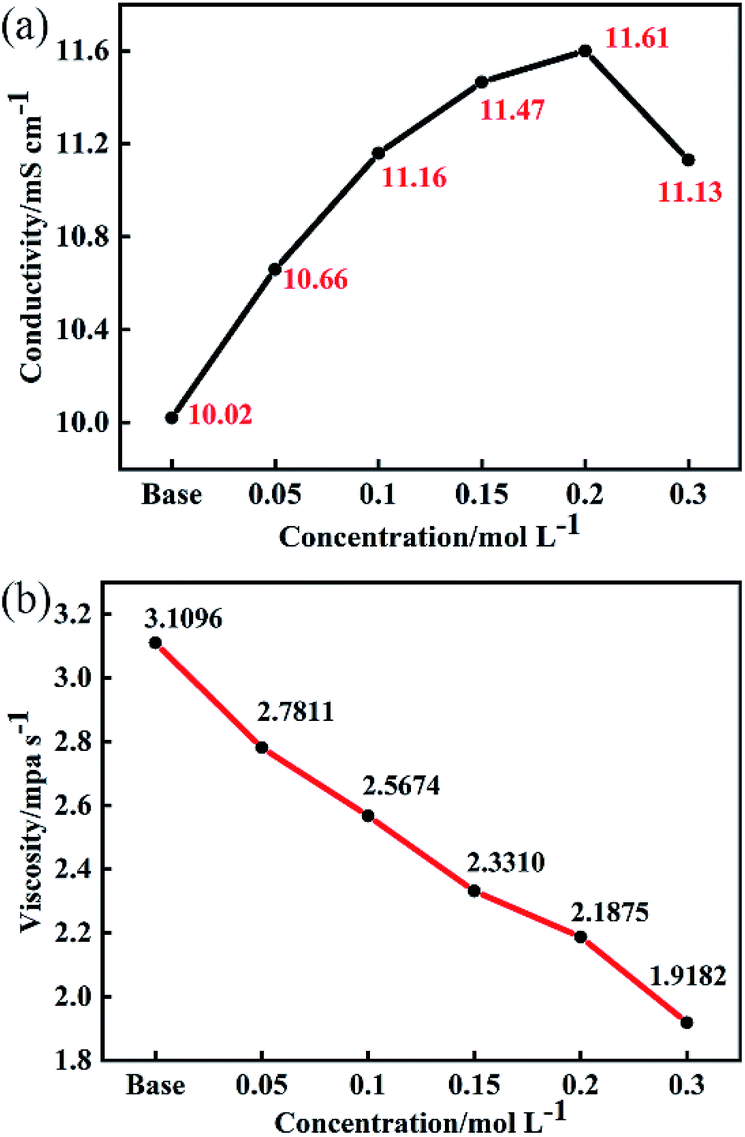 | ||
| Fig. 1 Effect of different concentrations of LiDFP on conductivity (a) and viscosity (b) of electrolyte. | ||
To investigate the electrochemical performance of the electrolyte, LSV and CV were tested by using Auto lab. The working electrode is made of stainless-steel electrode, and the counter electrode and the reference electrode are lithium foils. As shown in Fig. 2a, the base electrolyte decomposes at about 4.24 V, and the electrolyte is decomposed at about 4.47 V after adding 0.2 mol L−1 of LiDFP, indicating that LiDFP can increase the electrochemical window and suppress the decomposition of the electrolyte. The initial cyclic voltammetry curve in Fig. 2b shows that the oxidation peak of the cell moves towards low potential in the dual-lithium salts electrolyte, which can be attribute to the decomposition of LiDFP reduces the decomposition of the electrolyte and the polarization of the interface between the electrolyte and the electrode.19,20
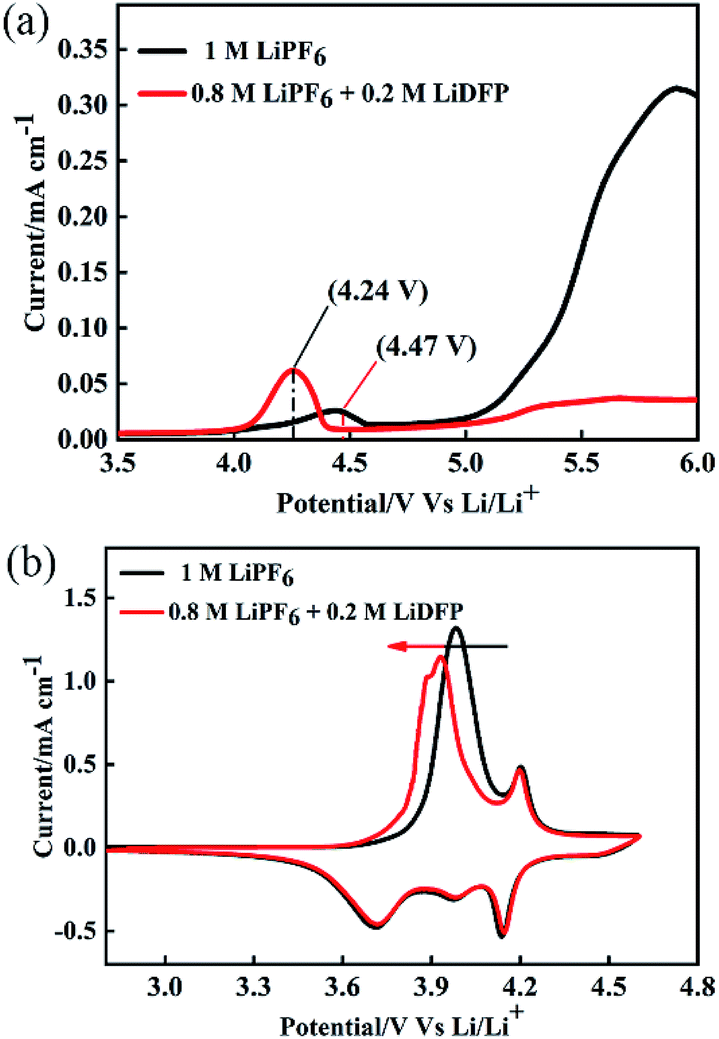 | ||
| Fig. 2 Electrochemical window (a) and redox stability (b) of different electrolytes. | ||
The cycling stability of the half-cells with the different concentrations LiDFP electrolyte was evaluated at a charge–discharge voltage of 2.8–4.5 V (Fig. 3). Based on the above analysis, the discharge capacity of the cell containing 0.2 M LiDFP was 198.5 mA h g−1, and that of the base electrolyte was only 108.1 mA h g−1 after 130 cycles. Then the capacity retention rate of the cell with 0.2 M LiDFP was as high as 92.1%, and that of the cell with base electrolyte was only 53.1%. The severe capacity degradation of the cell with base electrolyte can be mainly caused by serious decomposition of the electrolyte at high voltage and the structural damage of electrode. However, the cycling stability of the cell with LiDFP can be attributed to the fact that the LiDFP can suppress the decomposition of the electrolyte and form a stable interface to protect the stability of the structure of electrode.
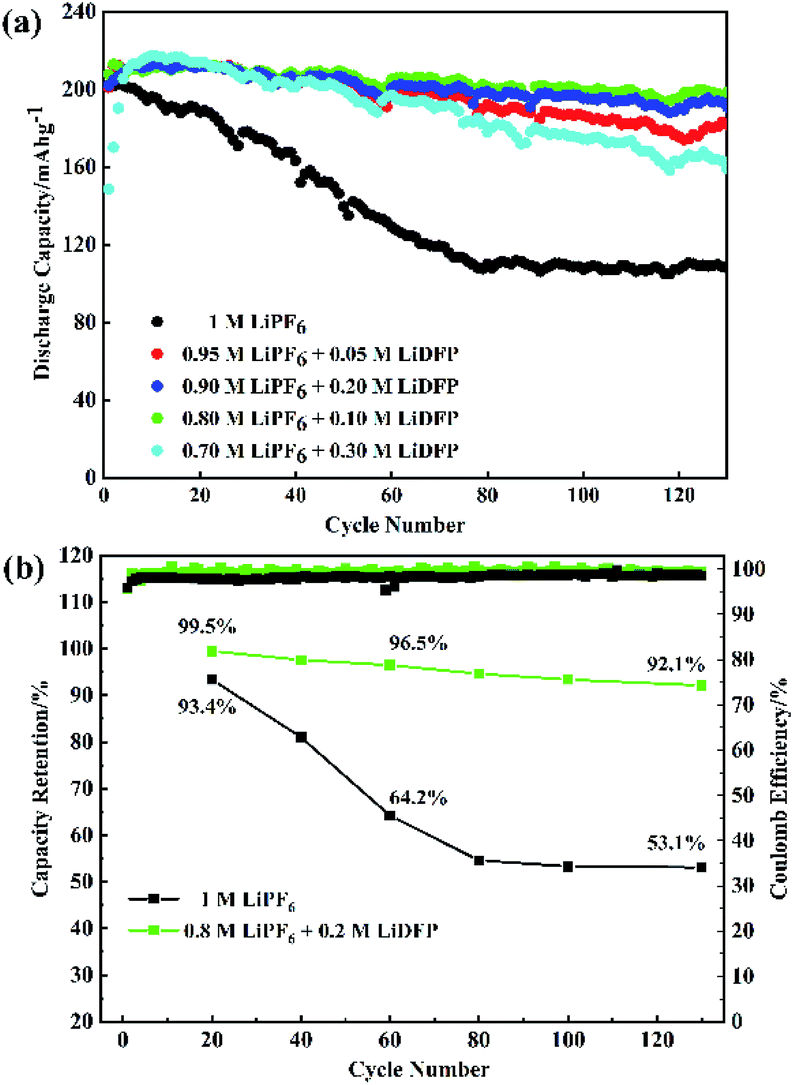 | ||
| Fig. 3 The cycling performance of NCM811/Li cells with different electrolytes. | ||
The rate performance of NCM811/Li cells with different electrolytes were tested at 0.1C, 0.5C, 1C, 3C, 5C, 10C. As shown in Fig. 4a, when the cell with base electrolyte is charged-discharged at low current, the discharge specific capacity is large. However, when using high current (more than 5C) to charge, the discharge specific capacity is reduced and the discharge voltage of the cell is seriously reduced. The increase of polarization voltage of the cell with base electrolyte under high current indicates that the increase of irreversible capacity, which leads to the serious decrease of discharge specific capacity. On the contrary, although the cell with dual-lithium salts electrolyte has a slightly lower coulomb efficiency, the discharge specific capacity is larger at high rate above 5C (Fig. 4b). This can be attributed to the decrease of polarization voltage of the cell with dual-lithium salts electrolyte, which indicates that the reversible capacity of the cell increases with a high capacity retention rate.
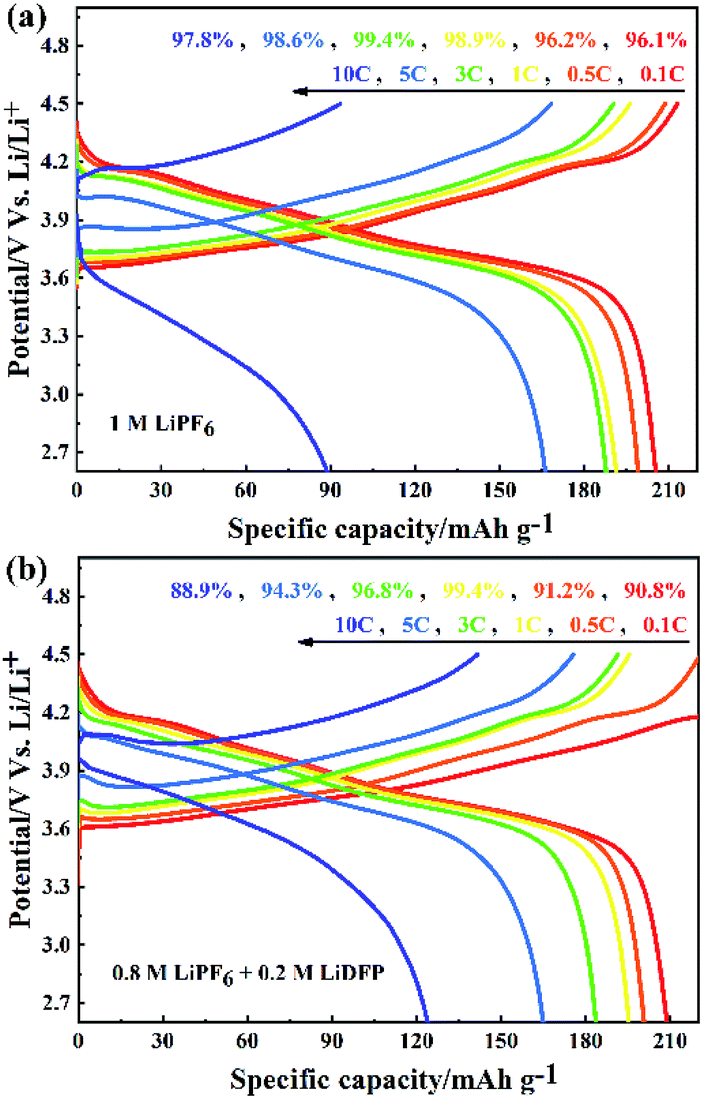 | ||
| Fig. 4 The rate performance of NCM811/Li cells with (a) the blank electrolyte containing 1 M LiPF6; and (b) the dual-salt electrolyte containing 0.8 M LiPF6 + 0.2 M LiDFP. | ||
The electrochemical impedance spectra of the cells with different number of cycles were also tested (Fig. 5). The high-frequency semicircle indicates the interface impedance, the mid-frequency semicircle stands for the charge transfer impedance, and the low-frequency region is the Li+ diffusion impedance.21–23 Different electrochemical impedance spectra reflect different variations within the cells. After 3 cycles, the interface impedance of the dual-lithium salts electrolyte system is very large, which can be attributed to the initial decomposition of LiDFP increases the inorganic component of the interface. With the progress of charging-discharging, the Li+ diffusion resistance changes in the different electrolytes. After 20 and 40 cycles, the Li+ diffusion resistance of the dual-lithium salts system decreases significantly, that is, the slope of the line increases, indicating that the Li+ diffusion kinetics is accelerated. This may be the reason for the increase of the specific discharge capacity of the dual-lithium salts system. After 70 cycles, the impedances of the two electrolyte systems were similar, and both decreased compared to the impedances at initial stage of discharge, indicating that the cathode-electrolyte interface of two systems forms a stable CEI film. The presence of this film avoids the continuous contact of the electrode material with the electrolyte and reduces the side reactions, which can be effectively enhance the cycling performance of the cells.
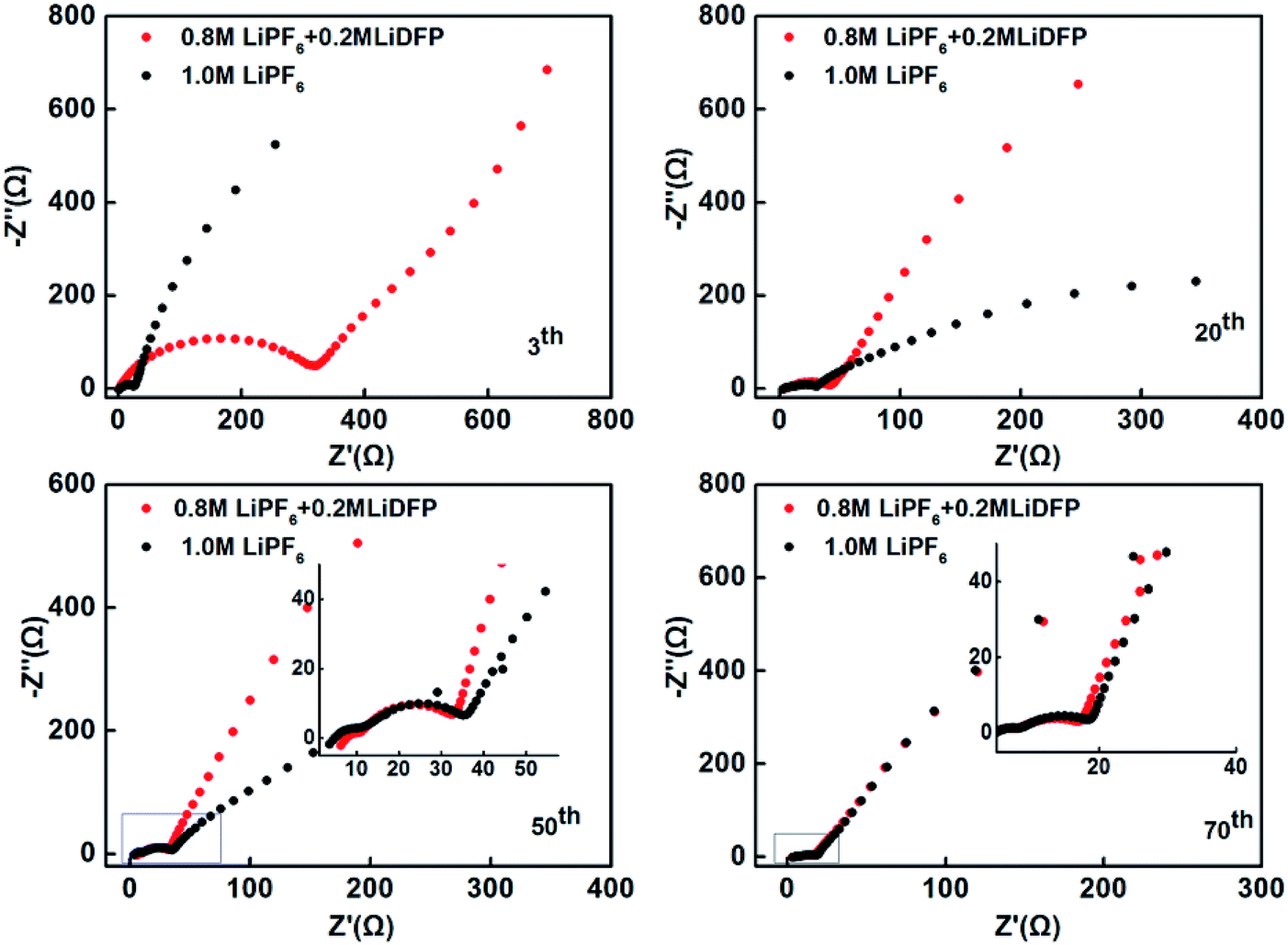 | ||
| Fig. 5 The electrochemical impedance spectra of the cells after 3, 20, 50, 70 cycles. | ||
Fig. 6 is the XPS spectrum of the surface of cathodes after 120 cycles in base and dual-lithium salts electrolytes. For C1s, on the two cathode surfaces, the peak at 286.4 eV comes from the C–O and the CO bond, respectively.24 These peaks are substances such as raw ROCO2Li, ROLi and Li2CO3, which are produced by decomposition of the electrolyte solvent.25,26 In addition, the peak at 291.9 eV corresponds to the C–F bond of the PVDF adhesive.27 In the dual-lithium salts system, the decrease of carbonate component on the surface indicates that the dual-lithium salts inhibit decomposition of the electrolyte. The peak at 532.5 eV in the O1s spectrum can be detected on both cathodes, indicating that both electrolytes were decomposed and involved in the formation of the CEI film. The C–F (687.9 eV) peak in the F1s spectrum is the PVDF component in the cathode. The peak of LiF in the dual-lithium salts system is obviously stronger than that in base electrolyte system, indicating that the decomposition of LiDFP produces LiF, which also proves that the impedance of the dual-lithium salts interface is large in the impedance spectrum. On the P2p spectrum, LixPFy is added to the surface of the dual-lithium salts system.28 These are the decomposition products of LiDFP. The intermediate decomposition product LixPOyFz indicates that both electrolytes decompose under high voltage. Although the interface inorganic component increases due to the increase of interface inorganic components, the interfacial impedance of the dual-lithium salts decreases in the later stage of discharge, indicating that the organic component is formed on the surface of the inorganic component as the progress of charge–discharge.29 These microscopic changes provide a theoretical basis for improving the cycling stability of the cell.
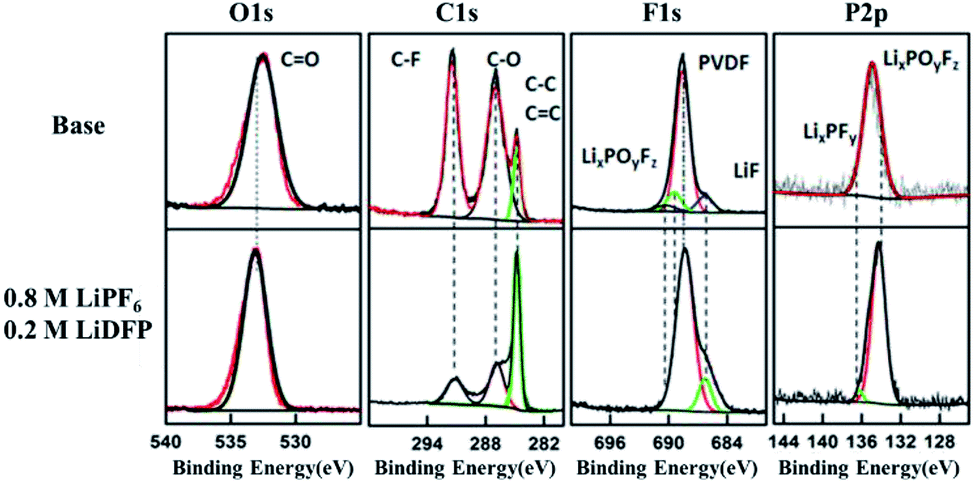 | ||
| Fig. 6 C1s, O1s, F1s, P2p XPS spectra of cathode surface in base and dual-lithium salts electrolytes. | ||
Under high voltage, the structure of NCM811 material will be destroyed, resulting in deterioration of the cycle stability of the battery. By analyzing the transition metal deposited on the cathode, the degree of material damage can be inferred. The contents of Ni, Co and Mn were analysed by ICP-AES, as shown in Fig. 7. It is found that the concentration of transition metal ions in the dual-lithium salts system is lower than that in the base electrolyte system, which proves that the structure of the material remains intact, and also shows that the dual-lithium salts has a protective effect on the electrode material.
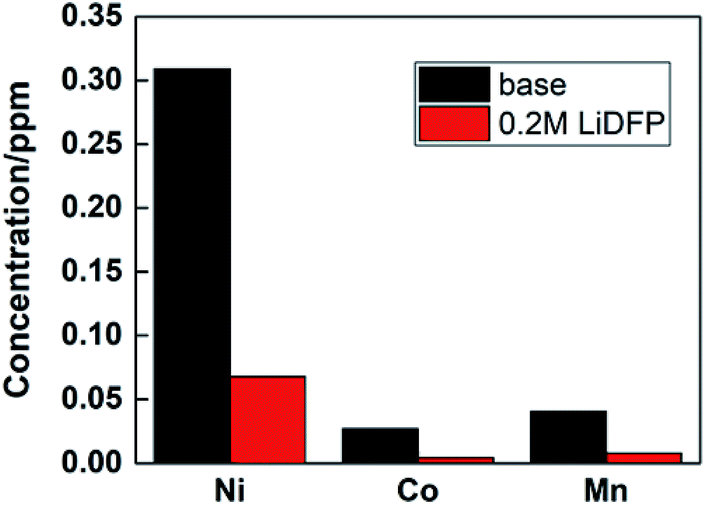 | ||
| Fig. 7 Analysis of dissolution of transition metal ions in electrolyte by ICP-AES. | ||
Fig. 8 shows the results of EDS analysis of SEM and surface elements of the electrodes after 130 cycles in different electrolytes. It can be clearly observed that the fresh cathode electrode material has a regular spherical structure (Fig. 8a), and the surface is clean and free of deposits. However, the surface of the material after cycling in the base electrolyte (Fig. 8c) is covered with a thick layer of component, which is usually caused by decomposition of the electrolyte. The surface of the electrode after cycling in the dual-lithium salts electrolyte (Fig. 8b) is relatively clean and the structure remains intact. From the above results, the addition of LiDFP inhibits the decomposition of the electrolyte and reduces the diffusion resistance of Li+. EDS analysed the elemental electrolyte (Fig. 8d) and the elements on the surface of the electrode after cycling in the dual-lithium salts. It was found that the addition of LiDFP increased the content of surface F and P elements. LiDFP has been integrated into the electrode–electrolyte interface to increase the inorganic component content. In addition, it can be seen that the content of Al decreased from 1.27 wt% to 0.41 wt% after the addition of LiDFP, indicating that LiDFP can inhibit the corrosion of the current collector by the acidic component in the electrolyte.
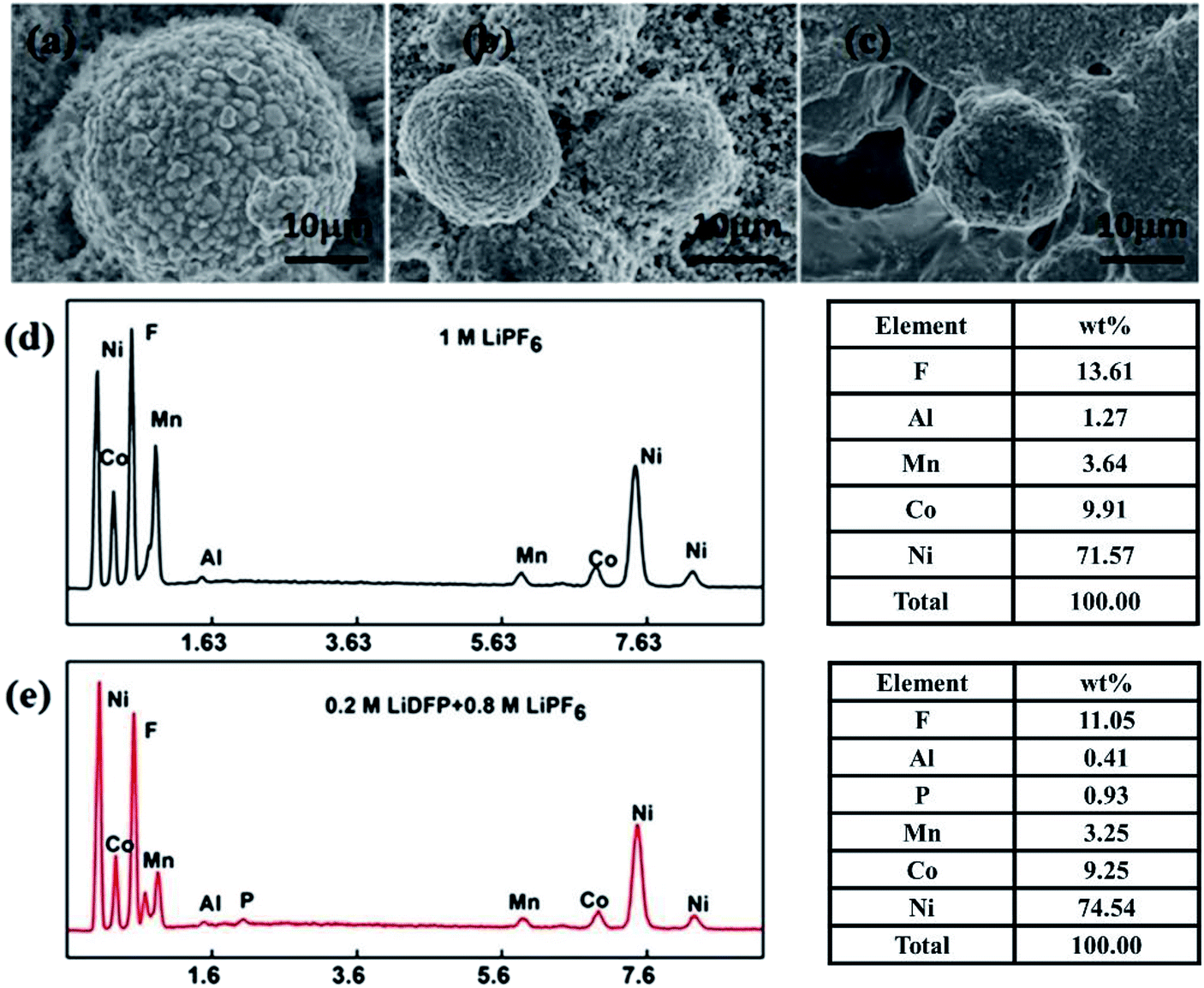 | ||
| Fig. 8 SEM images of the NCM 811 electrode: fresh (a) and after 130 cycles in (b) the dual-salt electrolyte containing 0.8 M LiPF6 + 0.2 M LiDFP, (c) the blank electrolyte containing 1 M LiPF6. The corresponding EDS analysis of surface element distribution of the NCM 811 electrode after 130 cycles in blank electrolyte (d) and dual-salt electrolyte (e). | ||
The overcharge protection performance of NCM811/Li cells with different electrolytes was further investigated by charging the cells to 5 V at the rate of 0.2C. As shown in Fig. 9, the curves of the two cells are similar before charging to 4.7 V. But after charging to 4.7 V, in the cells with 1 M LiPF6, the charge voltage rises sharply to 5 V, while the cell with the dual-lithium salts rises relatively more slowly. The time of the cell charging to 5 V are 7.2 h and 28.8 h, respectively. This shows that the combination of LiPF6 and LiDFP can enhance the overcharge protection performance, which provides a basis for improving the stability of the electrolyte and the safety performance of the battery.
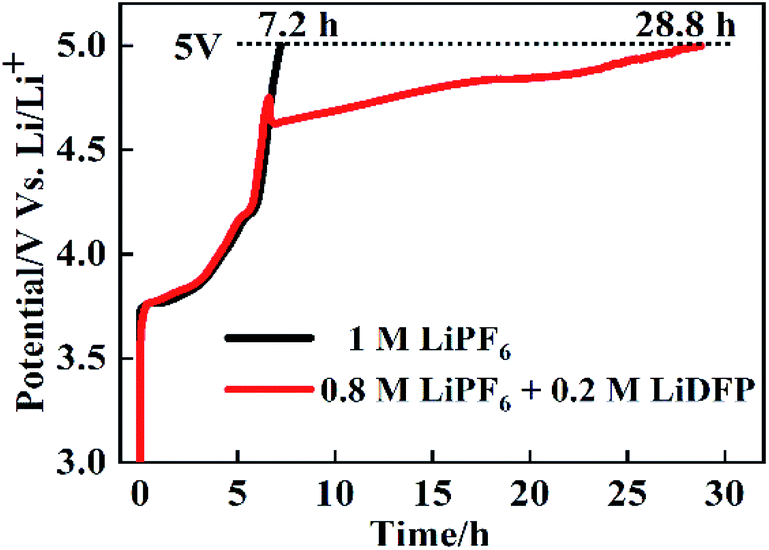 | ||
| Fig. 9 The overcharge protection performance of NCM811/Li cells with different electrolytes. | ||
4. Conclusion
The combination of LiPF6 and LiDFP can reduce the viscosity of the electrolyte and improve the ionic conductivity of the electrolyte. At the same time, it can improve the cycling stability of the NCM811/Li cells at high voltage (4.5 V). It was found that the capacity retention rate of the cell with LiPO2F2/LiPF6 dual-lithium salts was as high as 92.1% after 130 cycles, which is much higher than that in the base electrolyte (only containing LiPF6). Electrochemical impedance spectroscopy indicated that the interfacial impedance of the cells with dual-lithium salts was large at the initial stage of discharge, and the interface impedance decreased at the end of the discharge. The CV test found that the cell with dual-lithium salts electrolyte cell has a lower polarization. XPS and SEM analysis found that the LiPO2F2 lithium salt can reduce the decomposition of the electrolyte. ICP-AES shows that the LiPO2F2 can be decomposed on the surface of the electrode to stabilize the structure of the electrode material. Our results show that the strategy of dual-lithium salts provides a new idea for developing high performance electrolyte of lithium ion batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 51922099 and 21503006) and the Research Foundation of Training Plan for Excellent Young Scholars of Beijing Technology and Business University.References
- Y. M. Lee, Y. G. Lee, Y. M. Kang and K. Y. Cho, Electrochem. Solid-State Lett., 2010, 13, A55 CrossRef CAS.
- A. Kraytsberg and Y. Ein-Eli, Adv. Energy Mater., 2012, 2, 922 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303 CrossRef CAS PubMed.
- A. M. Haregewoin, A. S. Wotango and B. J. Hwang, Energy Environ. Sci., 2016, 9, 1955 RSC.
- L. Wang, B. Chen, J. Ma, G. Cui and L. Chen, Chem. Soc. Rev., 2018, 47, 6505 RSC.
- N. S. Choi, J. G. Han, S. Y. Ha, I. Park and C. K. Back, RSC Adv., 2015, 5, 2732 RSC.
- H. Kuriyama, H. Saruwatari, H. Satake, A. Shima, F. Uesugi, H. Tanaka and T. Ushirogouchi, J. Power Sources, 2015, 275, 99 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503 CrossRef CAS PubMed.
- G. D. Chen, J. An, Y. M. Meng, C. Z. Yuan, B. Matthews, F. Dou, L. Y. Shi, Y. F. Zhou, P. G. Song, G. Wu and D. S. Zhang, Nano Energy, 2019, 57, 157 CrossRef CAS.
- T. Chen, F. Wang, X. Li, X. X. Yan, H. Wang, B. W. Deng, Z. W. Xie and M. Z. Qu, Appl. Surf. Sci., 2019, 465, 863 CrossRef CAS.
- J. An, L. Y. Shi, G. R. Chen, M. S. Li, H. J. Liu, S. Yuan, S. M. Chen and D. S. Zhang, J. Mater. Chem. A, 2017, 5, 19738 RSC.
- Y. Y. Liu, Z. Yang, J. L. Li, B. B. Niu, K. Yang and F. Y. Kang, J. Mater. Chem. A, 2018, 6, 13883 RSC.
- Z. Wang, L. Xing, J. Li, B. Li, M. Xu, Y. Liao and W. Li, Electrochim. Acta, 2015, 184, 40 CrossRef CAS.
- P. Dong, D. Wang, Y. Yao, X. Li, Y. Zhang, J. Ru and T. Ren, J. Power Sources, 2017, 344, 111 CrossRef CAS.
- W. Zhao, L. Zou, J. Zheng, H. Jia, J. Song, M. H. Engelhard, C. Wang, W. Xu, Y. Yang and J. G. Zhang, ChemSusChem, 2018, 11, 2211 CrossRef CAS PubMed.
- X. Li, J. Zheng, M. H. Engelhard, D. Mei, Q. Li, S. Jiao, N. Liu, W. Zhao, J. G. Zhang and W. Xu, ACS Appl. Mater. Interfaces, 2018, 10, 2469 CrossRef CAS PubMed.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190 CrossRef CAS PubMed.
- J. Shi, Y. Xia, Z. Yuan, H. Hu, X. Li, H. Jiang, H. Zhang and Z. Liu, J. Mater. Chem. A, 2015, 3, 7006 RSC.
- H. Zhang, C. Li, M. Piszcz, E. Coya, T. Rojo, L. M. Rodriguez-Martinez, M. Armand and Z. Zhou, Chem. Soc. Rev., 2017, 46, 797 RSC.
- B. Zhao, R. Ran, M. Liu and Z. Shao, Mater. Sci. Eng., R, 2015, 98, 1 CrossRef.
- A. Funabiki, M. Inaba and Z. Ogumi, J. Power Sources, 1997, 68, 227 CrossRef CAS.
- M. D. Levi, G. Salitra, B. Markovsky, H. Teller, D. Aurbach, U. Heider and L. Heider, J. Electrochem. Soc., 1999, 146, 1279 CrossRef CAS.
- D. Lu, W. Li, X. Zuo, Z. Yuan and Q. Huang, J. Phys. Chem. C, 2007, 111, 12067 CrossRef CAS.
- W. Li and B. L. Lucht, J. Electrochem. Soc., 2006, 153, A1617 CrossRef CAS.
- R. Dedryve`re, D. Foix, S. Franger, S. Patoux, L. Daniel and D. Gonbeau, J. Phys. Chem. C, 2010, 114, 10999 CrossRef.
- J. Li, H. Li, Z. Wang, L. Chen and X. Huang, J. Power Sources, 2002, 107, 1 CrossRef CAS.
- L. Zhang, Y. Ma, X. Cheng, P. Zuo, Y. Cui, T. Guan, C. Du, Y. Gao and G. Yin, Solid State Ionics, 2014, 263, 146 CrossRef CAS.
- J. H. Park, J. S. Kim, E. G. Shim, K. W. Park, Y. T. Hong, Y. S. Lee and S. Y. Lee, Electrochem. Commun., 2010, 12, 1099 CrossRef CAS.
- Y. M. Song, C. K. Kim, K. E. Kim, S. Y. Hong and N. S. Choi, J. Power Sources, 2016, 302, 22 CrossRef CAS.
Footnote |
| † These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |