
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion effects on the solvation structure and properties of imide lithium salt-based electrolytes†
Li Wang *ab,
Zhen Luoa,
Hong Xua,
Nan Piaoa,
Zonghai Chenc,
Guangyu Tianb and
Xiangming He*ab
*ab,
Zhen Luoa,
Hong Xua,
Nan Piaoa,
Zonghai Chenc,
Guangyu Tianb and
Xiangming He*ab
aInstitute of Nuclear & New Energy Technology, Tsinghua University, Beijing 100084, China. E-mail: wang-l@tsinghua.edu.cn; hexm@tsinghua.edu.cn
bState Key Laboratory of Automotive Safety and Energy, Tsinghua University, Beijing, 100084, China
cChemical Sciences and Engineering Division, Argonne National Laboratory, Argonne, IL 60439, USA
First published on 17th December 2019
Abstract
The anion effect on Li+ solvation structure and consequent electrochemical and physical properties was studied on the basis of LiFSI-DMC (lithium bisfluorosulfonyl imide-dimethyl carbonate)- and LiTFSI-DMC (lithium bis(trifluoromethanesulfonyl imide)-dimethyl carbonate)-based dilute electrolytes, highly concentrated electrolytes, and localized concentrated electrolytes. With different anions, the electrolytes are different in possible solvation structures and charge distributions, leading to differences in terms of thermal properties, viscosity, ionic conductivity, electrochemical oxidation and reduction behaviors as well as LiNi0.6Mn0.2Co0.2|Li cell performances. The results indicate that the electronic structure of anions contributes greatly to the charge distribution of the Li+ solvation sheath, and consequently extends to the thermodynamics of the carbonate molecules, affecting reduction, oxidation reaction and products on the interface between electrolytes and electrodes. The comprehensive understanding of the solution structure and properties is necessary for the rational design of advanced electrolytes.
Introduction
Rapid progress in the energy industry leads to the wide application of lithium-ion batteries (LIBs) as portable or stationary energy storage devices due to their advantages in terms of energy density, energy efficiency and moderate working temperature.1–4 The demands on higher energy density stimulate the development of LIBs. For example, the energy density of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 650 cells, based on a graphite anode and a 4.2 V cathode, has increased from 80 W h kg−1 to 300 W h kg−1 in the past three decades. However, it is a great challenge to further increase the energy density of LIBs, as engineering of the state-of-the-art battery chemistry approaches the limit.5 It is well known that employing high-voltage cathode materials is an ideal way to further increase the energy density of LIBs, either by increasing the charge cut-off voltage of the classic 4.2 V cathode materials6 or by developing new cathode materials.7 However, the charging cut-off voltage of higher than 4.2 V challenges the stability of the electrolytes.8–13 The conventional dilute carbonate-based electrolytes, with EC (ethylene carbonate) and 1 M LiPF6 as the skeleton composition, are easily decomposed at a potential higher than 4.3–4.4 V vs. Li/Li+ depending on the cathode chemistry.9–11 The common practice is to coat the surface of the cathode materials or to construct a cathode-electrolyte interphase (CEI) by adding suitable additives.14 However, these defensive strategies fail in long time cycling, since coating defects are avoidable during engineering and battery cycling. Simultaneously, LiPF6 in the electrolyte tends to decompose at temperatures above 60 °C, and EC oxidation releases active protons at a charge cut-off voltage higher than 4.3 V.12 Both cases may induce electrolyte deterioration. Then, new electrolyte chemistries have to be explored to match the next-generation battery chemistry.
650 cells, based on a graphite anode and a 4.2 V cathode, has increased from 80 W h kg−1 to 300 W h kg−1 in the past three decades. However, it is a great challenge to further increase the energy density of LIBs, as engineering of the state-of-the-art battery chemistry approaches the limit.5 It is well known that employing high-voltage cathode materials is an ideal way to further increase the energy density of LIBs, either by increasing the charge cut-off voltage of the classic 4.2 V cathode materials6 or by developing new cathode materials.7 However, the charging cut-off voltage of higher than 4.2 V challenges the stability of the electrolytes.8–13 The conventional dilute carbonate-based electrolytes, with EC (ethylene carbonate) and 1 M LiPF6 as the skeleton composition, are easily decomposed at a potential higher than 4.3–4.4 V vs. Li/Li+ depending on the cathode chemistry.9–11 The common practice is to coat the surface of the cathode materials or to construct a cathode-electrolyte interphase (CEI) by adding suitable additives.14 However, these defensive strategies fail in long time cycling, since coating defects are avoidable during engineering and battery cycling. Simultaneously, LiPF6 in the electrolyte tends to decompose at temperatures above 60 °C, and EC oxidation releases active protons at a charge cut-off voltage higher than 4.3 V.12 Both cases may induce electrolyte deterioration. Then, new electrolyte chemistries have to be explored to match the next-generation battery chemistry.
Superconcentrated15–23 and localized concentrated24–28 electrolytes are emerging towards high-voltage cathodes, as well as lithium metal and silicon-based anodes. It is found that when the molar ratio of lithium salts to solvents is high enough, the electrolyte will show superior performances in terms of thermal stability,16,25 redox stability,17–19,26–28 metal corrosion20 and reaction kinetics,21 resulting in high cycle stability and rate performance of the batteries.22–28 In these electrolytes, the interactions between anions and solvents via Li+ coordination are the wire-puller of all the advantages or disadvantages. The study of anion effect on Li+ solvation structure is important for us to understand the insight information and further design new electrolytes.29
Imide lithium salts are employed in these new electrolytes, not only due to their higher solubility and better thermal and chemical stability than LiPF6,30–32 but also because their anions are prone to participation in Li+ solvation even in 1 M LiFSI-DME electrolytes.33 This unique behaviour of imide anions is also responsible for the formation of 3D solvation clusters of hundreds of nano-meters in the super-concentrated electrolytes. In addition, the Li+ transference number and migration manner are different in super-concentrated electrolytes when compared with the electrolytes with the same components but normal concentrations. Moreover, they are poor in binding with solvents directly, due to their large size and low charge density, which facilitates the oxidation stability of solvents29 and may even change the reactions at the electrode interface.34 Among them, lithium bis(trifluoromethanesulfonyl imide) (LiTFSI) and lithium bisfluorosulfonyl imide (LiFSI) are widely studied. There are many super-concentrated electrolytes based on LiTFSI and LiFSI, with different solvents including DME (dimethyl ether),35 TMP (trimethyl phosphite),36,37 AN (acetonitrile),38,39 DMSO (dimethyl sulfoxide),40 and DMC (dimethyl carbonate).41 However, so far, there is no systematic comparison on the insight differences of these two salts in dilute, highly concentrated electrolytes and localized concentrated electrolytes.
Herein, LiTFSI, LiFSI and DMC are employed to construct dilute, highly concentrated and localized concentrated electrolytes respectively. To build the localized concentrated electrolyte, 1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropyl ether (HFRE) is selected as the non-solvent. The differences in solvent structures, physical and chemical properties, and electrochemical performances between these electrolytes are well compared to understand the effects of anions on the solution structure and properties.
Experimental
Preparation of electrolytes and electrodes
LiFSI (Suzhou Yake Chemical Reagent Co., Ltd.), LiTFSI (Suzhou Yake Chemical Reagent Co., Ltd.), LiPF6 (Canrd Co., Ltd) and all solvents (DMC, EC:DMC = 1:1 by volume, Canrd Co., Ltd) were of battery grade. The lithium salts were all used without further treatment. All the solvents were dried using a molecular sieve before use. The electrolytes were prepared by mixing lithium salts and the solvents in a defined ratio in an Ar-filled glove box. The water vapour and oxygen content in the glove box were both controlled to be less than 2 ppm. The water content in the prepared electrolytes was less than 15 ppm as detected using a Karl Fischer moisture titrator (V30S, METTLER TOLEDO).
The cathodes were prepared using a blade-coating machine. First, active materials LiNi0.6Co0.2Mn0.2O2 (NCM622), polyvinylidene difluoride (PVdF, Canrd Co., Ltd), and carbon black (Aladdin Industrial Corporation) were mixed in a weight ratio of 90:5:5 (NCM622:PVdF:AB) in N-methylpyrrolidone (NMP, Canrd Co., Ltd) by strong agitation to from a slurry. Then the slurry was casted on an Al foil (20 mm thickness, Shenzhen Kejing Star Technology Co., LTD) and dried at 120 °C under vacuum for 12 h. Finally, the as-prepared electrode was cut into discs with a diameter of 7 mm. NCM622 mass loading of the as-prepared cathodes was 3–4 mg cm−2. The graphite electrodes were purchased from Chilwee Group Co., Ltd, of which the active material mass loading was about 11.13 mg cm−2.
Characterization
The density and viscosity of the electrolyte samples were measured using a glass density meter and a rotary rheometer (MCR301, Anton Paar GmbH), respectively. The ionic conductivity was measured by AC impedance spectroscopy at 1 kHz (CHI600e, CH Instruments, Inc.) in a symmetric cell (Pt|electrolyte|Pt). A nonporous polypropylene membrane, with a round opening of 7 mm Φ in the middle, was used between the electrodes to provide a fixed space for ion transport. The electrolyte impedance can be read in the Nyquist plots, and the ionic conductivity of the electrolytes was calculated according to the equation σ = L/(RS), where L is the thickness of the nonporous polypropylene membrane and S is the area of the round opening.The solution structure was studied by a Raman spectrometer (HR-800) with an excitation laser of 514 nm. A Netzsch DSC-TG (STA 449F5) analyzer was used to investigate the thermal behaviour of the electrolytes. In sample preparation, 3–5 mg of the material was put in an Al pan in an argon-filled glove box. The heating and cooling rates during the measurement were both 10 °C min−1.
Cell assembly
NCM622|Li, Li|Gr, Li|Pt and SS (Stainless steel)|Li cells were assembled with the standard 2032-type coin cell hardware in an Ar-filled glove box. A Celgard 3501 separator was used. The amount of electrolytes in all coin cells was 80 μl to fully wet the separators and electrodes.Electrochemical measurements
The electrochemical stability of different electrolytes was measured by linear sweep voltammetry (LSV, CHI600e, CH Instruments, Inc.). In detail, the voltage window for Li|Gr or Li|Pt cells is from 3 V to 0 V to determine the reduction potential and from 2.5 V to 6 V to evaluate the oxidation potential. The corrosion capability of the electrolyte was evaluated using SS|Li cells in the voltage range of 2.5–5 V, where the polished stainless steel (SS, SUS-304, purchased from Shenzhen Kejing Star Technology Co., Ltd) discs was used as the working electrode. Galvanostatic charge/discharge cycling and rate capability tests were conducted on a charge/discharge unit (LAND CT2001A). All the charge/discharge were performed in a constant-current mode accompanied by 30 min rest after every charge or discharge end, and the cell cyclability was evaluated at 1C-rate.DFT simulations
DFT (density functional theory) calculations were performed using DMol3 module under Accelrys Materials Studio 7.0 at National Supercomputing Center in Shenzhen. The structures of solvents, Li+–solvents, and Li+–anion–solvent complexes, as well as their corresponding charge distributions were calculated using a density-functional theory42,43-based conductor-like screening model (DFT-COSMO)44–47 implemented in the DMol3 module48,49 of Accelrys Materials Studio 2017. The generalized gradient approximation (GGA) in the form of Perdew–Burke–Ernzerhof (PBE)37 was selected as the exchange-correlation functional. Double numerical basis with polarization functions (DNP) was selected as the basis set. The convergence criteria for energy, force, and displacement are 1 × 10−6 Ha, 1 × 10−4 Ha Å−1, and 5 × 10−4 Å, respectively. In dilute solutions, Li+ ions are much less than DMC molecules, so Li+ ions should be fully coordinated with DMC molecules in a ratio of 1:4. We calculated the complexes of Li+ and DMC molecules in dilute solutions, as well as different Li+–DMC–FSI− and Li+–DMC–TFSI− complexes in different ratios.
Results and discussion
Solvation structures in different electrolytes
LiTFSI/DMC and LiFSI/DMC electrolytes in different molar ratios were studied by Raman spectroscopy to find the structure information. In particular, the O–CH3 stretching mode of the DMC solvent is presented in the range of 900–950 cm−1, and the S–N stretching mode of the FSI− anion or TFSI− is presented in the range of 700–780 cm−1.41 The S–N stretching mode is sensitive to both the anion and the coordination structures, and the coexistence of multiple solvation structures makes the identification complex. As shown in Fig. 1a and b, DMC shows a single peak at about 910 cm−1, which can be considered as the characteristic peak for free DMC molecules. This peak weakens gradually with the increase in concentration, and disappears completely when the molar ratio of LiTFSI:DMC and LiFSI:DMC is more than 1:2. At the same time, a peak emerges at a higher wavelength (930–935 cm−1) in both LiTFSI- and LiFSI-based electrolytes, which is attributed to the DMC molecules bonded in the Li+ solvation sheath.50 Due to the large size and low charge density, FSI− and TFSI− are poor in the anion solvation.29,33 Therefore, solvation in this paper generally refers to cation solvation. The intensity of the peak at 930–935 cm−1 increases with the increase in concentration of the electrolyte. At the same time, the peak representing free DMC molecules deceases with the increase in concentration, and disappears when (lithium salt):DMC molar ratio is higher than 1:2. This phenomenon indicates that there are no free DMC molecules in the LiTFSI/DMC and LiFSI/DMC electrolytes in a (lithium salt):DMC molar ratio higher than 1:2. That is, all the DMC molecules are involved in the Li+ solvation structure. Though the most probable coordination number in the Li+ solvation structure is debatable,33 quantum chemistry calculations and MD simulations suggest that 4 is energetically favourable.51,52 However, the electrolyte with Li+:DMC = 1:4 still shows a considerably intensive peak for free DMC molecules, and the peaks for free DMC and coordinated DMC are even similar in intensity. This observation reveals that FSI− and TFSI− anions may be present in the Li+ solvation sheath even when DMC molecules are enough to solvate Li+ cations, which coincides with the experimental result.33 Therefore, when the molar ratio of Li+:DMC is less than 1:2, there may be a considerable amount of Li+ cations coordinating not only with DMC molecules but also with anions to form solvent structures in different ratios.37,41,51 It is worth noting that the electrolyte with LiTFSI:DMC = 1:3 still shows the peak of free DMC, and the peak intensity is similar to that of the peak for Li+-coordinated DMC. However, in the electrolyte with LiFSI:DMC = 1:3, the peak for free DMC is only half the height of the peak for Li+-coordinated DMC. This shows that at the same Li+:anion:DMC molar ratio, there are more free DMC molecules in LiTFSI/DMC than in the LiFSI/DMC electrolyte. As the energy preferable coordination number of Li+ is 4,51,52 the above-mentioned result indicates that TFSI− shows more tendency to coordinate with Li+ than with FSI− anion. Fig. 1c and d shows the comparison between super-concentrated electrolytes and localized concentrated electrolytes with LiFSI and LiTFSI, respectively. It can be seen that free DMC can be observed in the localized concentrated electrolytes in a molar ratio of 1:3:3 for both LiFSI- and LiTFSI-based electrolytes, which are similar to the case without HFRE. For HFRE, its characteristic peaks maintain well even when the concentration is high up to 1:1.5 for both LiFSI- and LiTFSI-based electrolytes, indicating that HFRE molecules are nearly free in the localized concentrated electrolytes. At the same time, the peak characteristics of DMC and anions are almost the same for both super-concentrated electrolytes and localized concentrated electrolytes though their apparent concentration are greatly different, indicating that the structural characteristics of the super-concentrated electrolytes can be preserved even after being diluted by a large amount of HFRE.
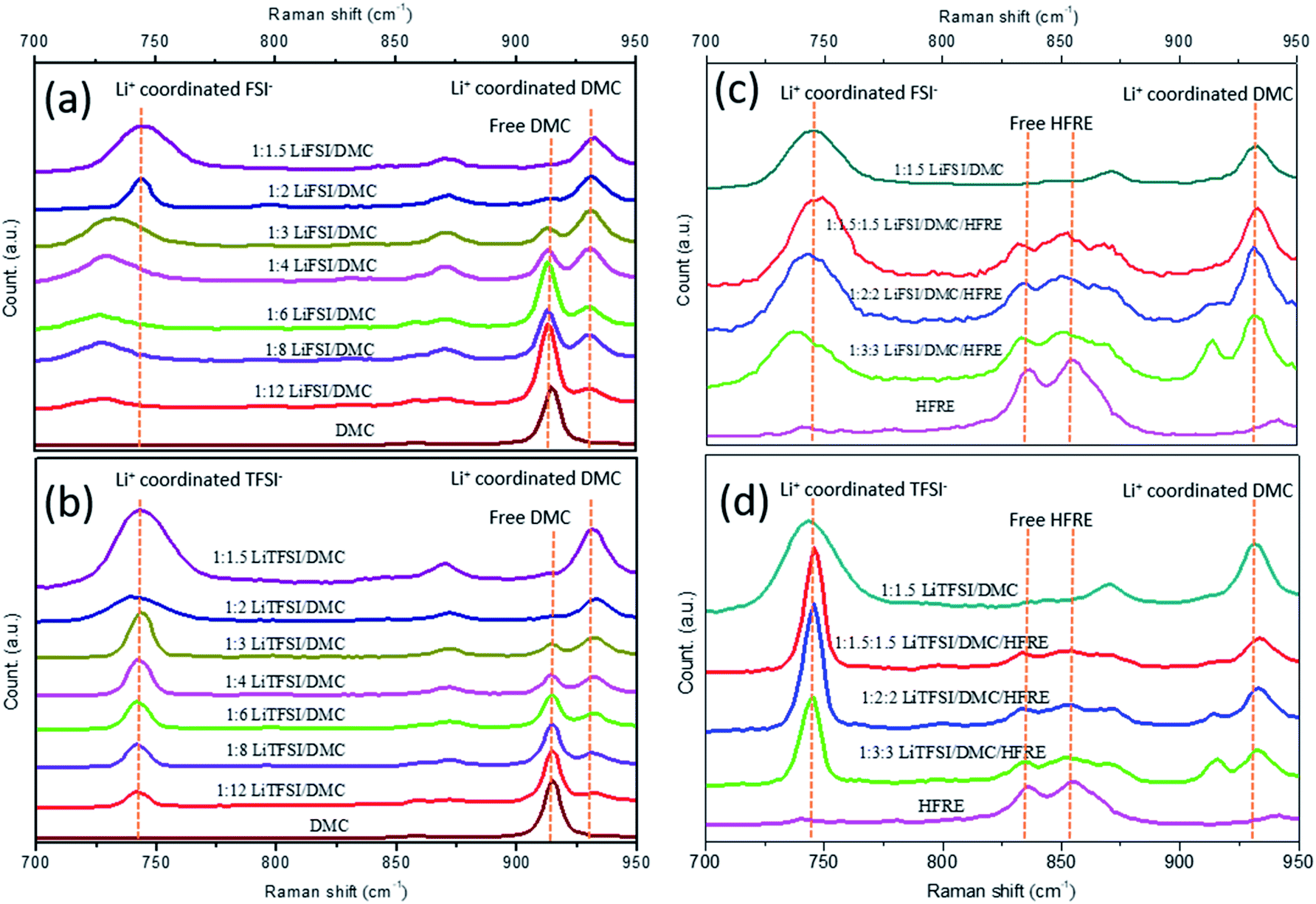 | ||
| Fig. 1 Raman spectra of different electrolytes in the range of 700–950 cm−1. (a) LiFSI/DMC electrolytes with different LiFSI/DMC molar ratios and pure DMC; (b) LiTFSI:DMC electrolytes with different LiTFSI:DMC molar ratios and pure DMC; (c) LiFSI:DMC:HFRE localized concentrated electrolytes with different molar ratios, highly concentrated LiFSI:DMC (molar ratio of 1:1.5) and pure HFRE; (d) LiTFSI:DMC:HFRE localized concentrated electrolytes with different molar ratios, highly concentrated LiTFSI:DMC (molar ratio of 1:1.5) and pure HFRE. | ||
The most possible conformations of Li+ solvation structures are screened by their total energies calculated by DFT, and their HOMO (highest occupied molecular orbital)/LUMO (lowest unoccupied molecular orbital) energy levels are calculated too (Table S1†). It is showed that the complex molecular energy of the full cis complex is always the lowest in different conformations of DMC and Li+–4DMC, which means that cis complexes are the most possible conformations in the dilute electrolytes. According to the calculation, Li+–(cis3-DMC)–(cis-FSI−) is the most possible conformation in the super-concentrated and localized concentrated electrolytes for LiFSI-based electrolytes, and Li+–(cis3-DMC)–(trans-TFSI−) is the most stable in the super-concentrated and localized concentrated electrolytes. The conformation difference between Li+–3DMC–FSI− and Li+–3DMC–TFSI− may be caused by the steric effect of TFSI−. A theoretical understanding of the electrochemical differences of the dilute and super-concentrated electrolytes is attempted through HOMO and LUMO distribution, as shown in Fig. 2. It is known that HOMO distribution indicates the active sites during oxidation, and LUMO distribution indicates the active sites during reduction. Then it can be observed that for Li+–(cis4-DMC) complexes (Fig. 2a and d), which is the dominated solvation structure in dilute electrolytes, the LUMO and HOMO all locate on DMC molecules. These results are coincided with the experimental results. That is, in the electrochemical reduction/oxidation, which generally happens on the surface of anode/cathode, DMC molecules in the solvation sheath will be reduced/oxidized in preference to free DMC molecules, contributing to the formation of the SEI (solid electrolyte interphase) layer or CEI (cathode electrolyte interphase) layer. For Li+–3DMC–FSI− (Fig. 2b and e), the energy-favourable conformation is Li+–(cis3-DMC)–(cis-FSI−), which may be a major solvation structure in the super-concentrated and localized concentrated electrolytes, according to the DFT-COSMO simulation. As shown in Fig. 1a, both LUMOs and HOMOs mainly locate on FSI− ions rather than DMC. The results indicate that both the reduction and oxidation reactions may occur on FSI− ions. FSI− ions even preferentially react before the DMC molecules to form an intermediate fragment containing F elements, and finally form a dense inorganic SEI film containing LiF.53 This has been observed in many research studies and intensively used to form LiF-rich SEI layers for lithium metal anodes19 or even graphite anodes.54
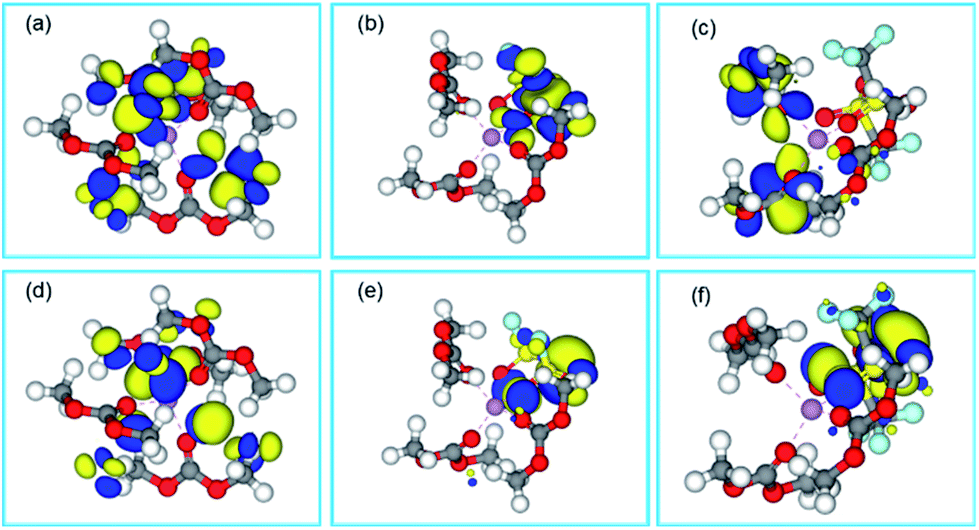 | ||
| Fig. 2 LUMOs of (a) Li+–(cis4-DMC); (b) Li+–(cis3-DMC)–(cis-FSI−); (c) Li+–(cis3-DMC)–(trans-TFSI−); HOMOs of (d) Li+–(cis4-DMC); (e) Li+–(cis3-DMC)–(cis-FSI−); (f) Li+–(cis3-DMC)–(trans-TFSI−). Grey sphere, C; red sphere, O; white sphere, H; magenta sphere, Li. The yellow and blue regions of HOMOs/LUMOs represent the positive and negative parts of orbitals, respectively. | ||
The case is different for Li+–3DMC–TFSI− complexes (Fig. 2c and f), whose energy-favourable conformation is Li+–(cis3-DMC)–(trans-TFSI−) according to the DFT-COSMO simulation. Most of the LUMOs are on DMC, while most of the HOMOs are on TFSI−. These observations imply that in the LiTFSI-based concentrated or localized concentrated electrolytes, TFSI− anions participate in the electrochemical oxidation rather than reduction.55 That is, FSI− shows higher activity than TFSI− in SEI formation. It is known that LiF-containing SEI and CEI films are beneficial to the cycle stability of the batteries, and this result indicates that LiFSI-based electrolytes will show superior performances to both anodes and cathodes, but LiTFSI-based electrolytes may only show advantages against cathodes.
In consideration that TFSI− and FSI− are bidentate ligands, we assume that lithium ions with two DMC molecules and one TFSI−/FSI− coordinated, though the possible conformations in the super-concentrated and localized concentrated electrolytes are in fact many. Besides the energy preferable conformation (Fig. S1†), surface charge distributions under the environment of electrolytes (Fig. S2†), LUMOs and HOMOs of Li+–2DMC–TFSI− and Li+–2DMC–FSI− are all simulated using DFT-COSMO (Fig. S3 and S4†). The conclusion results are similar to those of Li+–3DMC–FSI− and Li+–3DMC–TFSI−. That is, LiFSI tends to be involved in electrochemical reduction and oxidation to form a dense fluoride layer, and will show superior performances to LiTFSI in super-concentrated electrolytes.
Physical and chemical properties
The ionic conductivity and viscosity of the LiFSI- and LiTFSI-based electrolytes, including dilute, super-concentrated and localized concentrated electrolytes, are compared in Fig. 3a. The ionic conductivity shows the peak distribution, while the viscosity shows a monotonic increase with the increase in Li+:DMC. For both lithium salts, the sharp decrease in ionic conductivity appears with a sharp increase in viscosity. This trend in conductivity versus salt concentration follows the typical behaviour of conventional non-aqueous electrolyte solutions, and could be explained by the combined results of the number of ions and pairs in the electrolyte.24 Specifically, the critical ratio, where the curves of ionic conductivity turn, is 1:6 for LiFSI-based electrolytes, while this is 1:8 for LiTFSI-based electrolytes. It is known that a high degree of salt dissociation is favourable for high concentrations of mobile ions and high conductivity, whereas a high viscosity and large anion size would impede ion transport, leading to low conductivity. Relatively, the LiTFSI-based electrolytes show higher viscosity and lower ionic conductivity than LiFSI-based electrolytes. This can be essentially attributed to the higher dissociation and the smaller size of FSI− anions when compared with those of TFSI− anions. Then it is reasonable that LiFSI-based electrolyte show superior rate performances in this sense. The detailed comparison will be discussed later. When Li+:DMC is higher than the critical ratio, the viscosity of the electrolyte increases sharply, and the diffusion capability of the ions decreases faster compared with the increase in the charge density. This may be attributed to the various solvation structures, such as contact ion pairs (CIPs, an anion coordinating to one Li+) and aggregates (AGGs, an anion coordinating to two or more Li+).15 After the addition of the diluent (to form localized concentrated electrolyte), the viscosity decreased dramatically, but the ionic conductivity does not significantly increase for both LiFSI- and LiTFSI-based electrolytes, indicating that the solvation sheath of Li+ cations does not change much but the aggregation of the solvated structure is dispersed by the diluent. Besides, it is noticeable that the variations in the ionic conductivity with HFRE addition are different for LiTFSI- and LiFSI-based electrolytes. Though it cannot be clearly explained at this time, it is indubitable that the differences can only be associated with the anions.
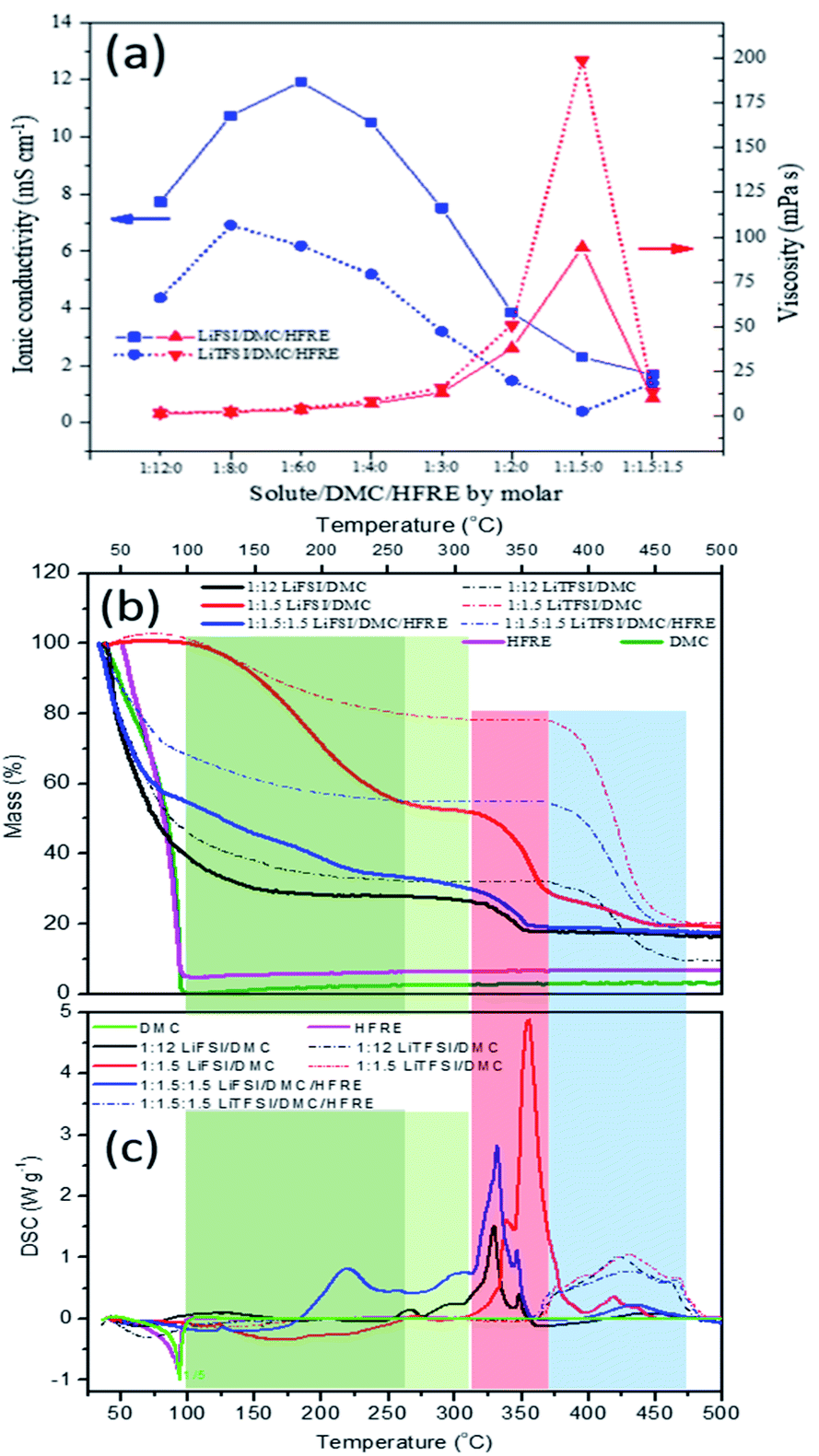 | ||
| Fig. 3 The ionic conductivities (blue, left y-axis) and viscosities (red, right y-axis) (a) of LiTFSI:DMC:HFRE and LiFSI:DMC:HFRE electrolytes at different molar ratios. The TGA (b) and DSC (c) curves of electrolytes with various concentrations and solvents. | ||
The differential scanning calorimetry (DSC) and thermogravimetric analysis (TG) results of LiTFSI- and LiFSI-based electrolytes, including dilute, super-concentrated and localized concentrated electrolytes, are shown in Fig. 3b and c to further investigate the states of DMC. Plain DMC and HFRE are confirmed to be fully volatilized below 100 °C (the purple and green curves in Fig. 3b). Accordingly, the measured residual mass and theoretical residual mass are compared to get further information about the solution structures and physical/chemical properties. Though the boiling point of HFRE is below 100 °C, it is highly thermally stable among the most studied non-solvents relatively (Table S2†), and it can also serve as a flame retardant. That is why, HFRE is chosen for this study. The detailed comparison based on Fig. 3a and b is listed in Table S3.† The mass loss can be separated into three regions. The first region is located in the temperature ranging from room temperature to 100 °C, which is associated with solvent evaporation. For both LiFSI:DMC = 1:12 and LiTFSI:DMC = 1:12 dilute electrolytes, their measured mass losses are higher than the theoretical mass losses at 100 °C. In theoretical calculation, the residue is assumed to be LiFSI:DMC = 1:4, while the measured residue species are calculated to be 1:3.55:0 and 1:3.49:0 respectively. This indicates that the solvation sheath with LiFSI:DMC = 1:4 will lose some DMC at 100 °C, and the interactions between DMC and LiFSI is stronger than that between DMC and LiTFSI. For LiFSI:DMC = 1:1.5 and LiTFSI:DMC = 1:1.5 super-concentrated electrolytes, no mass loss happens in this temperature range. It is reasonable as the residue species of dilute electrolyte at 100 °C is LiFSI:DMC = 1:3.55 and LiTFSI:DMC = 1:3.49, and the initial (lithium salt):DMC ratios of the concentrated electrolytes are both higher than 1:3. As a non-solvent, HFRE is supposed to be totally vaporized as there is no interactions between HFRE and lithium salt. However, both LiFSI:DMC:HFRE = 1:1.5:1.5 and LiTFSI:DMC:HFRE = 1:1.5:1.5 localized concentrated electrolytes show HFRE residue at 100 °C (1:1.5:0.2 and 1:1.5:0.44, respectively). This can only be illustrated by the enhanced interactions between solvated DMC and HFRE. When the temperature goes higher than 100 °C (from 100 °C to Td, Td is the onset temperature of salt decomposition, which is 315 °C for LiFSI-based electrolytes54 and about 370 °C for LiTFSI-based electrolytes), the mass loss slows down for both dilute electrolytes and localized concentrated electrolytes. The process happening in this temperature range may be the continuous loss of solvent molecules from the solvation sheath. The residual mass at Td is over the mass percentage of neat lithium salts for both LiFSI- and LiTFSI-based electrolytes, indicating that the DMC in the solvation sheath is stable even when the temperature is 200 °C higher than its boiling point. As decomposition is the main reaction when temperature goes higher than Td, it can be deduced that DMC will be involved in salt decompositions either for LiFSI or for LiTFSI. By comparison, it can be observed that the thermal stability of the solvent will be greatly enhanced in super-concentrated electrolytes and localized concentrated electrolytes due to the solvation effect. Besides, the DMC molecules in the Li+ solvation sheath are bonded more strongly with FSI− than with TFSI−, which is consistent with the results of Raman spectroscopy. Furthermore, the decomposition starting temperature of LiFSI in the electrolytes is about 45–60 °C lower than that of LiTFSI, and larger amount of heat is released when LiFSI is thermally decomposed. These are detrimental to the safety of the battery. Therefore, LiTFSI-based electrolytes are more advantageous than LiFSI-based electrolytes in terms of thermal safety.
Electrochemical performance
The electrochemical measurements of the electrolytes based on LiFSI and LiTFSI are performed to understand the effect of the solvation structure on electrochemical properties. The reduction (Fig. 4a and c) and oxidation (Fig. 4b and d) behaviours are studied using LSV. Graphite (Fig. 4a and b) or metal platinum (Fig. 4c and d) is used as the working electrode to verify the effect of surface chemistry on electrochemical behaviours, and a lithium foil is used as the counter electrode.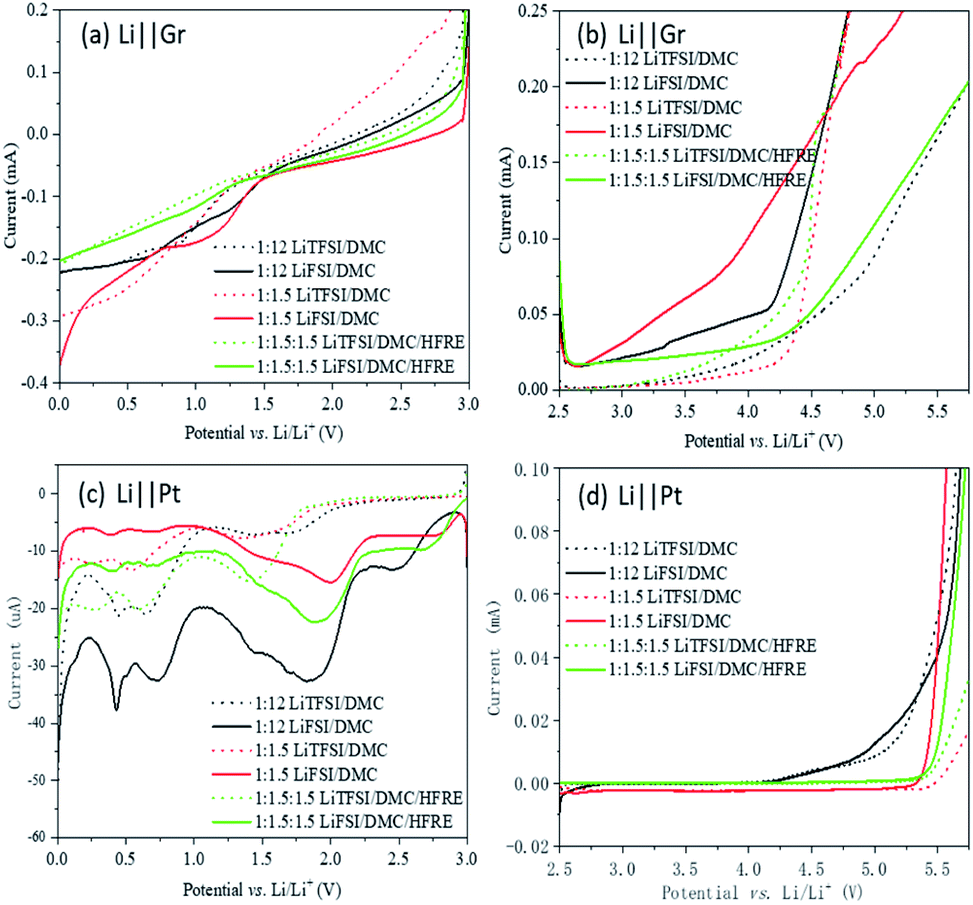 | ||
| Fig. 4 LSV curves of different electrolytes on the graphite-based electrode (a and b) and on the Pt foil electrode (c and d). | ||
The reduction current is about ten times larger on the graphite electrode than on platinum for both LiFSI- and LiTFSI-based electrolytes. However, the reaction on the platinum electrode is distinctive, while the reduction on graphite shows no obvious onset potential for both LiFSI- and LiTFSI-based electrolytes. Though the reaction mechanisms are still in investigation, the results show clearly that surface chemistry can affect the electrochemical reduction greatly. Besides, on both electrodes, LiFSI-based electrolytes react at higher potential than LiTFSI-based electrolytes. This coincides with the DFT results, indicating that the active sites locate on FSI− in LiFSI-based concentrated electrolytes, while it is located on DMC in LiTFSI-based concentrated electrolytes. For LiFSI-based electrolytes, the reduction behaviours of the super-concentrated electrolyte and localized concentrated electrolyte are similar, which are both reduced at higher potentials, compared to the dilute electrolyte. On the platinum electrode, LiFSI-based electrolytes are reduced around 1.75–2 V vs. Li/Li+, which may help to form protective films on the anode material.15 The LUMOs of the complex of Li+–3DMC–FSI− mainly locate on FSI− (Fig. 2b). Then in the super-concentrated and localized concentrated electrolytes, FSI− will be preferentially reduced. However, for LiTFSI-based electrolytes, cases are totally the opposite. The dilute electrolyte is the most reductive. This result also coincides with the DFT simulation results. The reducible sites in LiTFSI-based super-concentrated electrolytes are solvated DMC, and the introduction of TFSI− anions will lower the reducibility of the solvated DMC, as confirmed by the surface charge distribution (Fig. S2†). Therefore, the LiTFSI-based electrolytes, regardless of the concentration, are not conducive to the formation of protective films during electrochemical reduction.
For traditional 1 M LiPF6-based carbonate electrolytes, the electrochemical oxidation generally occurs at potentials higher than 4.3 V vs. Li/Li+. The oxidation potentials of both LiFSI- and LiTFSI-based dilute electrolytes on the platinum electrode are similar (Fig. 4d). However, LiFSI- and LiTFSI-based super-concentrated electrolytes and localized concentrated electrolytes remain stable even at a potential of 5.25 V vs. Li/Li+. This is because the HOMOs of Li+–3DMC–FSI− and Li+–3DMC–TFSI− are mostly on the anion. When the oxidation reaction occurs, the anion will react before the solvent molecules to form a fluoride passive layer, thereby inhibiting further oxidation of the electrolyte and widening the electrochemically stable window of the electrolytes.15,53,54 The LiTFSI-based super-concentrated electrolytes and the localized concentrated electrolytes have slightly higher electrochemically stable windows than those of LiFSI-based electrolytes, but the difference is not significant.
In addition to electrochemical oxidation, current collector corrosion is also an important factor to determine the application of an electrolyte. Unfortunately, metal corrosion is a serious problem for the LiFSI- and LiTFSI-containing batteries. As the cathode current, a corroded aluminum foil will lead to an increase in internal resistance and electronic contact loss of active materials, which is, in turn, manifested as decay in the rate capability and capacity.56 The common dilute (around 1 M) amide salt-based organic electrolytes cannot form passivation films on the surface of the aluminum foils to inhibit the corrosion, while LiPF6 can provide F− to form insoluble AlF3 and LiF.57–59 In this case, metal corrosion is highly anion sensitive, so it is essential to investigate the behaviors of LiTFSI- and LiFSI-based electrolytes to understand the anion effect on the chemical properties of the electrolytes. Moreover, many studies prove that LiTFSI- and LiFSI-based electrolytes will lose the corrosion ability for the aluminum foil as it reaches a certain concentration.60,61 However, the corrosion ability for other metals is seldom reported, though the metal components in a battery that are in contact with the electrolyte are many, such as the Ni tab and stainless steel case. Herein, the corrosion behaviors of stainless steel in LiTFSI- and LiFSI-based electrolytes are studied by cyclic voltammetry analysis.
As shown in Fig. 5, stainless steel corrosion occurs at 3.65 V, 3.55 V and 3.55 V vs. Li/Li+ in LiFSI:DMC = 1:12, LiFSI:DMC = 1:3 and LiFSI:DMC = 1:1.5, respectively, indicating that LiFSI-based electrolytes still corrode stainless steel even at high concentrations. By comparison, electrolytes with LiTFSI:DMC = 1:12 and LiTFSI:DMC = 1:3 show corrosion of stainless steel at 3.90 V vs. Li/Li+, while LiTFSI/DMC (1:1.5) shows little corrosion against stainless steel. This difference indicates that the solvation sheath in high-concentration electrolytes hinders the property of TFSI−, while showing little effect on FSI−. This may be related to their difference in the bonding strength with Li+ cations. LiFSI- and LiTFSI-based electrolytes have different corrosion characteristics for different metals, which limits the wide applications of new electrolytes such as super-concentrated electrolytes and affects the material selection and structural design of these batteries.
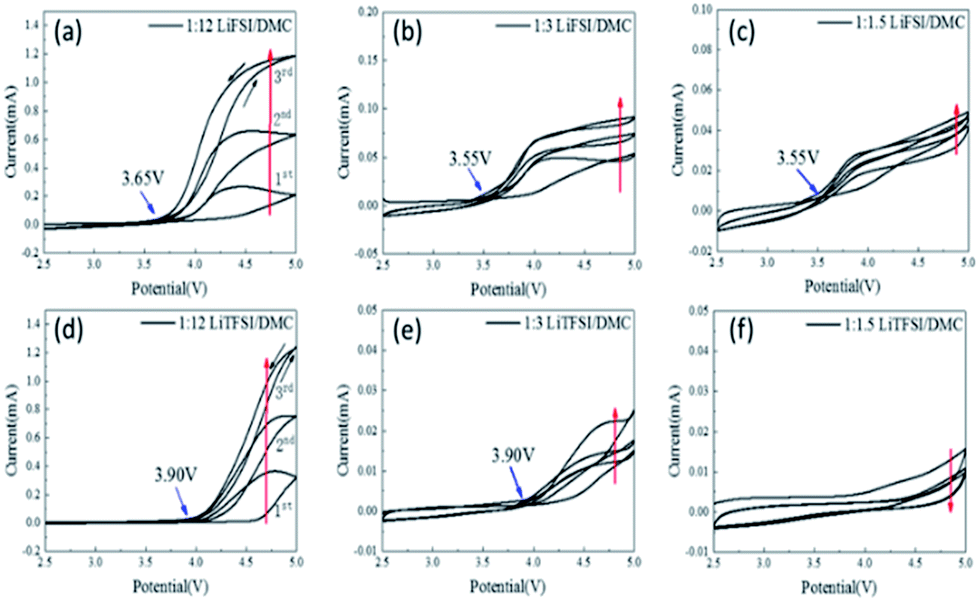 | ||
| Fig. 5 Anodic oxidation curves of stainless steel foil in LiFSI- and LiTFSI-based electrolytes. | ||
To verify the performances in advanced LIB performances, the localized concentrated electrolytes (LiTFSI:DMC:HFRE = 1:1.5:1.5 and LiFSI:DMC:HFRE = 1:1.5:1.5), super-concentrated electrolytes (LiTFSI:DMC = 1:1.5 and LiFSI:DMC = 1:1.5) and traditional 1 M LiPF6 in EC:DMC = 1:1 are compared using NCM622|Li half-cells. The rate capability (Fig. 6a and b) and cyclability (Fig. 6c and d) are intensively studied at 25 °C. 0.5C cycling tests at a normal temperature (25 °C) (Fig. 6a and d) are carried out with the cut-off voltage of 3 V and 4.5 V. Fig. 6a and b shows that the reversible capacity of NCM622 varies with electrolytes. To be specific, 1 M LiPF6 in EC:DMC = 1:1 electrolytes delivers the lowest capacity, even at 0.1C-rate, and the differences in capacity increase with the increase in C-rate. The capacity retention is only 35% at 5C-rate, while compared with that at 0.1C-rate. Both LiFSI- and LiTFSI-based super-concentrated electrolytes and localized concentrated electrolytes deliver higher reversible capacity and higher capacity retention at a high C-rate, compared to 1 M LiPF6-based electrolytes. Relatively, LiFSI-based electrolytes are superior to LiTFSI-based electrolytes, though the differences are very small. The localized concentrated electrolytes and the super-concentrated electrolytes show very close performance under low-rate conditions, for either LiFSI- or LiTFSI-based electrolytes. However, under high-rate conditions (5C), the localized concentrated electrolytes had better cycle performance. Specifically, LiTFSI:DMC:HFRE = 1:1.5:1.5 shows a capacity retention of 67% at 5C-rate. The lower viscosity and higher ionic conductivity of the localized concentrated electrolytes contribute to better wettability with a separator and a porous electrode and fast cation transference between the cathode and the anode, which are essential for high utility of the active materials under high-rate conditions.
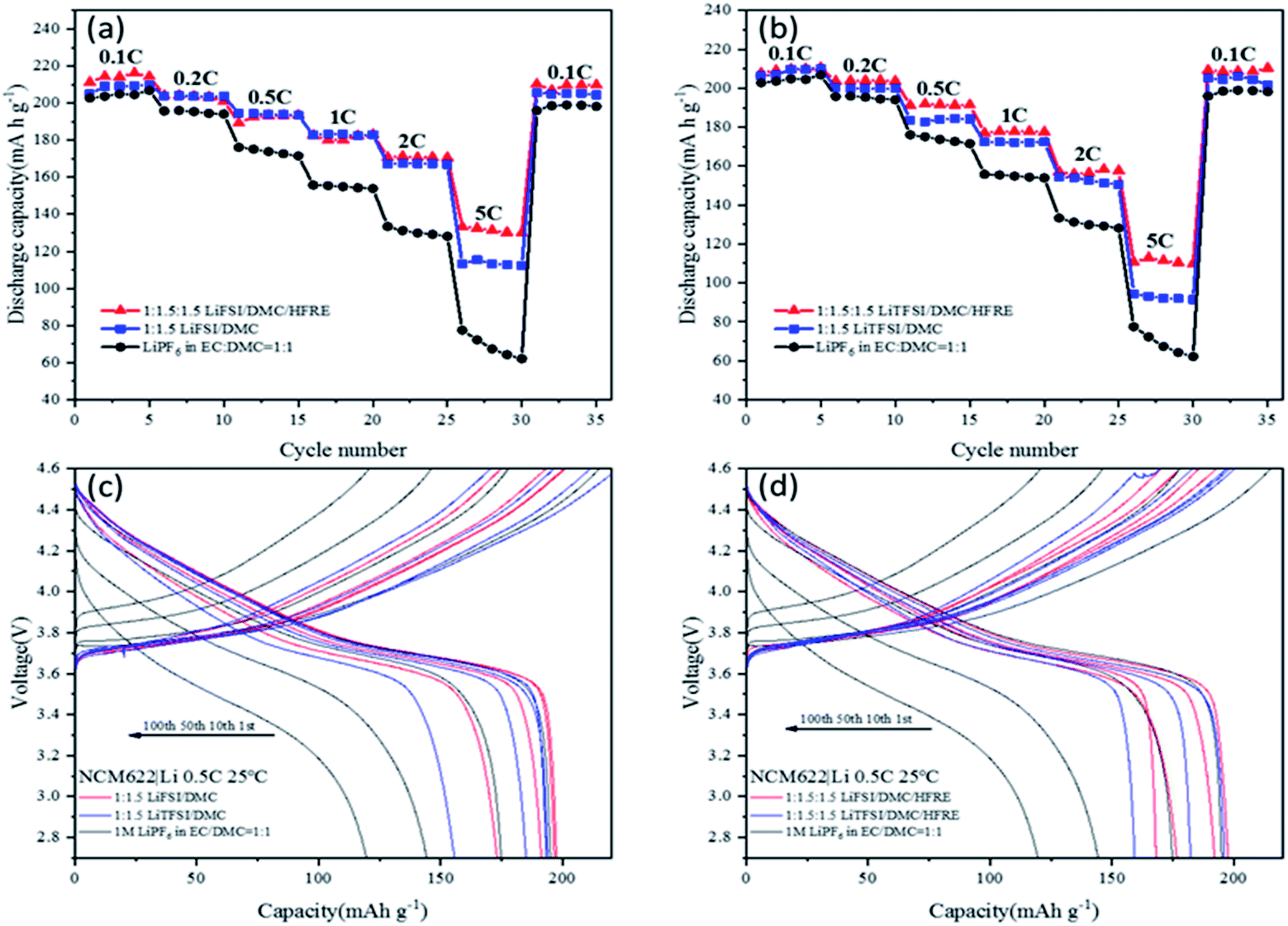 | ||
| Fig. 6 Rate capability measurements of NCM 622‖Li batteries at 25 °C using (a) LiFSI:DMC:HFRE = 1:1.5:1.5, LiFSI:DMC = 1:1.5, 1 M LiPF6 in EC:DMC = 1:1 electrolytes and (b) LiTFSI:DMC:HFRE = 1:1.5:1.5, LiTFSI:DMC = 1:1.5, 1 M LiPF6 in EC:DMC = 1:1 electrolytes. Long-term cycling of NCM 622‖Li batteries at 0.5C-rate at 25 °C, using (c) 1 M LiPF6 in EC:DMC = 1:1 electrolytes and highly concentrated electrolytes (LiFSI:DMC = 1:1.5, LiTFSI:DMC = 1:1.5), and (d) 1 M LiPF6 in EC:DMC = 1:1 electrolyte and localized concentrated electrolytes (LiFSI:DMC:HFRE = 1:1.5:1.5, LiTFSI:DMC 1:1.5:1.5). The voltage window for all above tests is from 3 V to 4.5 V. | ||
In long-term cyclability, the cell using 1 M LiPF6-based electrolytes fades rapidly, and the capacity retention is only 64% at 100th cycle (Table S4†). Both LiFSI- and LiTFSI-based concentrated electrolytes show better cyclability than 1 M LiPF6-based electrolytes, and the capacity retention of LiFSI-based super-concentrated electrolytes is higher (89.7% at 100th cycle, Table S4†) than that of LiTFSI-based super-concentrated electrolytes. For the localized concentrated electrolytes, they are also superior to 1 M LiPF6-based electrolytes, and LiFSI-based electrolytes are better than LiTFSI-based electrolytes. Considering that the anode is a thick lithium metal, the fading capacity may mainly indicate the fading of the NCM622 material at a high charging potential (4.5 V vs. Li/Li+). This proves that FSI− facilitates more stable CEI formation when compared to TFSI− and PF6−.
Conclusions
In this study, LiFSI- and LiTFSI-based electrolytes were investigated in terms of solvent structure, physicochemical properties and electrochemical performances to understand the anion effects on the performances of electrolytes. The differences in ionic polarizability and size between FSI− and TFSI− result in different interaction strengths with Li+ cations, and non-solvent and charge distribution, leading to different physical and chemical behaviours under the same condition. FSI− ions are more active in participating in Li+ solvation, reduction and oxidation than TFSI−. This makes it superior in forming effective SEI and CEI layers, indicative of good cycling stability. In addition, the smaller FSI− anion endows better cation mobility, indicative of good rate performance. The active FSI−, however, is also the cause for severe metal corrosion and poor thermal stability, which may lead to safety issues of lithium ion batteries. In general, anions may show great effect on the solvation sheath of Li+ cations, as well as the solvation structure and the solution structure. Their tuneable ionic polarizability, symmetry, size and composition provide many possibilities for tailoring the performances of the electrolytes.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work is supported by Ministry of Science and Technology of China (No. 2019YFE010186) and the National Natural Science Foundation of China (No. U1564205). Research at Argonne National Laboratory was supported by U.S. Department of Energy (DOE), Vehicle Technologies Office. Argonne National Laboratory is operated for the US Department of Energy by U Chicago Argonne, LLC, under contract DE-AC02-06CH11357. The authors thank Joint Work Plan for Research Projects under the Clean Vehicles Consortium at U.S. and China – Clean Energy Research Center (CERC-CVC2.0, 2016-2020).Notes and references
- S. Kondou, M. L. Thomas, T. Mandai, K. Ueno, K. Dokko and M. Watanabe, Phys. Chem. Chem. Phys., 2019, 21, 5097–5105 RSC.
- L. Xia, L. Yu, D. Hu and G. Z. Chen, Mater. Chem. Front., 2017, 1, 584–618 RSC.
- D. Aurbach, J. Power Sources, 2000, 89, 206–218 CrossRef CAS.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 RSC.
- M. Li, J. Lu, Z. H. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- J. N. Zhang, Q. H. Li, E. Y. Hu, C. Ma, H. G. Yu, X. Q. Yang, S. F. Li, R. J. Xiao, C. Y. Ouyang, X. Yu, W. Yang, M. Ge, Y. Chu, X. Huang, L. Q. Chen and H. Li, Nat. Energy, 2019, 4(7), 594–603 CrossRef CAS.
- M. Hu, X. Pang and Z. Zhou, J. Power Sources, 2013, 237, 229–242 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- H. J. Noh, S. J. Youn, C. S. Yoon and Y. K. Sun, J. Power Sources, 2013, 233, 121–130 CrossRef CAS.
- D. Streich, C. Erk, A. Gueguen, P. Muller, P. F. Chesneau and E. J. Berg, J. Phys. Chem. C, 2017, 121, 13481–13486 CrossRef CAS.
- X. Q. Zeng, G. L. Xu and Y. Li, ACS Appl. Mater. Interfaces, 2016, 8, 3446–3451 CrossRef CAS PubMed.
- Y. Y. Xie, H. Gao, J. H. Gim, A. T. Ngo, Z. F. Ma and Z. H. Chen, J. Phys. Chem. Lett., 2019, 10(3), 589–594 CrossRef PubMed.
- M. Gauthier, T. J. Carney, A. Grimaud, L. Giordano, N. Pour, H. H. Chang, D. P. Fenning, S. F. Lux, O. Paschos, C. Bauer, F. Maglia, S. Lupart, P. Lamp and Y. S. Horn, J. Phys. Chem. Lett., 2015, 6, 4653–4672 CrossRef CAS PubMed.
- Y. H. Xiao, L. J. Miary, Y. Wang and G. Ceder, Joule, 2019, 3, 1–24 CrossRef.
- Y. Yamada and A. Yamada, J. Electrochem. Soc., 2015, 162, A2406–A2423 CrossRef CAS.
- K. Yoshida, M. Tsuchiya, N. Tachikawa, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2011, 115, 18384–18394 CrossRef CAS.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 13121–13129 CrossRef CAS PubMed.
- Y. Yamada, K. Furukawa, K. Sodeyama, K. Kikuchi, M. Yaegashi, Y. Tateyama and A. Yamada, J. Am. Chem. Soc., 2014, 136, 5039–5046 CrossRef CAS PubMed.
- R. Tatara, D. G. Kwabi, T. P. Batcho, M. Tulodziecki, K. Watanabe, H.-M. Kwon, M. L. Thomas, K. Ueno, C. V. Thompson and K. Dokko, J. Phys. Chem. C, 2017, 121, 9162–9172 CrossRef CAS.
- K. Matsumoto, K. Inoue, K. Nakahara, R. Yuge, T. Noguchi and K. Utsugi, J. Power Sources, 2013, 231, 234–238 CrossRef CAS.
- V. A. Nikitina, M. V. Zakharkin, S. Y. Vassiliev, L. V. Yashina, E. V. Antipov and K. J. Stevenson, Langmuir, 2017, 33(37), 9378–9389 CrossRef CAS PubMed.
- K. Takada, Y. Yamada, E. Watanabe, J. Wang, K. Sodeyama, Y. Tateyama, K. Hirata, T. Kawase and A. Yamada, ACS Appl. Mater. Interfaces, 2017, 9, 33802–33809 CrossRef CAS PubMed.
- H. Zhang, S. Jeong, B. Qin, D. Vieira Carvalho, D. Buchholz and S. Passerini, ChemSusChem, 2018, 11, 1382–1389 CrossRef CAS PubMed.
- T. Doi, Y. Shimizu, M. Hashinokuchi and M. Inaba, J. Electrochem. Soc., 2017, 164, A6412–A6416 CrossRef CAS.
- J. Wang, Y. Yamada, K. Sodeyama, E. Watanabe, K. Takada, Y. Tateyama and A. Yamada, Nat. Energy, 2017, 3, 22–29 CrossRef.
- S. Chen, J. Zheng, D. Mei, K. S. Han, M. H. Engelhard, W. Zhao, W. Xu, J. Liu and J. G. Zhang, Adv. Mater., 2018, 30, 1706102 CrossRef PubMed.
- X. Ren, S. Chen, H. Lee, D. Mei, M. H. Engelhard, S. D. Burton, W. Zhao, J. Zheng, Q. Li, M. S. Ding, M. Schroeder, J. Alvarado, K. Xu, Y. S. Meng, J. Liu, J. G. Zhang and W. Xu, Chemistry, 2018, 4, 1877–1892 CrossRef CAS.
- L. Yu, S. Chen, H. Lee, L. Zhang, M. H. Engelhard, Q. Li, S. Jiao, J. Liu, W. Xu and J. G. Zhang, ACS Energy Lett., 2018, 3, 2059–2067 CrossRef CAS.
- A. Von Wald Cresce, M. Gobet, O. Borodin, J. Peng, S. M. Russell, E. Wikner, A. Fu, L. B. Hu, H. S. Lee, Z. C. Zhang, X. Q. Yang, S. Greenbaum, K. Amine and K. Xu, J. Phys. Chem. C, 2015, 119(49), 27255–27264 CrossRef CAS.
- H. B. Han, S. S. Zhou, D. J. Zhang, S. W. Feng, L. F. Li, K. Liu, W. F. Feng, J. Nie, H. Li and X. J. Huang, J. Power Sources, 2011, 196, 3623–3632 CrossRef CAS.
- A. Abouimrane, J. Ding and I. J. Davidson, J. Power Sources, 2009, 189, 693–696 CrossRef CAS.
- L. F. Li, S. S. Zhou, H. B. Han, H. Li, J. Nie, M. Armand, Z. B. Zhou and X. J. Huang, J. Electrochem. Soc., 2011, 158(2), A74–A82 CrossRef CAS.
- Y. Zhang, M. Su, X. Yu, Y. Zhou, J. Wang, R. Cao, W. Xu, C. Wang, D. R. Baer, O. Borodin, K. Xu, Y. Wang, X. L. Wang, Z. Xu, F. Wang and Z. Zhu, Anal. Chem., 2018, 90(5), 3341–3348 CrossRef CAS PubMed.
- L. M. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. L. Fan, C. Luo, C. S. Wang and K. Xu, Science, 2015, 350(6263), 938–943 CrossRef CAS PubMed.
- L. E. Camacho-Forero, T. W. Smith and P. B. Balbuena, J. Phys. Chem. C, 2017, 121, 182–194 CrossRef CAS.
- P. Shi, H. Zheng, X. Liang, Y. Sun, S. Cheng, C. Chen and H. Xiang, Chem. Commun., 2018, 54, 4453–4456 RSC.
- Y. Yamada and A. Yamada, J. Electrochem. Soc., 2015, 162(14), A2406–A2423 CrossRef CAS.
- Y. Yamada and A. Yamada, Chem. Lett., 2017, 46, 1056–1064 CrossRef.
- K. Sodeyama, Y. Yamada, K. Aikawa, A. Yamada and Y. Tateyama, J. Phys. Chem. C, 2014, 118, 14091–14097 CrossRef CAS.
- R. Tatara, D. G. Kwabi, T. P. Batcho, M. Tulodziecki, K. Watanabe, H. M. Kwon, M. L. Thomas, K. Ueno, C. V. Thompson, K. Dokko, Y. Shao-Horn and M. Watanabete, J. Phys. Chem. C, 2017, 121, 9162–9172 CrossRef CAS.
- J. Wang, Y. Yamada, K. Sodeyama, C. H. Chiang, Y. Tateyama and A. Yamada, Nat. Commun., 2016, 7, 12032 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- J. Andzelm, C. Kölmel and A. Klamt, J. Chem. Phys., 1995, 103, 9312–9320 CrossRef CAS.
- A. Klamt, V. Jonas, T. Bürger and J. C. Lohrenz, J. Phys. Chem. A, 1998, 102, 5074–5085 CrossRef CAS.
- E. Mullins, R. Oldland, Y. Liu, S. Wang, S. I. Sandler, C.-C. Chen, M. Zwolak and K. C. Seavey, Ind. Eng. Chem. Res., 2006, 45, 4389–4415 CrossRef CAS.
- E. Mullins, Y. Liu, A. Ghaderi and S. D. Fast, Ind. Eng. Chem. Res., 2008, 47, 1707–1725 CrossRef CAS.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- J. Katon and M. Cohen, Can. J. Chem., 1975, 53, 1378–1386 CrossRef CAS.
- O. Borodin, M. Olguin, P. Ganesh, P. R. C. Kent, J. L. Allen and W. A. Henderson, Phys. Chem. Chem. Phys., 2016, 18, 164–175 RSC.
- W. Cui, Y. Lansac, H. Lee, S. T. Hong and Y. H. Jang, Phys. Chem. Chem. Phys., 2016, 18, 23607–23612 RSC.
- L. E. Camacho-Forero, T. W. Smith and P. B. Balbuena, J. Phys. Chem. C, 2017, 121, 182–194 CrossRef CAS.
- H. B. Han, S. S. Zhou, D. J. Zhang, S. W. Feng, L. F. Li, K. Liu, W. F. Feng, J. Nie, H. Li, X. J. Huang, M. Armand and Z. B. Zhou, J. Power Sources, 2011, 196, 3623–3632 CrossRef CAS.
- L. Suo, O. Borodin, W. Sun, X. Fan, C. Yang, F. Wang, T. Gao, Z. Ma, M. Schroeder and A. von Cresce, Angew. Chem., Int. Ed., 2016, 55, 7136–7141 CrossRef CAS PubMed.
- E. Krämer, T. Schedlbauer, B. Hoffmann, L. Terborg, S. Nowak, H. J. Gores, S. Passerini and M. Winter, J. Electrochem. Soc., 2013, 160, A356–A360 CrossRef.
- X. Zhang and T. Devine, J. Electrochem. Soc., 2006, 153, B344–B351 CrossRef CAS.
- X. Zhang and T. Devine, J. Electrochem. Soc., 2006, 153, B375–B383 CrossRef CAS.
- S. T. Myung, Y. Sasaki, S. Sakurada, Y. K. Sun and H. Yashiro, Electrochim. Acta, 2010, 55, 288–297 CrossRef.
- H. Moon, T. Mandai, R. Tatara, K. Ueno, A. Yamazaki, K. Yoshida, S. Seki, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2015, 119, 3957–3970 CrossRef CAS.
- Y. Yamada, C. H. Chiang, K. Sodeyama, J. Wang, Y. Tateyama and A. Yamada, ChemElectroChem, 2015, 2, 1687–1694 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07824j |
| This journal is © The Royal Society of Chemistry 2019 |