
dPCR – the digital polymerase chain reaction
Analytical Methods Committee AMCTB No. 79
First published on 7th July 2017
Abstract
Quantitative real-time PCR (qPCR) is based on the assumption that the amplification of DNA target molecules is exponential. DNA targets may be quantified by comparing the number of amplification cycles required to achieve a predetermined signal threshold to that obtained for a calibrant. However, many factors complicate this calculation, creating uncertainties and inaccuracies. Digital PCR (dPCR) is a modification of the qPCR method that can be employed to quantify precisely defined nucleic acid targets. The technique is based on the concept of limiting dilutions, which involves the partitioning of a PCR reaction into multiple sub-reactions such that each sub-reaction either contains none or one or more DNA targets. Following thermal cycling, reactions are classified as either positive (target detected) or negative (no target detected), hence providing the basis for a digital output format. By determining the proportion of empty partitions, Poissonian statistics can be applied and the initial number of target molecules present can be estimated.
The digital polymerase chain reaction (dPCR) is a modification of the conventional PCR method that can be used to quantify precisely-defined nucleic acid targets, including DNA, cDNA, and RNA, and offers an alternative to conventional qPCR for quantifying the amount of DNA present in a sample.
Conventional qPCR is made more complicated by a number of factors that can create additional uncertainties and inaccuracies in the process of quantification. Among the most important is that the results are highly sensitive to small changes in amplification efficiency; for example, over 20 amplification cycles, a 2% difference in amplification efficiency between reference materials and test sample results in up to 50% change in the estimated amount of DNA.

How dPCR works
The dPCR technique is based on the concept of limiting dilutions, where the PCR reaction is split into a large number of sub-reactions such that some reactions will not contain any target at all. Sample partitioning can be achieved by the use of micro well plates, oil emulsion, and arrays of miniaturised chambers with nucleic acid binding surfaces. Because the quantification of the target is based on counting the number of positive PCR reactions obtained, the number of PCR reactions required to perform the analysis has to be such that the Poisson distribution of molecules results in the average number of molecules in each reaction being less than approximately five. In practice this means that a minimum of several hundred partitions is needed for an acceptable level of uncertainty in the estimate. More recent technology uses hundreds of thousands or even millions of reactions.On completion of the thermal cycling process reactions are classified as either positive (target detected) or negative (no target detected), thus providing the basis for a digital output format. The only prerequisite for this is that sufficient amplification occurs to allow detection. Further, if amplification is sufficiently reliable, almost every molecule present will be detected, allowing a direct count of the number of molecules present. As a direct consequence of this, dPCR can be defined as an absolute method for quantification, and differs significantly from conventional qPCR where quantitation is based on the proportionality between the numbers of amplification cycles required for a sample to exceed a predetermined sample signal threshold and relative to that obtained for a calibrant of known analyte concentration.
Quantification of dPCR
When a solution of homogenously-distributed DNA targets is partitioned, some of the partitions will contain more than one target molecule. Unfortunately, a partition that contains more than one DNA target that has been amplified by PCR is not distinct from a partition that initially contains only one DNA target. Fortunately, the distribution of target molecules across the multiple partitions will be Poissonian in nature (targets distribute between partitions independently and at a fixed rate). By using the Poisson distribution, the initial number of target molecules present can be estimated from the number of positive and negative partitions observed following thermal cycling (Fig. 1). Absolute quantification of the total number of target molecules present in a test sample using dPCR is then relatively straightforward, and can be achieved with application of eqn (1):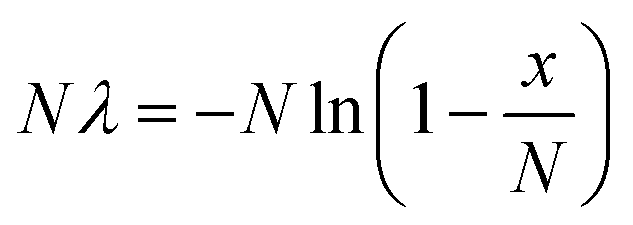 | (1) |
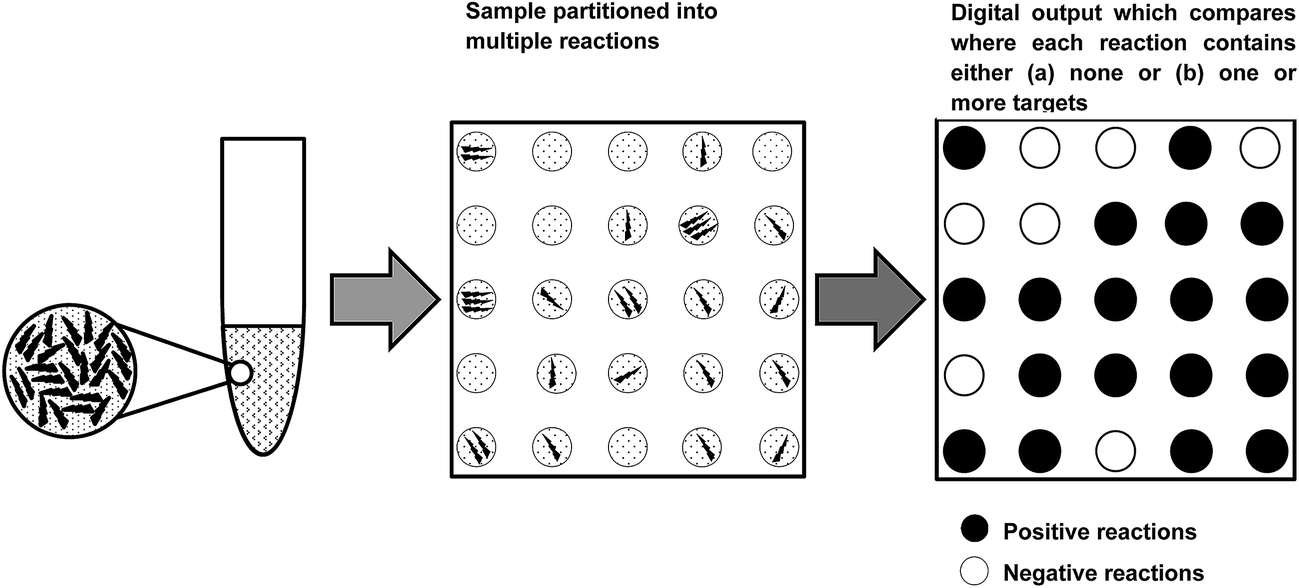 | ||
| Fig. 1 Generation of a digital output from the application of a limiting dilution. | ||
Conversion of the result to a concentration of the target can be achieved by factoring-in the total volume of all of the partitions into the equation, such that:
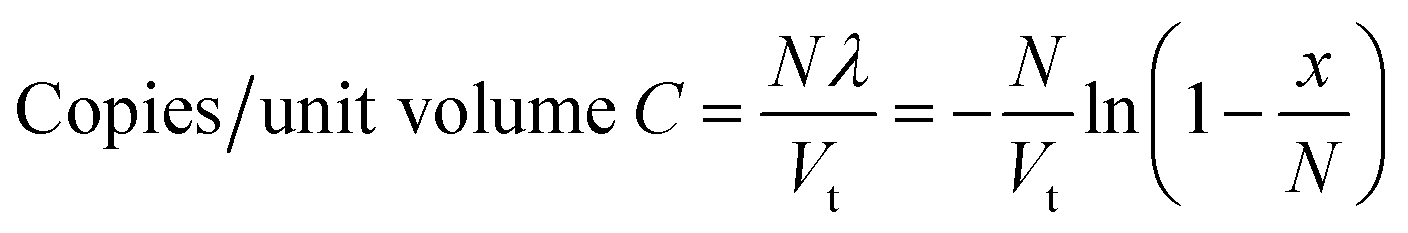 | (2) |
The technique offers several key advantages over conventional qPCR. The principal advantages are that there is no requirement for the use of a calibration curve, and the method is reportedly much less sensitive to PCR inhibition effects, giving greatly improved trueness and precision.
A further advantage is that the extreme partitioning of the sample reduces the ratio between different molecular species present in a single PCR reaction, reducing mutual inhibition (Fig. 2). This can considerably improve the capability for detecting rare mutant alleles, or foreign DNA sequences (for example, foetal DNA in maternal blood) and allow accurate quantitation of both targets where traditional qPCR might fail to detect the rare species.
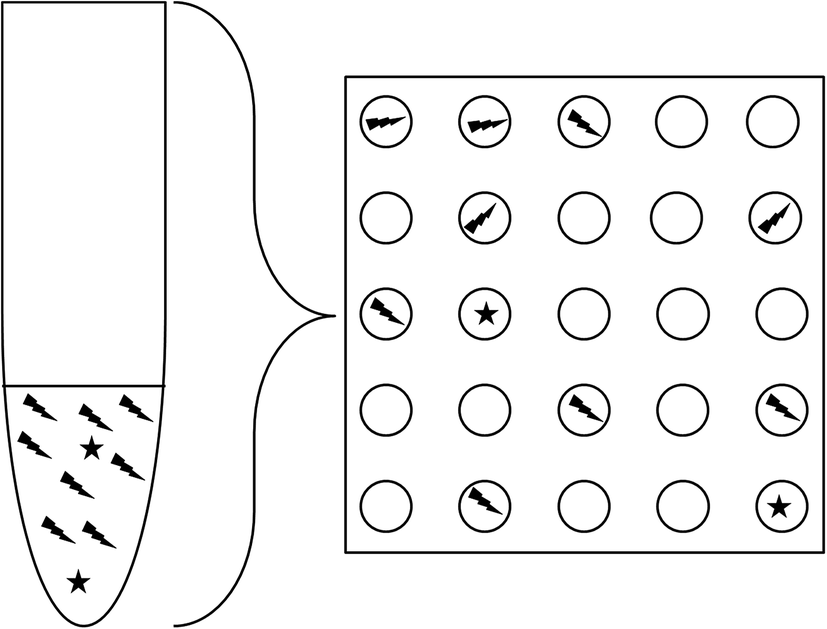 | ||
| Fig. 2 Dilution to very small counts per reaction improves the proportion of rare species in a given reaction volume, even though the average proportion remains the same. | ||
Applications
Digital PCR is particularly useful for applications where validity and precise determination are essential. Examples include the following:Absolute quantitation
• Production of certified reference materials (e.g. SRM 2366 from NIST);• Viral load determination (e.g. level of virus in 1 mL of blood).
Relative quantitation
• Copy Number Variation (CNV);• Relative expression levels.
Rare allele detection
• Tumour cells in background of wild-type cells;• Detection of circulating foetal DNA in maternal blood.
Conclusions
The current gold standard for measuring specific DNA amounts is quantitative PCR (qPCR). However, qPCR is subject to limitations that can be overcome by the application of dPCR. The dPCR approach can typically employ the same assays as used for conventional qPCR with minor modification, but reports the total number of individual target molecules in a digital format, whilst enabling a greater degree of precision and accuracy to be achieved. The approach is becoming increasingly adopted for applications that require high sensitivity and is rapidly becoming the standard method employed by international metrology institutes for assigning absolute DNA copy number values to reference materials. With continued technical development within the field, instrument performance will continue to improve and associated costs fall, which will enable access to the technology by an increasing number of laboratories.Timothy Wilkes and Steven Ellison (LGC Ltd).
This Technical Brief was prepared for the Analytical Methods Committee and approved by the Committee on 16/05/17.

| This journal is © The Royal Society of Chemistry 2017 |