
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regioselective routes to orthogonally-substituted aromatic MIDA boronates†
Adam J.
Close
a,
Paul
Kemmitt
b,
S.
Mark Roe
c and
John
Spencer
*a
aDept of Chemistry, School of Life Sciences, University of Sussex, Falmer, BN1 9QJ, UK. E-mail: j.spencer@sussex.ac.uk
bAstraZeneca, Mereside Alderley Park, Macclesfield, SK10 4TG, UK
cDept of Biochemistry, School of Life Sciences, University of Sussex, Falmer, BN1 9QJ, UK
First published on 14th June 2016
Abstract
A series of tetrasubstituted aromatics has been synthesized, many of which are based on elaborated N-methyliminodiacetic acid (MIDA)-boronates. A sequence employing nitration, bromination, stepwise Suzuki–Miyaura (SM) coupling with a boronic acid, then base-mediated unmasking of the boronic acid from the MIDA-boronate and a second SM-coupling has led to our desired, mainly 1,2,4,5-substituted tetrasubstituted aromatic targets.
Introduction
Tetrasubstituted aromatics are present in a number of bioactive molecules including kinase inhibitors such as Iressa, Tagrisso and Vemurafenib.1–3 Carey et al. outlined that these seemingly simple, desired scaffolds are not readily available or are highly expensive; thus new methodology towards these molecules is required.4 In order to enable the generation of libraries of related 1,2,4,5- and 1,2,3,4-substituted systems, we sought novel, selective, efficient synthetic routes to the crucial generic building blocks I and II (Fig. 1).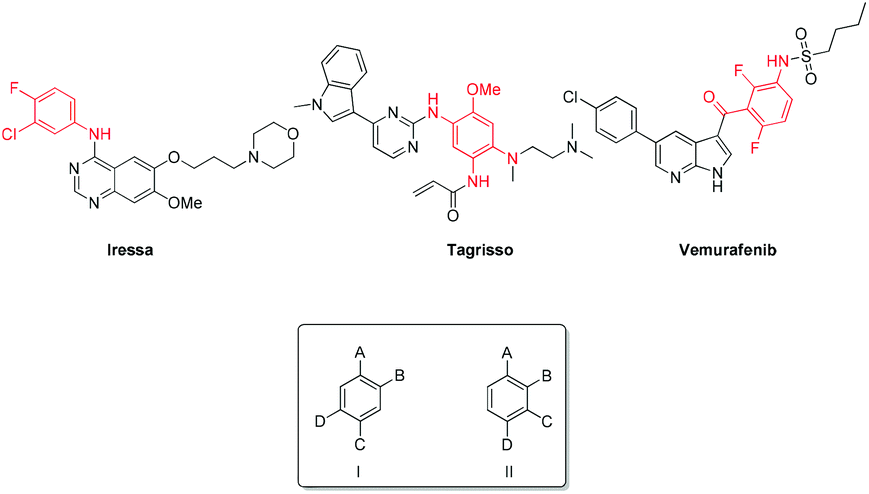 | ||
| Fig. 1 Examples of 1,2,4,5- and 1,2,3,4-substituted aromatic scaffolds. | ||
MIDA-boronates (Fig. 2) are versatile intermediates in a host of coupling reactions. Of note is their orthogonality with boronic acids in metal-catalyzed coupling chemistry, whereupon a subsequent aqueous base-mediated reaction can unmask the boronic acid functionality to enable a second coupling reaction.5,6 This iterative methodology has been used in a variety of palladium cross-coupling reactions e.g. Buchwald–Hartwig, Negishi, Sonogashira, Heck and Stille couplings.7–11 Indeed, these steps can be combined in environmentally-friendly telescopic couplings.12
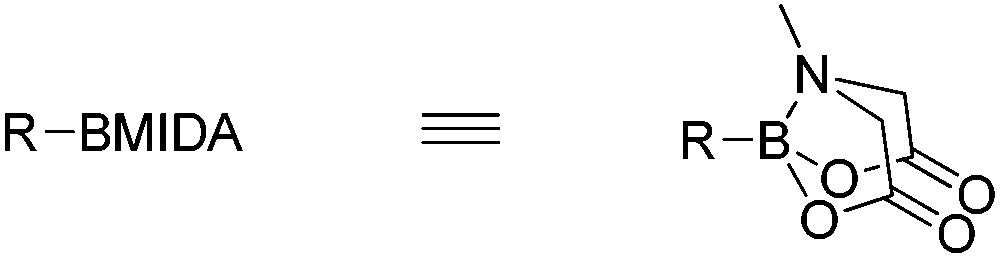 | ||
| Fig. 2 Bicyclic form of MIDA boronate. | ||
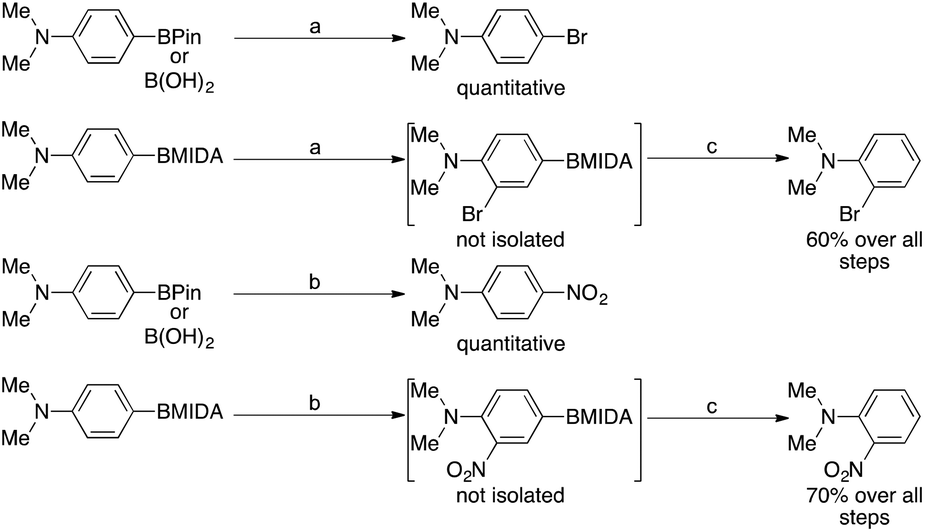
Burke et al. published a paper in 2015, that outlines a machine that enabled the synthesis 14 distinct classes of small molecules in a fully automated process.13 This uses a catch and release method of purification and shows the potential for MIDA boronates to enable quick library generation. However, they stated that the current drawbacks included the low availability of interesting MIDA boronate starting materials outlining the importance of new methodologies towards polysubstituted aromatic MIDA boronate cores.
Cheon et al. showed that under mild conditions, MIDA boronates can be used to block the para position in an aromatic group in order to enable selective ortho electrophilic substitution i.e. bromination or nitration. These intermediates were never isolated, as the MIDA boronate moiety was removed in situ (Scheme 1).14 Conversely, they showed that the corresponding boronic acids or pinacol esters underwent ipso substitution under these conditions. This was also observed as early as 1959 by Soloway15 who showed that if the directing effect of the substituents in a given phenylboronic acid would present nitration at the carbon ipso to the boronic acid, ipso nitration would be observed, displacing the boron.
 | ||
Scheme 1 Reaction conditions: a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) NBS 1 eq., DCM, rt, 10 min. bNBS 1 eq., AgNO3 1 eq., EtCN, reflux, 3 h. c(i) NaOH, THF, 10 min, (ii) wet DMSO 100 °C, 24 h. NBS 1 eq., DCM, rt, 10 min. bNBS 1 eq., AgNO3 1 eq., EtCN, reflux, 3 h. c(i) NaOH, THF, 10 min, (ii) wet DMSO 100 °C, 24 h. | ||
Our recently reported microwave-mediated synthesis of MIDA-boronates enabled us to quickly access a number of templates on which to attempt electrophilic nitration and bromination chemistry (Scheme 2).16,17
 | ||
| Scheme 2 Formation of MIDA boronates. | ||
It has also been reported that MIDA boronates are highly stable under acidic Jones oxidations conditions.18 We found that nitration could be easily achieved using classical conditions e.g. nitric acid in sulfuric acid (Table 1).19
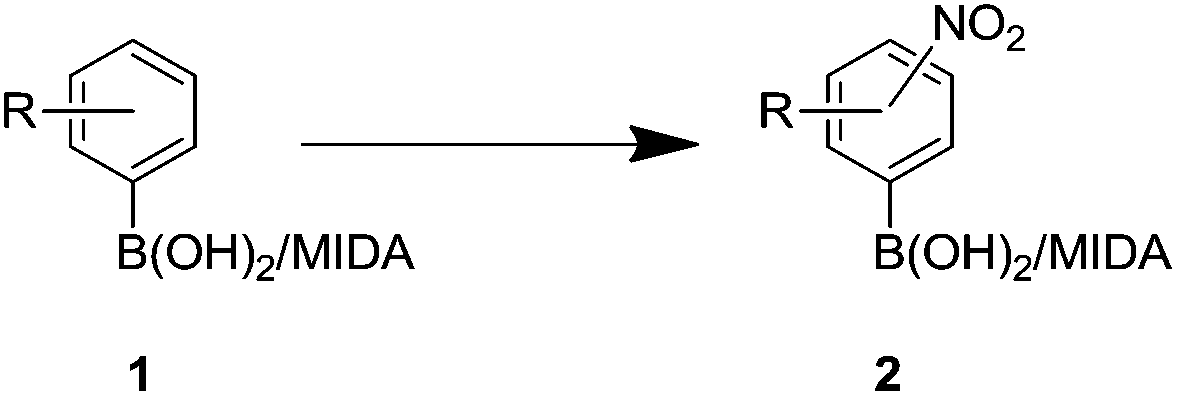

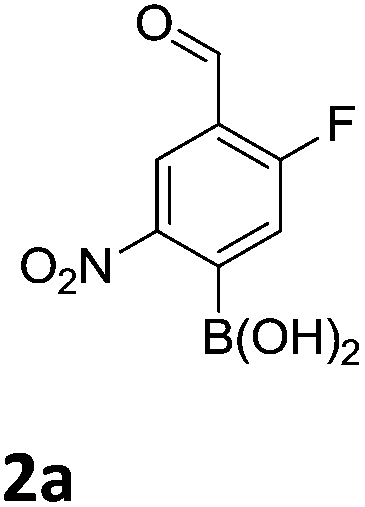
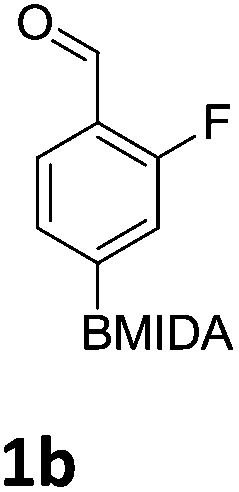
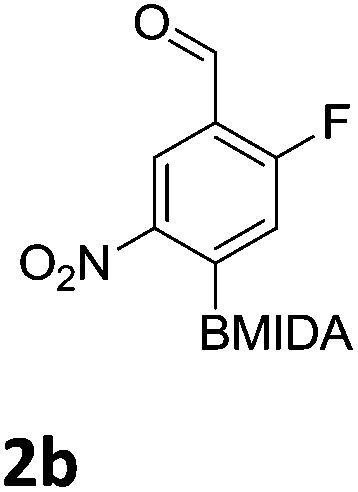
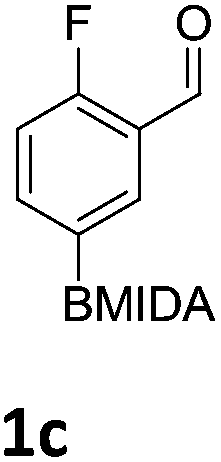
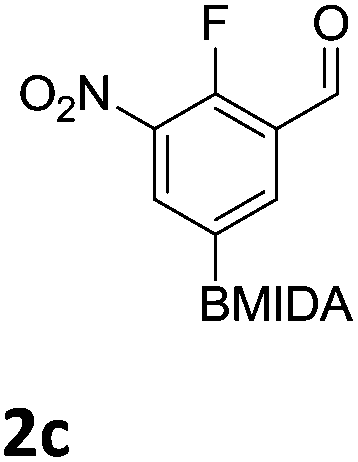
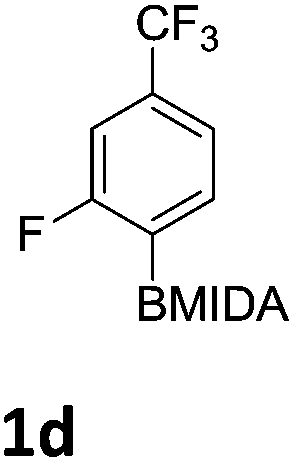
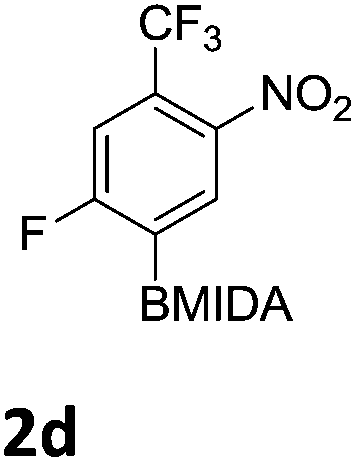
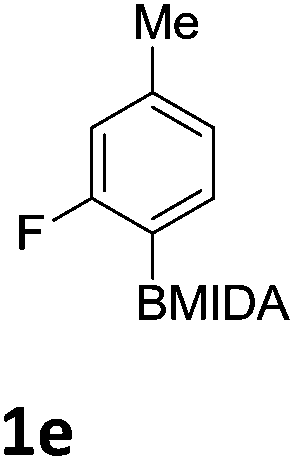
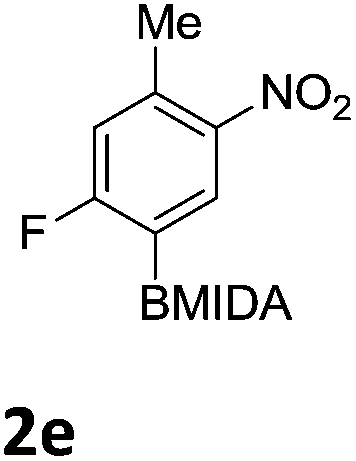
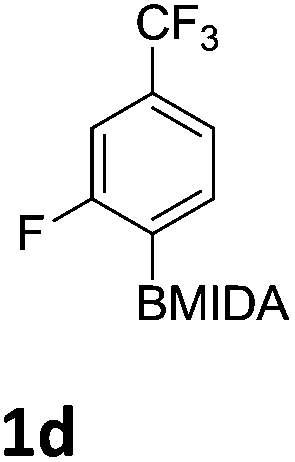
The nitration of poly-substituted phenylboronic acids has previously been achieved; hence we were not overly surprised when the nitration of 1a was high yielding, scalable and conveniently regioselective, directed by the halogen and formyl substituents (12 mmol scale, Table 1).20 Gratifyingly, comparable yields were observed when its corresponding MIDA boronate 1b was nitrated under these harsh conditions yielding 2b. It was also found that the MIDA “handle” aided the solubility of 1b in the reaction mixture. Ipso nitration at the boron was observed in the nitration of 1f (Scheme 3), due to the directing effect discussed above. Nevertheless we were pleased to see that its corresponding MIDA boronate 1c was somewhat resistant to ipso nitration at the boron center (Table 1, entry 3). The nitration of 1d and 1e required the nitric acid to be added in approximately stoichiometric amounts, affording 2d and 1e respectively (Table 1, entries 4 and 5).
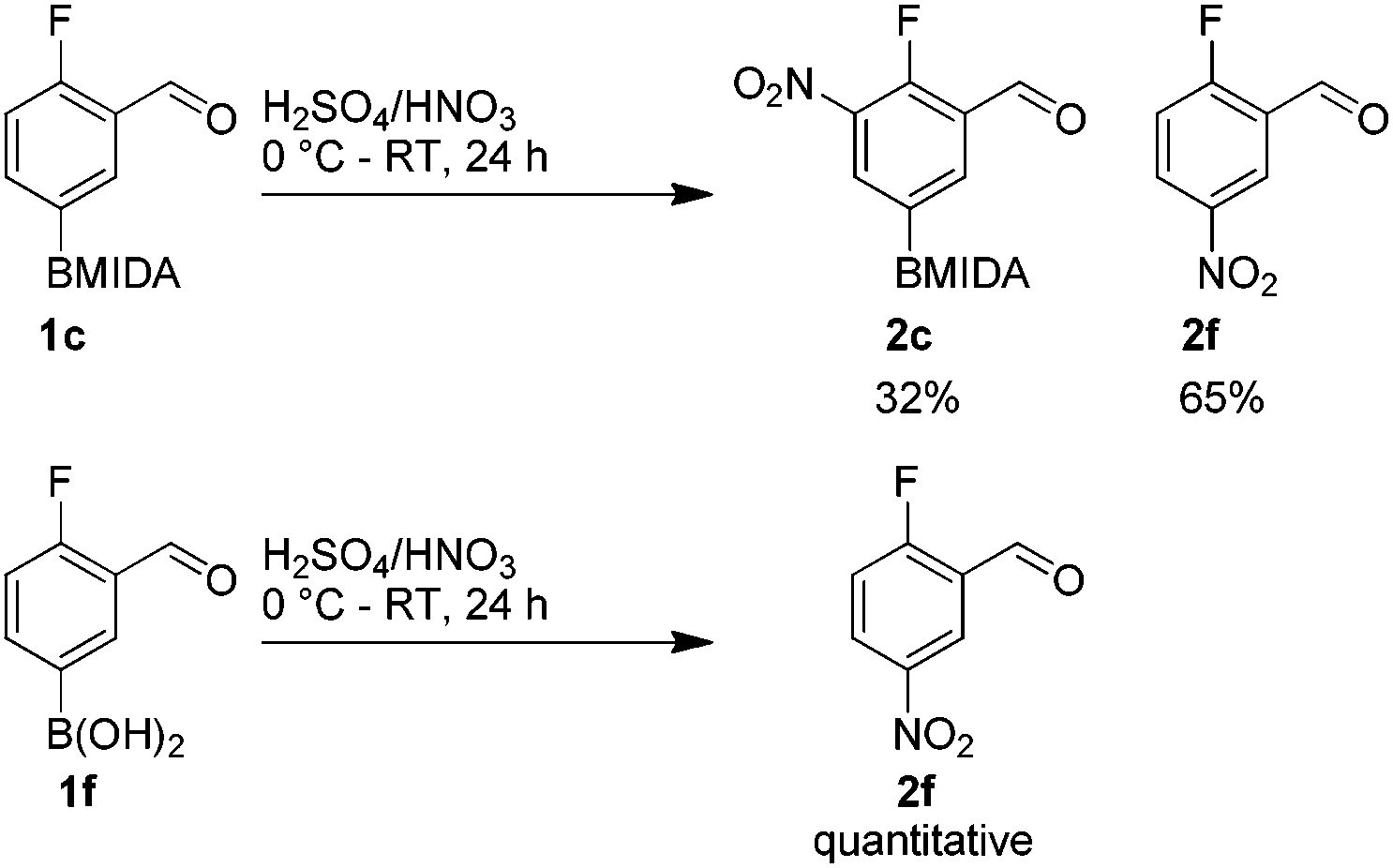 | ||
| Scheme 3 Nitration of 1c and its corresponding parent boronic acid 1f. | ||
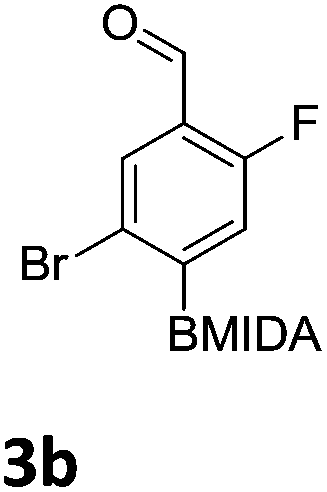
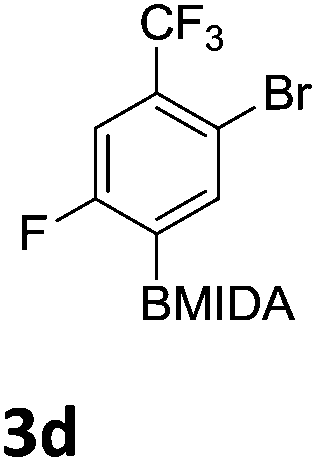
After the success of the initial nitration studies our efforts moved on to the bromination of phenyl MIDA boronates. We did not attempt the bromination of 1a since we preferred to adhere to the orthogonality afforded by the MIDA protecting group. We note that Hall et al. described the orthogonality of iodides with BPin.21 This first foray in to bromination again used harsh conditions described by Saiganesh et al. e.g. H2SO4 with N-bromosuccinimide (NBS).22 Adopting this methodology, 3b and 3d were synthesized (Table 2) and we were able to acquire a single crystal X-ray diffraction structure of 3b (Fig. 3). Higher yields were observed in the case of 3d when the reaction temperature was reduced to 30 °C.
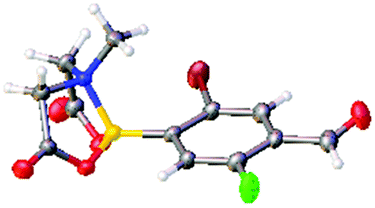 | ||
| Fig. 3 Solid state structure of 3b. | ||

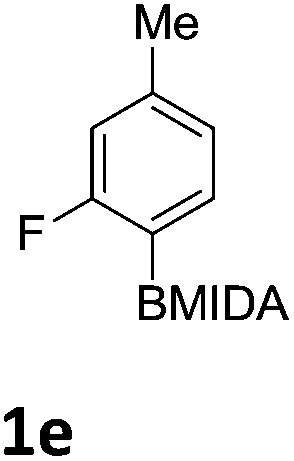
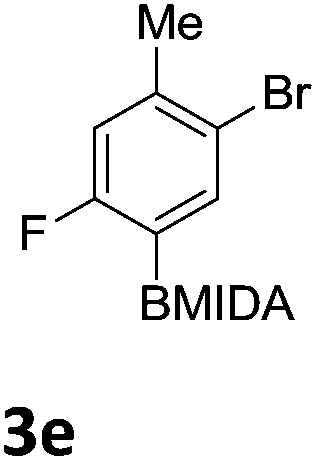
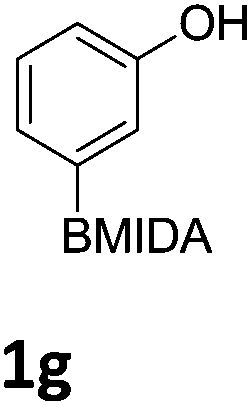
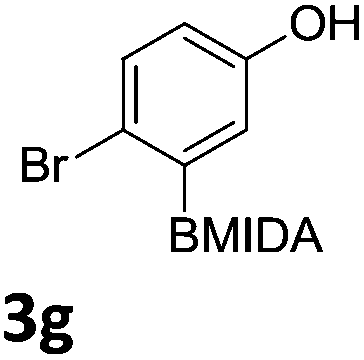
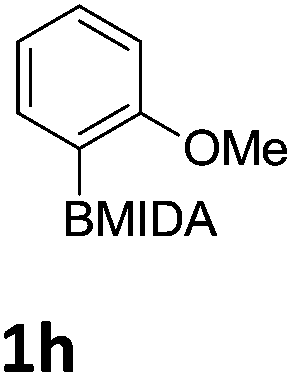
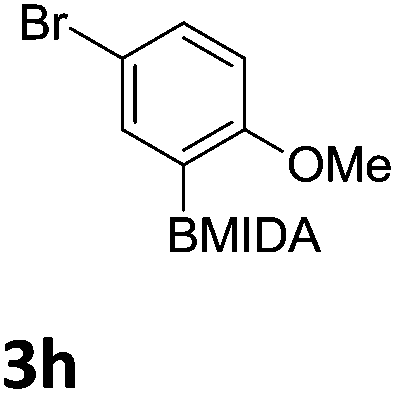
We found that activated compounds e.g.Table 2, entries 3–5, required milder conditions. Bovonsombat et al. demonstrated that NBS could be used to brominate phenols in the presence of stoichiometric PTSA.23 Adopting a similar methodology, utilizing H2SO4 instead of PTSA, led to the synthesis of 3e, 3g, and 3h, all in good yields. The bromination of 1c unfortunately gave an inseparable mixture of products, including the product of ipso bromination (Scheme 4).
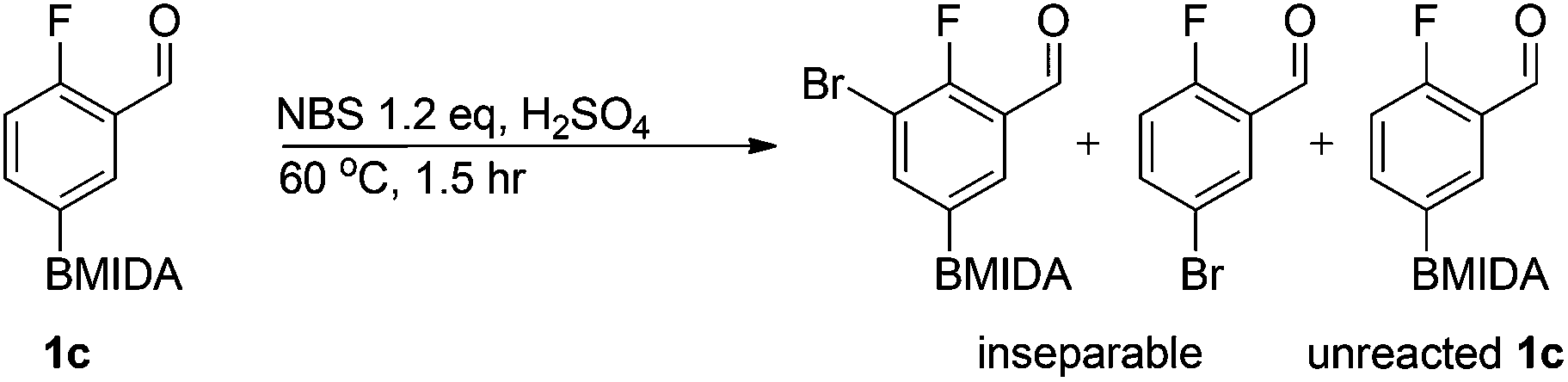 | ||
| Scheme 4 Bromination of compound 1c. | ||
Bromination, via ortho-selective C–H activation, with respect to the acetamide, using arylacetamide MIDA boronates was attempted adopting a similar protocol to Bedford et al., (Scheme 5).24
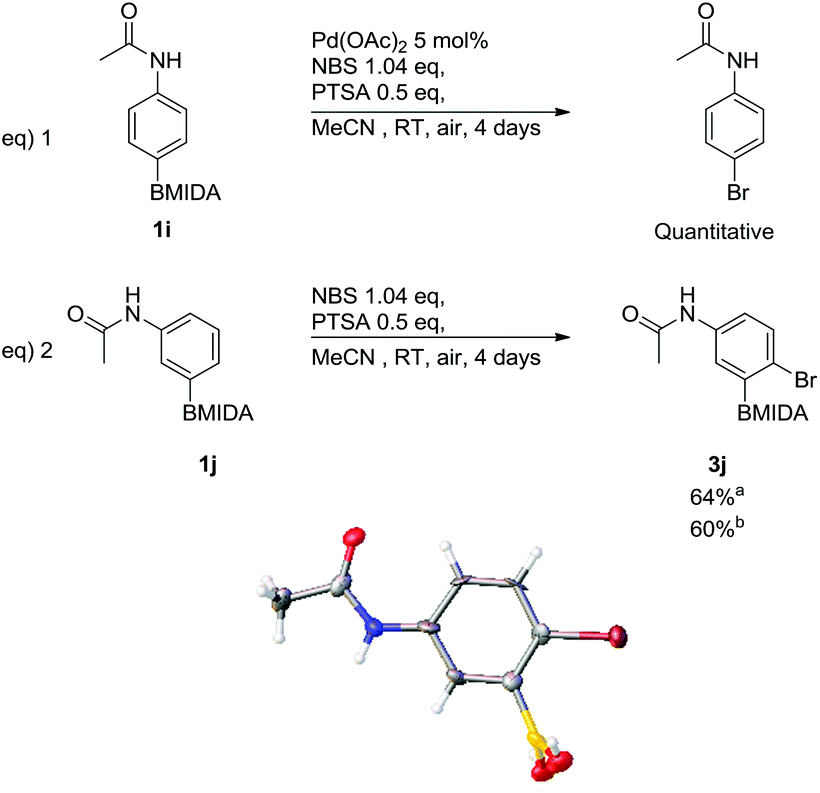 | ||
| Scheme 5 Attempted bromination via ortho-selective C–H activation, including X-ray structure of 3j′ (MIDA protection was cleaved in crystallisation process). aWith 5 mol% Pd(OAc)2, bwithout Pd(OAc)2. | ||
The only modification from Bedford's original protocol was changing the solvent from toluene to MeCN due to the poor solubility of the MIDA boronate. The reaction of 4-acetamidophenyl MIDA boronate, (1i) led to complete ipso-bromination of the MIDA boronate, forming N-4-bromophenylacetamide in a quantitative yield (Scheme 5, eqn (1)). This shows that the acetamide was unable to ortho direct the reaction. 3-Acetamidophenyl MIDA boronate was also subjected to these conditions yet in this case only substitution para to the acetamide occurred leading to 3j (Scheme 5, eqn (2)). These same conditions were also trialled using DMF as the solvent giving 3j in a comparable yield, hence MeCN was kept as the solvent of choice due to its relative ease of handling i.e. low boiling point. The reaction was also attempted without palladium(II) acetate in MeCN (Scheme 5, eqn (2)); 3j was again obtained in a comparable yield. This was expected due to the lack of C–H activation observed in the earlier reactions. This regiochemistry was confirmed via single crystal X-ray diffraction (Scheme 5). During the crystallization process the MIDA group was cleaved and the structure of the resulting free boronic acid 3j′ is shown. NOESY NMR also confirmed this regiochemistry of the parent MIDA boronate 3j.
It is pertinent to reflect upon failed bromination attempts on aryl MIDA derivatives (Table 3). These results show one of the drawbacks of this technique; if more than one major regioisomer can be produced then these reactions produce inseparable mixtures of compounds; evidenced by LCMS and 1H NMR analysis of crude reaction mixtures.25
|
|
||
|---|---|---|
| R | Conditions | Results |
| a NBS 1.2 eq., H2SO4, 60 °C, 2 h. b NBS 1.2 eq., H2SO4, 30 °C, 2 h. c NBS 1 eq., H2SO4 0.5 eq., MeCN, 50 °C, 36 h. d NBS 1 eq., H2SO4 1 eq., MeCN, 50 °C, 36 h. e NBS 1.2 eq., FeCl3, MeCN, 1 eq., 80 °C, 2 h. f NBS 1.2 eq., NH4NO3, MeCN 1 eq., 80 °C, 2 h. | ||
| m-Formyl | Multiple products | |
| Multiple products | ||
| p-Formyl | Multiple products | |
| Multiple products | ||
| No reaction | ||
| No reaction | ||
| m-Methyl | Multiple products | |
| Multiple products | ||
| Ph | Multiple products | |
| m-Fluroro | No reaction | |
| Multiple products | ||
| o-Fluroro | Multiple products | |
| m-Fluroro-p-methyl | Multiple products | |
Notwithstanding the latter unsuccessful examples this methodology is still a very simple, cheap and powerful tool for producing new interesting scaffolds when directing groups are propitiously placed to enable the formation of a major or single regioisomer.
To show the synthetic value of these intermediates we attempted some rapid Suzuki cross coupling reactions. A rapid screen of catalysts and bases was undertaken under anhydrous conditions (to maintain the MIDA boronate) in the absence of a glovebox, since we desired a method that was more widely applicable for medicinal chemists and industrialists. To do this an electronically challenging model reaction system was selected as (Scheme 6) where we took the relatively electron poor 4-acetylphenylboronic acid 1l.
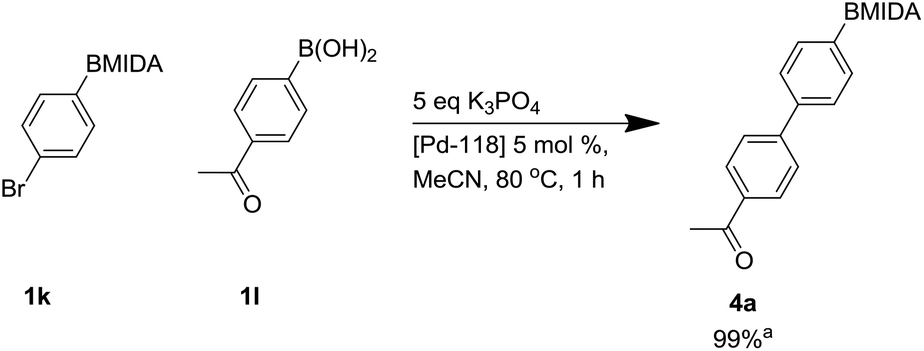 | ||
| Scheme 6 Suzuki reaction using Pd-118. aConversion calculated by LCMS. | ||
Initially this was attempted using microwave heating as in previous studies it has shown to reduce reaction times.26–29 Under the anhydrous conditions this cross-coupling gave low yields, using a variety of catalysts and bases. Camera monitoring of these microwave-mediated processes revealed that stirring never initiated due to flocculation of the base under these anhydrous conditions (Fig. 4).30–32
 | ||
| Fig. 4 Flocculation of the potassium phosphate, demonstrating that no stirring occurs in the microwave vessel while the solvent is boiling. | ||
Stirring is key to uniform heating in microwave reactions; studies have shown that reaction temperatures can differ up to 30 °C between the middle and outside of the reaction vessel in the absence of efficient stirring.33 We observed boiling of the MeCN (boiling point 82 °C) in these reactions before the microwave heated to its maximum temperature setting of 80 °C, as measured by an external IR thermometer. Also the microwave continued to heat to almost 90 °C after the irradiation was removed due to it overshooting the maximum temperature. As the heating in these reactions was monitored via an IR thermometer on the outside of the vessel, we could conclude that the internal temperature of these unstirred reactions was much higher than the set parameters. This was hypothesized to be the cause of the low yields, in these microwave reactions.
During microwave studies the most successful catalyst trialled was Pd 118 ([1,1′-bis(di-tert-butylphosphino)ferrocene]dichloro palladium(II) or PdCl2(dtbpf)) with potassium phosphate (see ESI†). Consequently, Pd-118 was used as catalyst with K3PO4, with standard thermal heating, giving a fast full conversion by LCMS to product 4a within 1 h.34
This methodology facilitated the generation of a small library of compounds, as shown in Scheme 7. As can be seen, this was tolerant of a number of substituents including heterocycles (4b, 4e, 4f) and fluoro substituents, which are important in a number of bioactive molecules.35
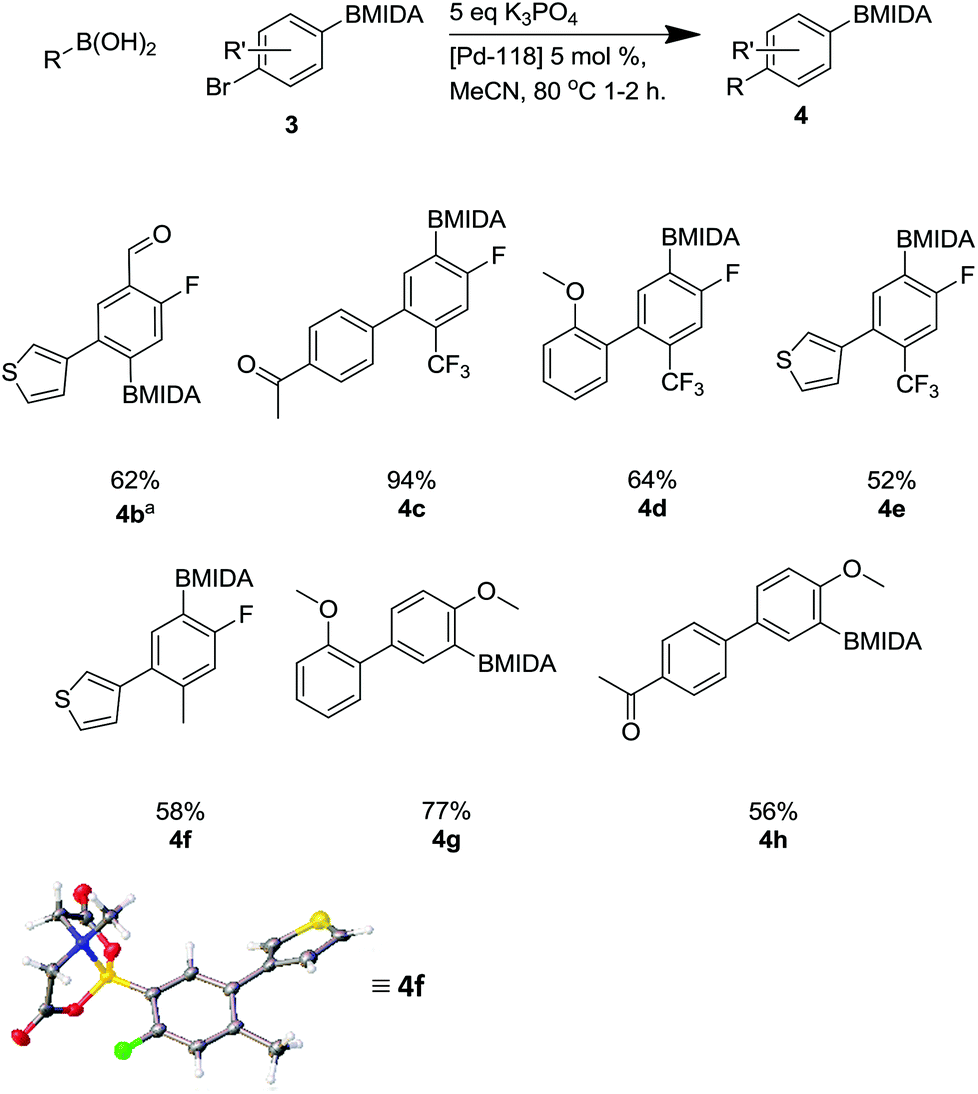 | ||
| Scheme 7 Suzuki reaction using Pd-118 including X-ray structure of 4f. aReaction required a 12 hour reaction time. | ||
Next, aqueous Suzuki couplings were attempted, with the aim of deprotecting the MIDA group in situ in order to perform a second C–C bond formation. This was optimized on the coupling of 4a with p-bromotoluene to form the known para-terphenyl 5a (Table 4).6,36 This enabled quick microwave-mediated optimization via crude 1H-NMR analysis on a small scale using a phase separator to facilitate efficient work-up.
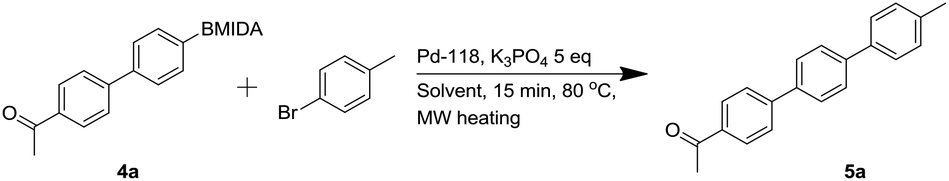
Initial optimization reactions with 5 mol% of Pd-118 catalyst, established that a THF/water mixture was superior to acetonitrile or dioxane/water mixtures although lowering the catalyst loading to 2.5 mol% led to a respectable 73% conversion to product.
This methodology was applied to the unoptimized synthesis of a small library of four representative final tetrasubstituted aromatic compounds with the required regiochemistry (Scheme 8).
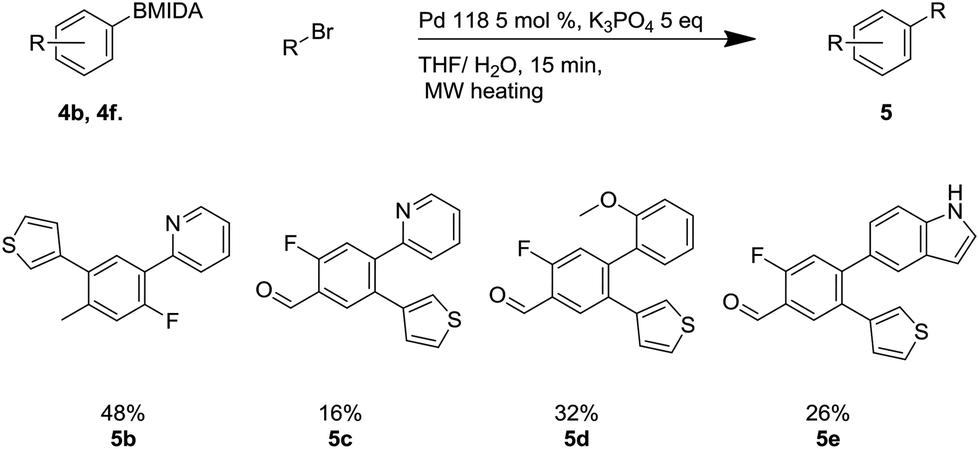 | ||
| Scheme 8 Aqueous deprotection Suzuki cross-couplings. | ||
Conclusions
In conclusion, examples of iterative Suzuki cross-couplings have been achieved, illustrating the potential of polysubstituted aromatic compounds derived from the electrophilic aromatic substitution of synthons containing orthogonal groups. This demonstrates how complex polyaromatics with tetrasubstituted cores can be synthesized in a simple four-step procedure i.e. MIDA protection, bromination, anhydrous cross-coupling and aqueous cross-coupling. This was performed using inexpensive reagents starting from commercially available trisubstituted boronic acid. The Suzuki cross-coupling reactions, both anhydrous and aqueous, suffered from low yields, particular the latter.Eleven polysubstituted MIDA-aromatics have been synthesized, ten of which are novel, via traditional bromination and nitration methodologies. These methods have shown to be scalable and give the desired products containing orthogonal groups, with specific regiochemistry.
Acknowledgements
AstraZeneca and the University of Sussex are acknowledged for funding (AJC). We would also like to thank the EPSRC UK National Mass Spectrometry Facility (University of Swansea) and Dr Alaa Abdul-Sada (University of Sussex) for performing mass spectrometry.Notes and references
- A. J. Barker, K. H. Gibson, W. Grundy, A. A. Godfrey, J. J. Barlow, M. P. Healy, J. R. Woodburn, S. E. Ashton, B. J. Curry, L. Scarlett, L. Henthorn and L. Richards, Bioorg. Med. Chem. Lett., 2001, 11, 1911–1914 CrossRef CAS PubMed.
- M. R. V. Finlay, M. Anderton, S. Ashton, P. Ballard, P. A. Bethel, M. R. Box, R. H. Bradbury, S. J. Brown, S. Butterworth, A. Campbell, C. Chorley, N. Colclough, D. A. E. Cross, G. S. Currie, M. Grist, L. Hassall, G. B. Hill, D. James, M. James, P. Kemmitt, T. Klinowska, G. Lamont, S. G. Lamont, N. Martin, H. L. Mcfarland, M. J. Mellor, J. P. Orme, D. Perkins, P. Perkins, G. Richmond, P. Smith, R. A. Ward, M. J. Waring, D. Whittaker, S. Wells and G. L. Wrigley, J. Med. Chem., 2014, 57, 8249–8267 CrossRef CAS PubMed.
- G. Bollag, P. Hirth, J. Tsai, J. Zhang, P. N. Ibrahim, H. Cho, W. Spevak, C. Zhang, Y. Zhang, G. Habets, E. A. Burton, B. Wong, G. Tsang, B. L. West, B. Powell, R. Shellooe, A. Marimuthu, H. Nguyen, K. Y. J. Zhang, D. R. Artis, J. Schlessinger, F. Su, B. Higgins, R. Iyer, K. D'Andrea, A. Koehler, M. Stumm, P. S. Lin, R. J. Lee, J. Grippo, I. Puzanov, K. B. Kim, A. Ribas, G. A. McArthur, J. A. Sosman, P. B. Chapman, K. T. Flaherty, X. Xu, K. L. Nathanson and K. Nolop, Nature, 2010, 467, 596–599 CrossRef CAS PubMed.
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 CAS.
- E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2007, 129, 6716–6717 CrossRef CAS PubMed.
- M. D. Burke and E. P. Gillis, US Pat., 0030238 A1, 2009 Search PubMed.
- N. Colgin, T. Flinn and S. L. Cobb, Org. Biomol. Chem., 2011, 9, 1864–1870 CAS.
- J. E. Grob, M. A. Dechantsreiter, R. B. Tichkule, M. K. Connolly, A. Honda, R. C. Tomlinson and L. G. Hamann, Org. Lett., 2012, 14, 5578–5581 CrossRef CAS PubMed.
- A. R. Burns, G. D. McAllister, S. E. Shanahan and R. J. K. Taylor, Angew. Chem., Int. Ed., 2010, 49, 5574–5577 CrossRef CAS PubMed.
- S. J. Lee, K. C. Gray, J. S. Paek and M. D. Burke, J. Am. Chem. Soc., 2008, 130, 466–468 CrossRef CAS PubMed.
- J. R. Struble, S. J. Lee and M. D. Burke, Tetrahedron, 2010, 66, 4710–4718 CrossRef CAS.
- M. C. Bryan, B. Dillon, L. G. Hamann, G. J. Hughes, M. E. Kopach, E. A. Peterson, M. Pourashraf, I. Raheem, P. Richardson, D. Richter and H. F. Sneddon, J. Med. Chem., 2013, 56, 6007–6021 CrossRef CAS PubMed.
- J. Li, S. G. Ballmer, E. P. Gillis, S. Fujii, M. J. Schmidt, A. M. E. Palazzolo, J. W. Lehmann, G. F. Morehouse and M. D. Burke, Science, 2015, 347, 1221–1226 CrossRef CAS PubMed.
- S. J. Ahn, C. Y. Lee, N. K. Kim and C. H. Cheon, J. Org. Chem., 2014, 79, 7277–7285 CrossRef CAS PubMed.
- A. H. Soloway, J. Am. Chem. Soc., 1959, 81, 3017–3019 CrossRef CAS.
- A. J. Close, P. Kemmitt, M. K. Emmerson and J. Spencer, Tetrahedron, 2014, 70, 9125–9131 CrossRef CAS.
- A. J. Close, V. Corden and J. Spencer, CEM microwave applications., URL: http://www.cemmicrowave.co.uk/assets/cameramida-boronate-app-note.pdf, Date accessed: 11-15-2015, 1–4.
- E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2008, 130, 14084–14085 CrossRef CAS PubMed.
- H. J. Breslin, M. J. Kukla, D. W. Ludovici, R. Mohrbacher, W. Ho, M. Miranda, J. D. Rodgers, T. K. Hitchens and G. Leo, J. Med. Chem., 1995, 38, 771–793 CrossRef CAS PubMed.
- F. R. Bean and J. R. Johnson, J. Am. Chem. Soc., 1932, 54, 4415–4425 CrossRef CAS.
- R. M. Al-Zoubi and D. G. Hall, Org. Lett., 2010, 12, 2480–2483 CrossRef CAS PubMed.
- K. Rajesh, M. Somasundaram, R. Saiganesh and K. K. Balasubramanian, J. Org. Chem., 2007, 72, 5867–5869 CrossRef CAS PubMed.
- P. Bovonsombat, R. Ali, C. Khan, J. Leykajarakul, K. Pla-on, S. Aphimanchindakul, N. Pungcharoenpong, N. Timsuea, A. Arunrat and N. Punpongjareorn, Tetrahedron, 2010, 66, 6928–6935 CrossRef CAS.
- R. B. Bedford, M. F. Haddow, C. J. Mitchell and R. L. Webster, Angew. Chem., Int. Ed., 2011, 50, 5524–5527 CrossRef CAS PubMed.
- K. Tanemura, T. Suzuki, Y. Nishida, K. Satsumabayashi and T. Horaguchi, Chem. Lett., 2003, 32, 932–933 CrossRef CAS.
- V. V. Namboodiri and R. S. Varma, Green Chem., 2001, 3, 146–148 RSC.
- P. Nun, J. Martinez and F. Lamaty, Synlett, 2009, 1761–1764 CAS.
- J. Spencer, C. B. Baltus, N. J. Press, R. W. Harrington and W. Clegg, Tetrahedron Lett., 2011, 52, 3963–3968 CrossRef CAS.
- J. Spencer, C. B. Baltus, H. Patel, N. J. Press, S. K. Callear, L. Male and S. J. Coles, ACS Comb. Sci., 2011, 13, 24–31 CrossRef CAS PubMed.
- S. V. Ley, R. J. Ingham, M. O'Brien and D. L. Browne, Beilstein J. Org. Chem., 2013, 9, 1051–1072 CrossRef CAS PubMed.
- M. D. Bowman, N. E. Leadbeater and T. M. Barnard, Tetrahedron Lett., 2008, 49, 195–198 CrossRef CAS.
- R. A. Skilton, R. A. Bourne, Z. Amara, R. Horvath, J. Jin, M. J. Scully, E. Streng, S. L. Y. Tang, P. A. Summers, J. Wang, E. Pérez, N. Asfaw, G. L. P. Aydos, J. Dupont, G. Comak, M. W. George and M. Poliakoff, Nat. Chem., 2014, 7, 1–5 CrossRef PubMed.
- M. A. Herrero, J. M. Kremsner, C. O. Kappe, K. V. Graz and A. Graz, J. Org. Chem., 2008, 73, 36–47 CrossRef CAS PubMed.
- J. D. Moseley, P. M. Murray, E. R. Turp, S. N. G. Tyler and R. T. Burn, Tetrahedron, 2012, 68, 6010–6017 CrossRef CAS.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- J. M. A. Miguez, L. A. Adrio, A. Sousa-Pedrares, J. M. Vila, K. K. M. Hii, K. Kuok and M. Hii, J. Org. Chem., 2007, 72, 7771–7774 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1474833–1474835. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob01141a |
| This journal is © The Royal Society of Chemistry 2016 |