
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thioether functionalised gallium and indium alkoxides in materials synthesis†
Felix
Biegger
a,
Christoph
Rameshan
a,
Alexander K.
Opitz
b,
Julian
Noll
c,
Thomas
Haunold
a,
Heinrich
Lang
c and
Sven
Barth
 *a
*a
aVienna University of Technology, Institute of Materials Chemistry, Getreidemarkt 9/BC/02, 1060 Vienna, Austria. E-mail: sven.barth@tuwien.ac.at
bVienna University of Technology, Institute of Chemical Technologies and Analytics, Getreidemarkt 9, 1060 Vienna, Austria
cTechnische Universität Chemnitz, Faculty of Natural Sciences, Institute of Chemistry, Inorganic Chemistry, 09107 Chemnitz, Germany
First published on 15th June 2016
Abstract
The thermolysis behaviour of a new class of metal alkoxides containing a thioether functionality in the alkyl chain is described. Homoleptic gallium alkoxides with sufficient volatility have been investigated in low pressure chemical vapour deposition (CVD) showing the potency of the thioether to act as a sulphidisation agent during decomposition of the precursor leading to Ga2O3−xSx films. Similar thermolysis experiments were conducted in high boiling point solvents leading to Ga2O3−xSx and In2O3−xSx particles. The thermolysis products have been investigated by SEM, EDX, XRD and XPS. Moreover, initial tests of the electrical transport properties of amorphous Ga2O3−xSx films have been conducted, showing increased conductivity and altered activation energies for the sulphur containing films.
Introduction
Alkoxides are popular molecular precursors for the preparation of oxides by gas and liquid phase processing.1 The volatility, solubility and decomposition temperature of the alkoxide can be altered by the nature of the organic ligand sphere. These modifications are usually performed to alter the reactivity and solubility in sol–gel reactions2 or the volatility and decomposition path in chemical vapour deposition (CVD).3,4 Popular modifications include the use of amino or ether functionalities in the organic backbone to alter the reactivity and to achieve a low nuclearity for gas phase synthesis.5,6In contrast to atomic layer deposition, where a following reaction step can cleave the existing metal–ligand bonds efficiently, the thermolysis requires a clean decomposition step to avoid carbon contamination.7–10 Reviews by C. Carmalt's group give a comprehensive overview about the state of the art of heteroleptic and homoleptic Ga and In alkoxide derivatives.11,12
For instance, gallium oxide and indium oxide coatings have been prepared using donor-functionalised and non-donor containing alkoxide precursors by CVD.13–17 The decomposition typically leads to oxides without the incorporation of other anionic species. The only well-described exceptions are perfluorinated alcoholates leading to the incorporation of fluoride ions in the oxides upon thermolysis.18,19 Nitrogen doping of oxides to alter their electronic properties and the associated bandgap usually requires the treatment of preformed oxides in ammonia atmospheres.20 These nitrogen containing oxides can be used as efficient visible-light photocatalysts and the incorporation of some sulphur has been investigated for these applications.21 However, the modification by sulphide ions is much less investigated and mostly limited to the oxidation of metal sulphides22 or systems of solid solutions, such as zinc oxysulphide, which can be prepared in situ.23 A rare report on solid state vapour transport synthesis of crystalline gallium oxysulphide with sulphur levels up to 5% showed fourfold enhancement of the photoluminescence signal.24 Attempts to grow thin films of indium oxysulphides for barrier coatings in thin film solar cells by atomic layer deposition still suffer from large carbon contamination.25
To the best of our knowledge, the thermolysis behaviour of alkoxides bearing a reactive side chain has not been described in literature. For instance, the potency of a thioether functionality to alter the product partially or fully in a thermolysis reaction using metal alkoxides was not investigated. The potency of thio- and selenoethers adducts of metal halides to form the respective chalcogenide films by low pressure CVD was described for several systems by G. Reid's group.26–28
In this study, the thermolysis reactions of gallium and indium alkoxides containing a thermolabile side-chain was investigated. These decomposition experiments are based on the synthesis of the first thioether functionalised gallium and indium alkoxide derivatives described recently.29 The here presented approach could be used to prepare sulphur containing oxide coatings and nanostructures with sulphur content depending on the thioether derivative, the process parameters and the metal centre of the alkoxide used.
Results and discussion
Fig. 1 shows the generic structures of symmetrical and asymmetrical secondary and tertiary alcohols, the schematic representation of the corresponding In and Ga alkoxides used in this study and the abbreviations of the different derivatives. The thioether functionalised alkoxides are prone to hydrolysis due to the weak sulphur–metal bonding, which can be easily monitored in solution by 1H NMR spectroscopy showing the free alcohol after less than a minute of exposure to ambient. In general, metal alkoxides are usually expected to form oxides upon thermal decomposition with varying carbon contamination, which depends on the effectiveness of the often observed γ-hydride elimination.1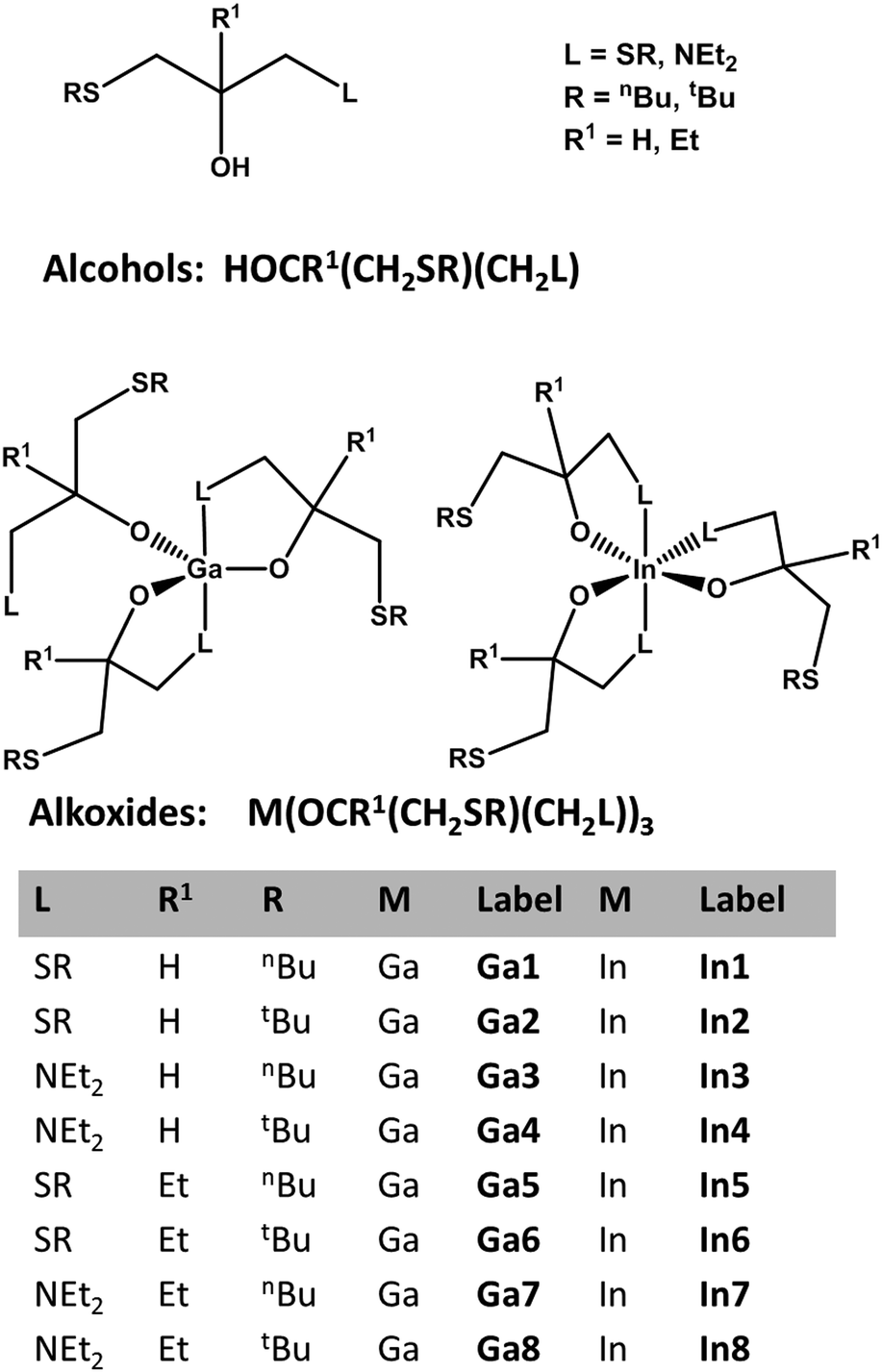 | ||
| Fig. 1 Schematic illustration of the used alcohol derivatives for the alkoxide synthesis, schematic representation of the metal alkoxide derivatives and labelling of the Ga and In alkoxides used in this study. | ||
On the other hand, organothiolates as well as thioether coordination to metal chlorides provide suitable precursors for the synthesis of different metal sulphides at a temperature range of 200–600 °C, which is reported to be significantly lower than the reaction of hydrogen sulphide with the metal halides.26,30–32
Thin film formation via LPCVD and chemical composition
The thermal decomposition of the homoleptic alkoxides via low pressure chemical vapour deposition (LPCVD) was investigated. The indium alkoxide derivatives were not suitable for LPCVD due to their insufficient volatility. In contrast, thin films of Ga2O3−xSx with varying sulphur content can be obtained from gallium alkoxides. The Ga1, Ga2 and Ga6 alkoxides did not provide efficient and reproducible decomposition characteristics and showed very slow film growth. This behaviour could be caused by the formation of oligomers due to the weak S-coordination resulting in a reduced volatility of Ga1, Ga2 and Ga6. The temperature for evaporation could not be increased any further, because the onset of decomposition as determined by thermogravimetry (165–175 °C) is very close to the temperatures already used for evaporation. Fig. 2 shows the sulphur content in thin films of the general formula Ga2O3−xSx determined by energy-dispersive X-ray (EDX) analysis. The film thickness was in the range of 0.2–0.5 μm and the coatings on glass or quartz were transparent.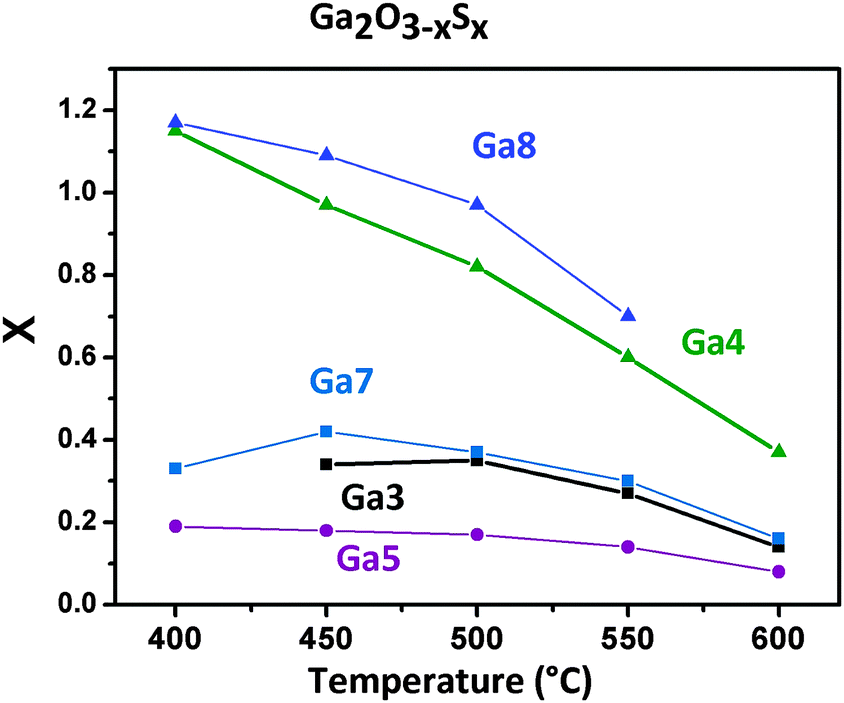 | ||
| Fig. 2 Sulphur content in the CVD deposits using the thioether functionalised gallium aminoalcoholates Ga3, Ga4, Ga7, Ga8 and a symmetric thioether functionalised gallium alkoxide Ga5 at various temperatures, which has determined by EDX analysis. X denotes the stoichiometric coefficient in Ga2O3−xSx assuming all the S in the films is sulphidic. | ||
The CVD using the n-butyl thioether aminoalcoholate Ga3 leads to a sulphur content in the deposits, which is fairly stable at x = 0.3 (6 at%) with decreasing content at higher temperatures (x = 0.14 at 600 °C). Coatings obtained using the tert-butyl derivative Ga4 contain the most S of the alkoxides based on secondary alcohols; however, the value drops from x = 1.17 (23.4 at%) at 400 °C to x = 0.37 (7.5 at%) at 600 °C. The lower S content at higher temperatures can be explained by the formation of the thermodynamically more stable Ga–O bonds.33 In addition, thermal evaporation of the gallium sulphide component could also contribute to the reduction of the sulphide content at 600 °C under low pressure conditions with a reported onset of vaporisation at ∼620 °C in literature.34 The carbon content decreases with increasing decomposition temperature with values below 1% for decomposition temperatures of 600 °C (Fig. S1a and S2a, ESI†). A higher carbon contamination in the films deposited at lower temperatures hints towards an incomplete decomposition of the ligands and their subsequent incorporation in the growing film. LPCVD using the tertiary alcohol derivatives Ga5–Ga8 showed higher carbon contamination (up to 20%) indicating a less-favourable decomposition mechanism due to the larger organic backbone (Fig. S1b, ESI†).
X-ray photoelectron spectroscopy (XPS) was performed on selected thin films derived from Ga3–Ga5 in order to investigate the chemical nature of the sulphur content in the Ga2O3−xSx films. All samples have been sputtered to eliminate the well-known higher carbon content at the surface of CVD-derived coatings.35 Spectral overviews after sputtering are shown in Fig. S2 of the ESI.† The S 2s peak was chosen for analysis due to the overlap of the Ga 3s peak and the S 2p peak position,36 which was often neglected in literature. In addition to the S 2s signal, the Ga 3p signal is also recorded in order to quantify the sulphur content.
We specifically focused on the asymmetric thioether precursors Ga3 and Ga4 with high sulphur contents and low carbon contamination at higher temperatures. Fig. 3 shows the S 2s peak region, which gives information about the chemical nature of the interesting sulphur content in the Ga2O3−xSx films. The single S 2s peak at 225.5–226 eV of the Ga2O3−xSx coatings show that a single sulphur species is present in the films at higher temperatures, which can be attributed to S2− in gallium sulphide.37 The additional peak at ∼230 eV can most likely be attributed to the undecomposed thioether moiety of the precursor and/or elemental sulphur with a binding energy of 229.2 eV,37 since no stable gallium sulphates are known and their binding energy is usually around 233 eV.38 The limited data on the S 2s peak does not provide any specific reference for thioethers. However, the only report on C–S–C bonds in sexithiophene is reported to have a binding energy of 228.3 eV.39 The films derived from Ga3 show only the specific sulphide signal with a small percentage of ∼3% signal at higher binding energy in the film deposited at 450 °C. This represents the effective decomposition of the precursor and also sulphur–carbon bond scission to form the sulphide species in the whole temperature range from 450–600 °C with a maximal sulphur content of 6.4 at% at lower temperatures. In contrast, the thermolysis reaction using Ga4 at temperatures below 550 °C does not lead to a single sulphur species in the XPS spectrum. The signal related to the high energy contribution of the S 2s peak is 1/3 of the total peak area and this allows us to calculate the sulphide content at 500 °C to be x = 0.53 (10.6 at%) in Ga2O3−xSx. The film grown at 600 °C using Ga4 contains 7.5 at% sulphur (11.6 at% at 550 °C), which is in good agreement with the EDX analysis described in Fig. 2. Prolonged sputtering leads to lower sulphur contents, which can be attributed to preferential sputtering of the sulphur species. Additional information on the alteration of the sulphur content can be observed for films with only one sulphur species present in the XPS before sputtering, which show sulphur contents in perfect agreement with the EDX data. The sulphur content in these films is reduced after sputtering for prolonged time.
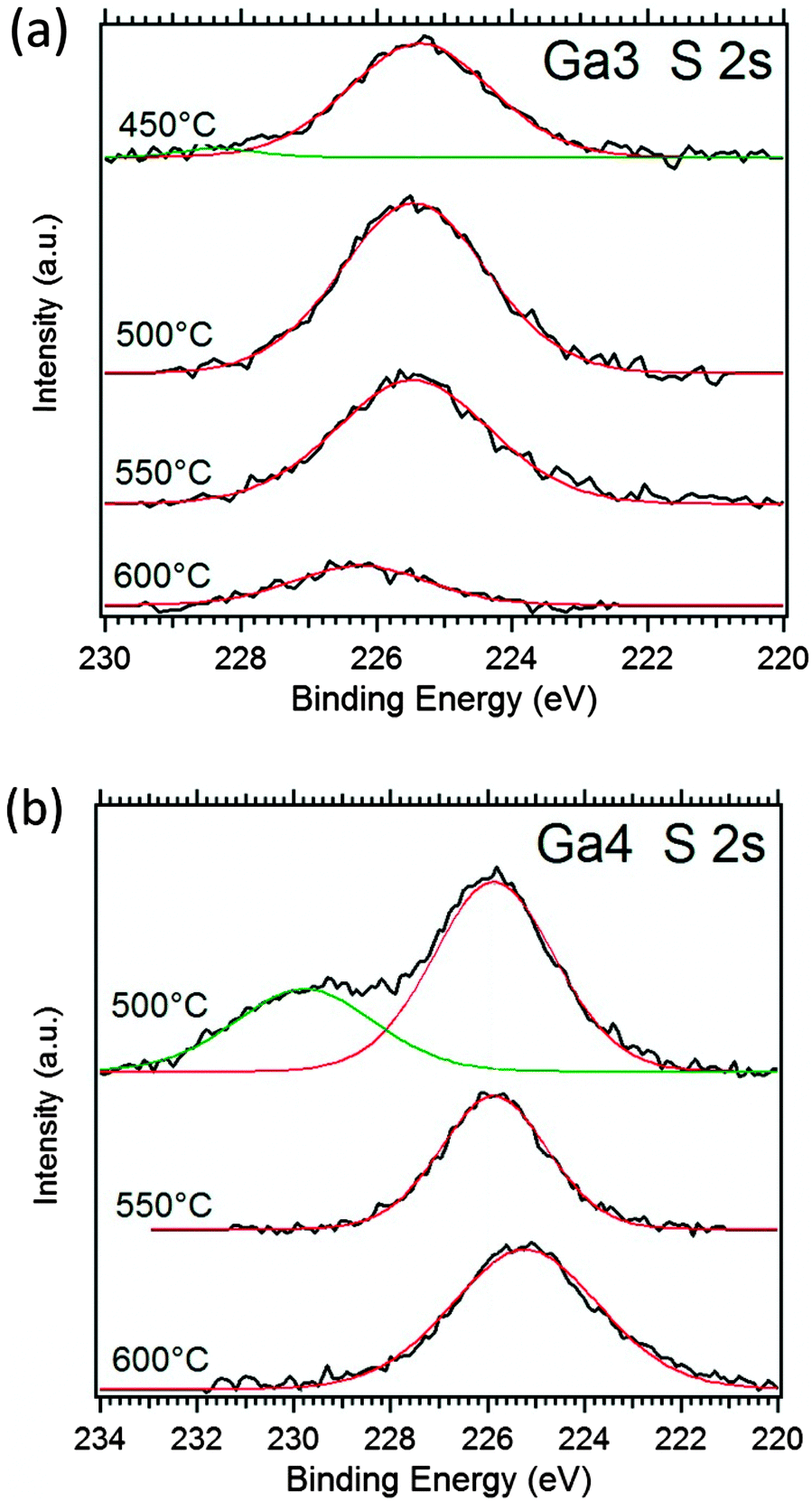 | ||
| Fig. 3 XPS spectra of the S 2s peak of CVD deposits using (a) Ga3 at temperatures of 450–600 °C and (b) Ga4 at 500–600 °C after sputtering recorded with an Al Kα lab source. | ||
In addition, depositions using Ga5 as a representative for derivatives without amino group have been investigated by XPS (Fig. S3, ESI†). As expected from EDX results in Fig. 2, the sulphide contents are lower (1 at% at 600 °C and 3 at% at 500 °C) than described for Ga3 and Ga4. Moreover, the sample already decomposes at 500 °C without the incorporation of the thioether in the film, which is 50 °C earlier than the Ga4 derivative (Fig. 3). The Ga5-derived coatings contain only one sulphide species when the surface-bound moieties, which are easily removed by sputtering from the sample grown at 500 °C (Fig. S3, ESI†), are not considered. These results clearly demonstrate that thioether functionalised alkoxides enable a partial sulphidisation even for oxophilic metals such as Ga.
In order to investigate the different sulphide content in the Ga-based films and also the cleanliness of the decomposition product, thermogravimetry (TG) coupled with mass spectrometry (MS) was performed. The decomposition is typically a two-step conversion for Ga1-4 and predominantly a single step decomposition for Ga5. However, the significantly higher mass in the decomposition product of the tertiary alcoholate Ga5 (6.1% at 250 °C; 1.25% at 500 °C above the theoretical mass for clean gallium oxide) coincidences with an insufficient ligand decomposition and resulting carbon contamination. In contrast, Ga3 and Ga4 showed only a slightly higher residual mass in the decomposition products at 500 °C than the theoretical mass for gallium oxide, which can be attributed to a considerable S content. This correlates with the high sulphur content in the thin films prepared by CVD, which contain up to 11.6% S as described above.
Indications for the effectiveness of thioethers in the sulphidisation of a metal alkoxide derived material/coating can be provided by MS data acquired during the decomposition of the metal alcoholates (Fig. S4, ESI†). The fragmentation pattern of the alkoxides with the asymmetric alcoholate ligands is much more complicated due to the additional fragments; however, each of the alcoholates produced significant amounts of hydrogen sulphide (m/z 34) during the decomposition. The mass spectrum of Ga5 as the only suitable Ga alkoxide derivative with symmetric thioether functionalised ligands for CVD reactions shows less hydrogen sulphide and butyl thiol (m/z 90), when related to the butane signal (m/z 56) and compared to the Ga3 spectrum (Fig. S4, ESI†). The presence of the two thioether groups per ligand in Ga5 does not translate in a higher signal of these species. The existence of hydrogen sulphide provides evidence of the complete thermal fragmentation and restructuring of the thioether, which is described to be a radical mechanism accompanied by a β-hydride elimination with hydrogen sulphide as a main product.40–42
As mentioned before, the thermal decomposition of metal chlorides with coordinated thioethers leads to metal sulphides at significantly lower temperatures than the reaction of hydrogen sulphide with the metal halides.26,30–32 Literature reports refer to a kinetically favoured path of thioether's sulphidisation efficiency due to an initial coordination;31 however, also the presence of reactive species formed in the boundary layer or at the substrate surface have to be considered for this sulphide formation. Thus the contribution of secondary decomposition products, such as radical species formed by thermal decomposition of thioethers, has to be taken into account.40
Hydrogen sulphide is a strong sulphidisation agent and described in literature to allow the transformation of oxides to the respective sulphides at elevated temperatures.43,44 The still moderate temperatures of 600 °C and below in the CVD process are enough to cause a modification of the growing oxide. However, the modification of a bulk oxide is quite slow and the reaction is limited to the surface. For instance, tempering micron sized Ga2O3 powder at 500 °C for 1h in a H2S/N2 flow leads to merely 1.8% of conversion to the sulphide according to EDX results.
We assume that hydrogen sulphide can take part in the sulphidisation during the growth; however, the formed radicals and thiols can also be involved in this process at the surface of the growing layer. The decomposition products recorded in the mass spectrum can only be an indication of the thermally induced decomposition and resulting intermediates in the boundary layer in the deposition area. In situ spectroscopic investigations are required to elucidate the decomposition mechanism in detail. At this stage, we can conclude that the thioether functionalised aminoalkoxides form significantly more hydrogen sulphide and therefore a more effective C–S–C bond scission can be expected. The here observed in situ reaction to Ga2O3−xSx leads to more homogeneous composition of the product than post-growth modifications of the oxide using sulphidisation agents.
Morphology of Ga2O3−xSx coatings and electrical conductivity
All Ga-based films of the here described alkoxides were amorphous, as expected in the investigated temperature range,15,45 and quite smooth up to 500 °C; however, a fine-grained surface structuring is already observed for the films with lower sulphur content including Ga3, Ga5 and Ga7. At higher temperatures and lower sulphur contents the films show a more pronounced nanostructuring of the surface. However, the films with the highest purely sulphidic sulphur content at 600 °C using Ga4 are still very smooth, when compared to the pure Ga2O3 coatings obtained from Ga(OtBu)3 as shown in Fig. 4. Post-growth annealing of a film derived from Ga4 at 600 °C for 2 hours under vacuum at 800 °C leads to crystallisation of the film with Ga2O3 in the monoclinic phase (ICDD 41-1103; Fig. S5, ESI†) and a small amount of a secondary phase which could not be assigned to either gallium oxide nor a sulphide phase. In addition, the absence of any sulphur signal in the EDX spectrum provides further evidence for a thermal desorption of gallium sulphide at elevated temperatures (>620 °C) as described in literature.34 The initially smooth homogeneous coating from Fig. 4a cracks during the annealing and appears to be partially porous in the SEM image (Fig. S5, ESI†). A comparison of the evolution of the morphology of Ga3 and Ga4 derived CVD coatings with increasing temperature is illustrated in Fig. S6 (ESI†). | ||
| Fig. 4 SEM images of LPCVD deposits using (a) Ga4 and (b) Ga(OtBu)3 as precursors at 600 °C substrate temperature. | ||
First measurements of the electrical conductivity of the Ga2O3−xSx (x = 0.37) thin films were performed between 200 and 500 °C by means of impedance spectroscopy using top coated interdigitated Pt electrodes and compared to Ga2O3 thin films (Fig. S7, ESI†). The most reliable results were obtained in reducing atmosphere (2.5% H2 in Ar), which prevents the sulphide content from oxidation during the measurement. The impedance spectra (Fig. S8 and S9, ESI†) suggest an electronic charge transport to be the most likely explanation for the conductivity of the samples. From the impedance data and the geometry of the samples conductivity values were calculated as described in the Experimental section. Gallium oxide can be used for hydrogen gas sensing at higher temperatures;46 however, the higher conductance values are a steady state and will not affect the overall tendencies described in this section.
The Arrhenius diagram in Fig. 5 compares the different electrical conductivities of the amorphous Ga2O3−xSx (x = 0.37) and Ga2O3 thin films. In case of both sample types – with and without sulphide content – the geometry of the current collector did obviously not affect the obtained conductivity values. Thus, contributions from a contact resistance as well as any artifacts originating from the contacting or from the Pt thin film current collector can safely be excluded. Since the capacitance of the thin film samples is mainly governed by the substrate46 a separation of electrode effects (if there are any significant) was not possible on this sample geometry. Consequently, the type of contact between Pt and the oxide film (i.e. ohmic or Schottky) cannot be evaluated from the data available so far. However, a detailed analysis of resistive contributions would be far beyond the scope of the present paper and may be covered in a forthcoming study. At temperatures between 200 and 350 °C the conductivity of the pure Ga2O3 films is significantly lower than of the sulphide containing ones. Moreover, in this temperature regime the activation energy of the conductivity (i.e. the slope in the Arrhenius plot) of the Ga2O3 samples is significantly higher (ca. 0.9 eV) than of the sulphide containing samples (ca. 0.1 eV). Above 400 °C the conductivity of both Ga2O3−xSx and Ga2O3 thin films becomes almost the same and interestingly also the activation energy of both sample types becomes rather similar.
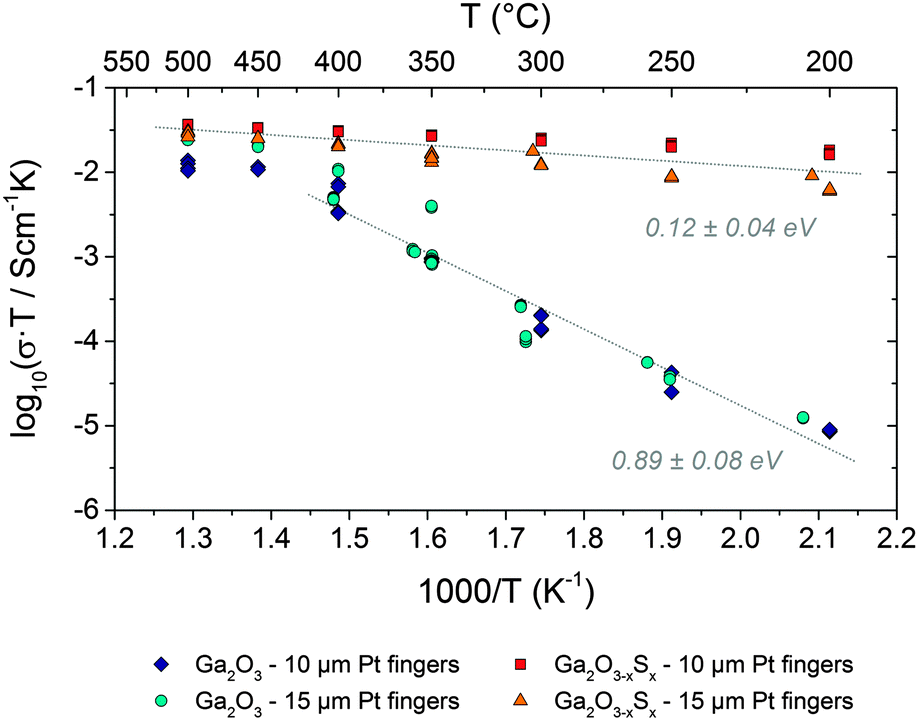 | ||
| Fig. 5 Arrhenius plot of the conductivity of both Ga2O3−xSx (x = 0.37) and Ga2O3 thin film samples. In order to obtain correct activation energy values σ × T values were plotted (a version of this diagram with σ values is shown in Fig. S10, ESI†). | ||
In an additional experiment one of the sulphide containing thin films was annealed at 500 °C in ca. 50 mbar oxygen for about one hour. As shown in Fig. S10 in the ESI† this treatment led to a strong decrease of the conductivity by about one order of magnitude. The activation energy, however, was virtually not affected. This experiment shows that sulphide incorporation has a significant but mechanistically highly complex influence on the electrical properties of Ga2O3 thin films. An unambiguous explanation for the effect of sulphide and especially for the rather low activation energy of the Ga2O3−xSx thin films can thus not be given so far and would also go beyond the scope of the present paper.
Particle synthesis via hot-injection method
The here described indium precursors were not suitable for LPCVD due to their insufficient volatility. For that reason, the precursors were decomposed in high boiling solvents by hot injection method without any additional surfactants.47 The resulting inorganic particles were dark beige to orange in colour instead of the colourless nature of indium oxide. However, in all cases sulphide-rich In2O3−xSx oxysulphides are formed as shown in Fig. 6. The sulphur content was determined by EDX and the influence of surface adsorbed species was not considered leading to a potential overestimation of the sulphur content.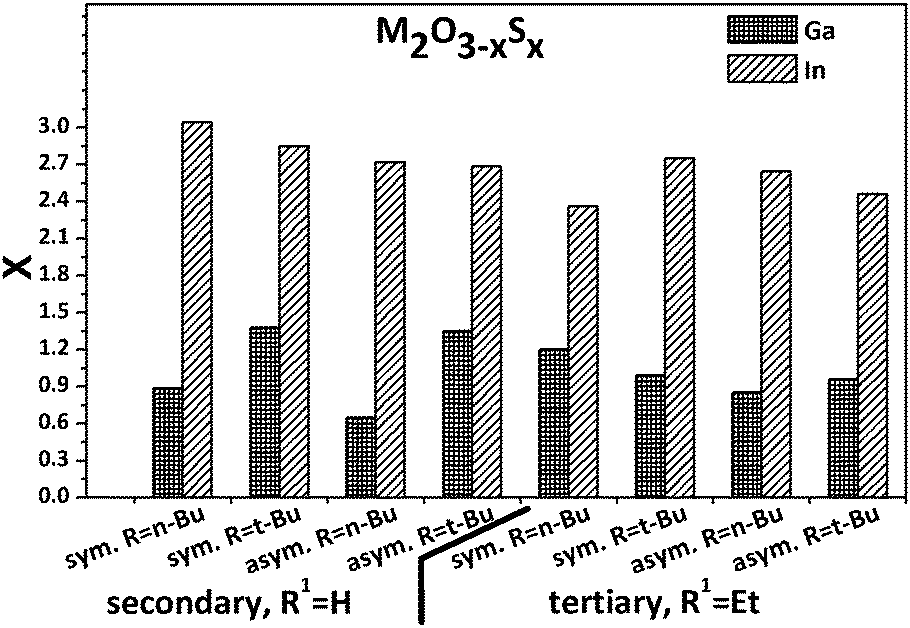 | ||
| Fig. 6 Overview about the sulphur content in M2O3−xSx (M = Ga, In) material obtained by thermal composition of Ga1–Ga8 and In1–In8 precursor derivatives in boiling squalane. | ||
The sulphur content is considerably higher in all cases for the In-based materials when compared to the Ga-based powders, which is not surprising when the bond energies between oxygen and the individual metals are considered.48 Indium oxide is easily converted to indium sulphide by thermal treatment at 500 °C for 60 min under diluted hydrogen sulphide in nitrogen (Fig. S11, ESI†), whereas gallium oxide under similar conditions converts only 1.8% to the sulphide according to EDX.
XRD analysis of the powders prepared from the thioether functionalised alkoxide derivatives revealed phase pure indium sulphides (tetragonal β-In2S3 (PDF 25-0390)) instead of In2O3, which is the material usually obtained from alkoxide precursors. Fig. 7 shows phase pure, microcrystalline β-In2S3 derived from In3, nanocrystalline β-In2S3 using In1 and a cubic α-In2O3 reference derived from the decomposition of In(OtBu)3, which is known to produce indium oxide upon thermal decomposition.16
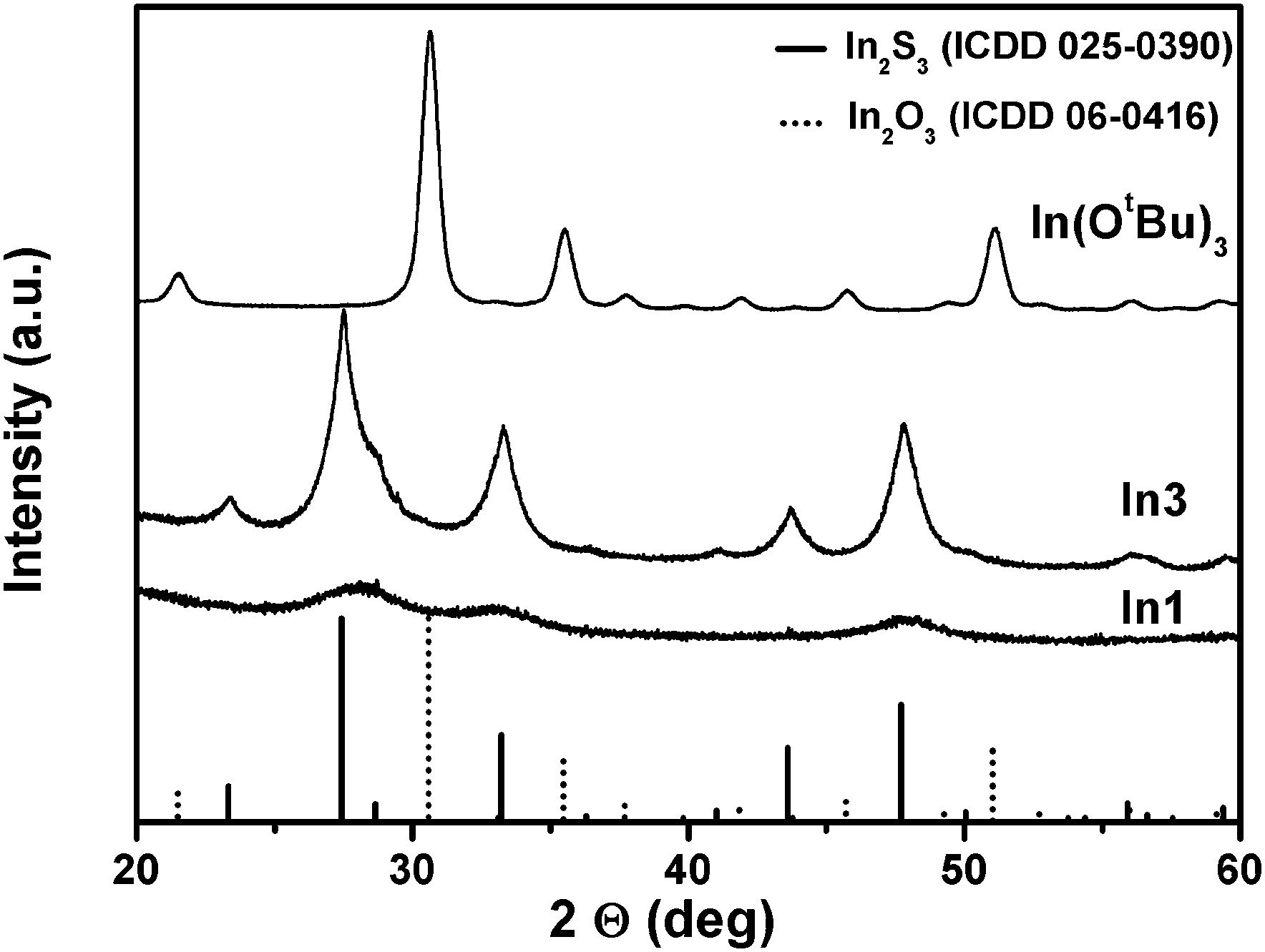 | ||
| Fig. 7 XRD pattern of decomposition products using thioether functionalised indium alkoxides In1 and In3 as well as a In(OtBu)3. The decomposition was carried out in boiling squalane by hot injection methods. | ||
All thioether functionalised indium alkoxides show reflexes of β-In2S3 with different crystal size, which is generally smaller for the symmetric alcoholate derivatives used in this study (Fig. S12, ESI†). The narrower reflexes of the solids prepared using thioether functionalised aminoalcoholates In3 + In4 and In7 + In8 reveal larger crystallite sizes, when compared to the symmetric thioether functionalised indium alkoxides. The higher crystallinity of the thioether functionalised aminoalcoholate derived materials points towards a cleaner decomposition path and easier formation of the active sulphidisation species, such as hydrogen sulphide and intermediates. Fig. S13 (ESI†) illustrates the higher amount of hydrogen sulphide in the mass spectrum of In3 when compared to In1 and related to the butene fragment of the thioether moiety. Comparing these data with the CVD results using the gallium derivatives, the conclusion can be drawn that a similar effect is obtained for the Ga2O3−xSx formation with the highest sulphur content in thioether functionalised aminoalcoholate derived coatings with very low carbon contamination using Ga3 and Ga4. Fig. S14 (ESI†) shows XPS spectra of the particles prepared by the thermal decomposition using In3 and In4. The data reveal signals in the expected energy range for In2S3 and the calculated In/S ratio matches the theoretical value of 0.67 quite well (0.68 for In3 and 0.71 for In4), leading to the conclusion that essentially indium sulphide is formed from the indium alkoxides.
The gallium alkoxide derivatives also lead to Ga2O3−xSx powders with only slightly higher sulphur content than the respective films formed via LPCVD (Fig. 6) and only one signal for sulphides is observed in the XPS spectra (Fig. S15, ESI†). Similar to the thin films, all gallium oxysulphide particles were amorphous as shown in Fig. S16 (ESI†).
Conclusions
The thermal decomposition of a new class of alkoxides with a thioether sidechain by low pressure chemical vapour deposition and also in high boiling solvent reveals the potency of the thioether functionality to form sulphides even though metal–oxygen bonds are pre-formed in the alkoxide precursors. The thioether-containing gallium aminoalcoholate precursors are the most suitable for the thin film studies as described herein due to their volatility, highest sulphur content obtained in the molecule-to-material conversion and also the reproducible film formation. The Ga and In alkoxide systems show a different behaviour with the partial conversion for gallium-based precursors to Ga2O3−xSx and a full conversion leading to phase pure indium sulphide particles. Gallium oxysulphide films show an altered electronic conductivity with low activation energy when compared to pure gallium oxide films, which could allow the tuning of the electronic properties in amorphous oxides by the incorporation of sulphide.Experimental
Metal alkoxide synthesis
The Ga alkoxides are prepared via alcohol exchange reaction using Ga(OtBu)3 and the respective alcohols, while the In derivatives are obtained using alcoholysis of In(N(Si(CH3)3)2)3. A detailed description of the synthesis procedure can be found in literature.29Low-pressure CVD
All experiments were conducted in a horizontal cold-wall reactor.49 Si(100) or SiO2 substrates were glued onto an inductively heated wedge shaped graphite susceptor with silver paste. 100 mg of the precursor species were used for the individual CVD experiments and attached to the CVD reactor, which has been desiccated under dynamic vacuum at 120 °C for 2 h. The deposition was performed under high vacuum using a turbo molecular pump at substrate temperatures of 400–600 °C and precursor temperatures ranging from 130–165 °C (155 °C Ga3; 142 °C Ga4; 165 °C Ga5; 150 °C Ga7; 130 °C Ga8). After cooling, all thin films were handled and stored under ambient atmosphere.Hot-injection pyrolysis
10 mL of anhydrous squalane were placed in a desiccated apparatus consisting of a 3-necked flask with gas-inlet, septum and air cooler. The apparatus was placed in a sand bath and after heating to reflux temperature (ca. 415 °C), 75 ± 5 mg of the metal alkoxide precursor dissolved in 1 mL of squalane were injected. After 30 min, the heating mantle was removed and the solution was allowed to cool to room temperature. The solution was centrifuged and subsequently sonicated and centrifuged four times with toluene to remove residues.SEM and XRD
The thin films and particles were analysed using a FEI Quanta 200 scanning electron microscope (SEM) with EDX detector. The X-ray diffraction (XRD) patterns were recorded on a PANalytical X-Pert PRO PW 3050/60 in Bragg–Brentano geometry and Cu-Kα radiation. Data analysis was carried out using the X-pert Highscore software.TG/MS
Thermogravimetric measurements (TG) were performed with a METTLER TOLEDO TGA/DSC1 1100 system equipped with a UMX1 balance. Thermogravimetric coupled mass spectrometry (TG-MS) experiments were performed with a METTLER TOLEDO TGA/DSC1 1600 system with an MX1 balance coupled with a Pfeifer Vacuum MS Thermostar GSD 301T2 mass spectrometer. The heating rate was 10 K min−1 with an argon flow rate of 60 mL min−1. The fragmentation of the ligand components were compared with literature data from the NIST chemistry webbook database.XPS analysis
XPS measurements were performed with a commercially available STM/XPS UHV setup from SPECS. The base pressure in the UHV system for XPS measurements was in the low 10−9 mbar regime. For the XPS measurements the system is equipped with a non-monochromatised dual anode X-ray gun with Al and Mg Kα anode (XRC 50, SPECS) and a hemispherical analyser PHOIBOS 100 (SPECS) with multi channeltron detector. The samples were introduced into the UHV system via a load lock after mounting them on a steel plate. The samples were measured at room temperature with Al Kα radiation in the as is state and after sputtering for 45 min at 5 × 10−6 mbar Ar and an acceleration voltage of 1 kV. The obtained data was analysed by using the commercial software CasaXPS. The peak positions were corrected via the signal for graphitic carbon and double checked by measuring the Fermi Edge. Peak positions were identified by using the NIST XPS database (http://srdata.nist.gov/xps/). Peaks were fitted after Shirley background subtraction using a Gaussian–Lorentzian mixed peak shape (30% L) without further constrains. For comparing Ga, C and S signals the intensity was corrected by the cross-sections obtained from the Elletra Trieste database (http://https://vuo.elettra.eu/services/elements/WebElements.html). Due to the overlap of the S 2p signal with the Ga 3s signal the calculations were performed via the S 2s and Ga 3p signal. The metal was set as the reference with 40 at% in the material.Electrical characterization
Impedance spectroscopy measurements were carried out on Ga2O3 and Ga2O3−xSx thin films with buried interdigitating Pt electrodes (cf. Fig. S7, ESI†); the substrate was quartz glass. The impedance of these thin film samples was measured in-plane by means of an Alpha-A High Performance Frequency Analyzer equipped with a POT/GAL 30 V 2A test interface (both Novocontrol, Germany). The common frequency range was 10 mHz–1 MHz but was adjusted if necessary; the AC voltage was 10 mV root mean square. The measurements were done in 800 mbar reducing atmosphere (2.5% H2/97.5% Ar) at temperatures between 200 °C and 500 °C. This atmosphere was chosen to avoid irreversible changes of the samples during the measurements, which might originate from a changing sulfide content caused by oxidation at elevated temperatures. The impedance spectra obtained on Ga2O3 and Ga2O3−xSx thin films are depicted in Fig. S8 and S9 (ESI†), respectively. On the Ga2O3 samples at temperatures between 200 and 300 °C an almost ideal semicircle could be observed in the measured frequency range – see Fig. S8 (ESI†). Since down to frequencies of 10 mHz no indication for an additional electrode feature could be observed, the resistive contribution is assumed to originate from an electronic conductivity of the thin film samples. The capacitance leading to the observed semicircle is typically caused by the substrate of the samples.50 At temperatures higher than 300 °C only a smaller part of a semicircle or a real axis intercept could be observed. For obtaining the conductivity of the thin films the low frequency axis intercept of the spectra – i.e. the DC resistance – was evaluated and the conductivity σ was calculated by| σ = b/(l × d × N × R) | (1) |
On the sulfide doped samples much smaller resistances were measured and no semicircle could be resolved even at 200 °C – cf. Fig. S9 (ESI†) (please note that the small inductive high frequency tail can be attributed to an artifact caused by the cables). As mentioned above the DC resistance was evaluated from the low frequency intercept of the spectra and the corresponding conductivity was calculated by means of eqn (1). (It is also worth mentioning that the resistance of the Pt fingers was always below 1% of the totally measured resistance and can thus safely be neglected.)
The obtained conductivity values are plotted in Arrhenius diagrams, which are shown in Fig. 5 as well as in Fig. S10 (ESI†). Fig. S10 (ESI†) contains data of an additional measurement, which was conducted on a sulfide containing sample after annealing at 500 °C in ca. 50 mbar oxygen for about 1 h. This treatment was done to oxidatively remove significant amounts of sulfur from the thin film. As a result the conductivity of the film is about one order of magnitude lower. The activation energy (i.e. the slope in the Arrhenius plot) is virtually not affected by this treatment. Reasons for this behavior cannot be evaluated from the presently available data. However, this measurement clearly shows the importance of the sulfide in the film for obtaining higher electric conductivities.
Acknowledgements
We would like to thank the X-ray center (XRC) for access to the powder diffractometers and the University Servicecenter for TEM (USTEM) for access to the scanning electron microscope at the Vienna University of Technology. In addition, N. Rüffer is acknowledged for her help with acquiring the TG/MS data.Notes and references
- D. C. Bradley, R. C. Mehrotra, I. P. Rothwell and A. Singh, Alkoxo and Aryloxo Derivatives of Metals, Academic Press, London, 2001 Search PubMed.
- R. Lichtenberger and U. Schubert, J. Mater. Chem., 2010, 20, 9287–9296 RSC.
- L. McElwee-White, Dalton Trans., 2006, 5327–5333 RSC.
- A. Devi, Coord. Chem. Rev., 2013, 257, 3332–3384 CrossRef CAS.
- F. Biegger and S. Barth, Monatsh. Chem., 2016, 147, 341–348 CrossRef CAS.
- W. A. Herrmann, N. W. Huber and O. Runte, Angew. Chem., Int. Ed. Engl., 1995, 34, 2187–2206 CrossRef CAS.
- M. Gebhard, M. Hellwig, H. Parala, K. Xu, M. Winter and A. Devi, Dalton Trans., 2014, 43, 937–940 RSC.
- S. Mathur, S. Barth, H. Shen, J. C. Pyun and U. Werner, Small, 2005, 1, 713–717 CrossRef CAS PubMed.
- A. L. Johnson, N. Hollingsworth, G. Kociok-Kohn and K. C. Molloy, Inorg. Chem., 2008, 47, 9706–9715 CrossRef CAS PubMed.
- S. Barth, R. Jimenez-Diaz, J. Sama, J. Daniel Prades, I. Gracia, J. Santander, C. Cane and A. Romano-Rodriguez, Chem. Commun., 2012, 48, 4734–4736 RSC.
- C. J. Carmalt and S. J. King, Coord. Chem. Rev., 2006, 250, 682–709 CrossRef CAS.
- L. G. Bloor, C. J. Carmalt and D. Pugh, Coord. Chem. Rev., 2011, 255, 1293–1318 CrossRef CAS.
- S. Basharat, C. J. Carmalt, R. Binions, R. Palgrave and I. P. Parkin, Dalton Trans., 2008, 591–595 RSC.
- C. E. Knapp, A. Kafizas, I. P. Parkin and C. J. Carmalt, J. Mater. Chem., 2011, 23, 1719 CrossRef CAS.
- M. Valet and D. M. Hoffman, Chem. Mater., 2001, 13, 2135–2143 CrossRef CAS.
- S. Barth, M. S. Seifner and J. Bernardi, J. Mater. Chem. C, 2014, 2, 5747–5751 RSC.
- S. Suh and D. M. Hoffman, J. Am. Chem. Soc., 2000, 122, 9396–9404 CrossRef CAS.
- L. A. Miinea and D. M. Hoffman, J. Mater. Chem., 2000, 10, 2392–2395 RSC.
- K. C. Molloy and J. E. Stanley, Appl. Organomet. Chem., 2009, 23, 62–67 CrossRef CAS.
- Y. P. Song, H. Z. Zhang, C. Lin, Y. W. Zhu, G. H. Li, F. H. Yang and D. P. Yu, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 075304 CrossRef.
- R. Marschall and L. Wang, Catal. Today, 2014, 225, 111–135 CrossRef CAS.
- P. Qin, G. Fang, F. Cheng, W. Ke, H. Lei, H. Wang and X. Zhao, ACS Appl. Mater. Interfaces, 2014, 6, 2963–2973 CAS.
- D. Lehr, M. Luka, M. R. Wagner, M. Bügler, A. Hoffmann and S. Polarz, Chem. Mater., 2012, 24, 1771–1778 CrossRef CAS.
- J. S. Kim, S. B. Lee, J. H. Bahng, J. C. Choi, H. L. Park, S. I. Mho, T. W. Kim, Y. H. Whang and G. C. Kim, Phys. Status Solidi A, 2001, 187, 569–573 CrossRef CAS.
- C. Bugot, N. Schneider, M. Bouttemy, A. Etcheberry, D. Lincot and F. Donsanti, Thin Solid Films, 2015, 582, 340–344 CrossRef CAS.
- S. D. Reid, A. L. Hector, W. Levason, G. Reid, B. J. Waller and M. Webster, Dalton Trans., 2007, 4769–4777 RSC.
- C. H. de Groot, C. Gurnani, A. L. Hector, R. Huang, M. Jura, W. Levason and G. Reid, Chem. Mater., 2012, 24, 4442–4449 CrossRef CAS.
- K. George, C. H. de Groot, C. Gurnani, A. L. Hector, R. Huang, M. Jura, W. Levason and G. Reid, Chem. Mater., 2013, 25, 1829–1836 CrossRef CAS.
- S. Barth, F. Biegger and M. Puchberger, Dalton Trans., 2015, 44, 16439–16445 RSC.
- C. H. Winter, T. S. Lewkebandara and J. W. Proscia, Chem. Mater., 1992, 4, 1144–1146 CrossRef CAS.
- T. Suren Lewkebandara, P. J. McKarns, B. S. Haggerty, G. P. A. Yap, A. L. Rheingold and C. H. Winter, Polyhedron, 1998, 17, 1–9 CrossRef.
- S. Ghoshal and V. K. Jain, J. Chem. Sci., 2007, 119, 583–591 CrossRef CAS.
- I. Barin, Thermochemical Data of Pure Substances, Wiley-VCH Verlag GmbH, 2008, pp. 728–780 Search PubMed.
- O. M. Uy, D. W. Muenow, P. J. Ficalora and J. L. Margrave, Trans. Faraday Soc., 1968, 64, 2998–3005 RSC.
- M. Kwoka, L. Ottaviano, M. Passacantando, S. Santucci and J. Szuber, Appl. Surf. Sci., 2006, 252, 7730–7733 CrossRef CAS.
- A. V. Naumkin, A. Kraut-Vass, S. W. Gaarenstroom and C. J. Powell, National Institute of Standards and Technology (NIST), 2012, vol. 20 Search PubMed.
- J. M. Thomas, I. Adams, R. H. Williams and M. Barber, J. Chem. Soc., Faraday Trans. 1, 1972, 68, 755–764 CAS.
- B. R. Strohmeier and D. M. Hercules, J. Phys. Chem., 1984, 88, 4922–4929 CrossRef CAS.
- D. Oeter, C. Ziegler and W. Göpel, Synth. Met., 1993, 61, 147–150 CrossRef CAS.
- E. H. Braye, A. H. Sehon and B. d. Darwent, J. Am. Chem. Soc., 1955, 77, 5282–5285 CrossRef CAS.
- X. Zheng, E. M. Fisher, F. C. Gouldin, L. Zhu and J. W. Bozzelli, Proc. Combust. Inst., 2009, 32, 469–476 CrossRef CAS.
- G. Martin, H. Martinez and J. Ascanio, Int. J. Chem. Kinet., 1989, 21, 193–206 CrossRef CAS.
- Y. Feldman, G. L. Frey, M. Homyonfer, V. Lyakhovitskaya, L. Margulis, H. Cohen, G. Hodes, J. L. Hutchison and R. Tenne, J. Am. Chem. Soc., 1996, 118, 5362–5367 CrossRef CAS.
- W. Klemm and H. U. v. Vogel, Z. Anorg. Allg. Chem., 1934, 219, 45–64 CrossRef CAS.
- R. Binions, C. J. Carmalt, I. P. Parkin, K. F. E. Pratt and G. A. Shaw, Chem. Mater., 2004, 16, 2489–2493 CrossRef CAS.
- R. Jones and H. Meixner, Sensors, Micro- and Nanosensor Technology: Trends in Sensor Markets, Wiley, 2008 Search PubMed.
- C. de Mello Donegá, P. Liljeroth and D. Vanmaekelbergh, Small, 2005, 1, 1152–1162 Search PubMed.
- N. Wiberg, Lehrbuch der Anorganischen Chemie, 2008 Search PubMed.
- S. Mathur, S. Barth and H. Shen, Chem. Vap. Deposition, 2005, 11, 11–16 CrossRef CAS.
- M. Gerstl, E. Navickas, M. Leitgeb, G. Friedbacher, F. Kubel and J. Fleig, Solid State Ionics, 2012, 225, 732–736 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional XPS spectra, TG and MS, impedance data, SEM images and XRD patterns are provided. See DOI: 10.1039/c6nj00402d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |