
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stability and geometry of silica nano-ribbons (SNRs): a first-principles study
C. M.
Fang
*ab,
A.
van Blaaderen
a and
M. A.
van Huis
*a
aSoft Condensed Matter (SCM), Debye Institute for Nanomaterials Science (DINS), Utrecht University, Princetonplein 5, 3584 CC Utrecht, The Netherlands. E-mail: M.A.vanhuis@uu.nl
bBrunel Centre for Advanced Solidification Technology (BCAST), IMM, Brunel University London, Uxbridge, Middlesex UB8 3PH, UK. E-mail: Changming.Fang@brunel.ac.uk
First published on 14th July 2016
Abstract
Silica based materials are attractive because of their versatility and their unique structures and properties, which have led to numerous applications of silica in a range of fields. Recently, various low-dimensional silica materials have been synthesized experimentally. Here we present a first-principles study on the geometry and stability of novel low-dimensional silica nano-ribbons (SNRs) using density-functional theory (DFT) with van der Waals interactions (optB88-vdW). SNRs of various widths with different surface groups, and with the geometry of hexagonal rings and squares, were taken into consideration. An atomically flat ribbon with mixing squares and rings is also included. The calculations showed high stability for the single layer and bilayer silica ribbons, both containing hexagonal rings. The calculations also revealed a high flexibility of silica chains. The local structure and chemical bonding were carefully analyzed. Electronic band structure calculations showed an insulating nature of the SNRs with energy gaps of about 5.0 to 6.0 eV, which are determined by nonbonding and anti-bonding O 2p states.
Introduction
Novel silica nano-ribbons (SNRs) are academically very interesting and provide opportunities for industrial applications as well. It is common knowledge that various forms of silica and silicates are abundantly present in the Earth's mantle and are used in many chemical processes and technological materials.1,2 Knowledge on silica is therefore of importance both for understanding of our natural environment and for developing novel applications. In the last decades, low-dimensional materials such as graphene have emerged as subjects of great interest.3,4 More recently, there have been intensive explorations on low-dimensional silica forms as well and different techniques have been employed to prepare thin silica sheets and related nanotubes and nanowires.5–12 Recently, Huang and co-workers prepared thin glassy silica sheets on graphene substrates. The prepared two-dimensional (2D) anhydrous silica sheets have a thickness of a few atoms.6,7 The 2D anhydrous bilayer silica sheets are composed of hexagonal rings.5–12 These successes have triggered our scientific curiosity and stimulated the investigation of these novel forms of silica, such as the low dimensional SNRs studied in this work.Historically, in the 1960's Wulff already proposed colloid-chemistry methods to prepare silica sheets of different thicknesses and shapes (lepidoid silica) by controlling the preparation conditions (temperature, pH-value, solution, etc.).13 Wulff's work provided possibilities of realization of silica SNRs. Therefore, knowledge about the stability and how this connects to the geometry and structure of different SNRs will be of use to experimentalists who focus on the synthesis and study of SNRs and related structures and compounds. As mentioned, different forms of silica are important building blocks for various materials with potential applications ranging from biosciences and medicine to the fields of catalysis and electronics.14–17 The different active sites on the SNRs provide possibilities to chemically or physically connect other atoms/ions/molecules/nanoparticles, such as transition metals, noble metals and semiconductor nanoparticles which can play a role as catalysts for chemical reactions, e.g. in water decomposition, etc. The insulating nature of low-dimensional SNRs provides a possibility for their applications as electronic conducting barriers with a thickness of atomic–level for nano-electronics. For these applications, knowledge about the geometry, chemical bonding as well as electronic properties of SNRs is of crucial importance.
Theoretical efforts on low-dimensional silica focus on anhydrous silica bilayer sheets,8–10 nanotubes and nanowires,18–22 and go beyond the computational publications on crystalline/amorphous silica and silicates formed under various conditions,16,23,24 and their applications, e.g. solar cells.5,26 Recently, we reported first-principles simulations for 2-dimensional (2D) (hydrated) silica sheets.27 Up to now there are no reports silica nanoribbons (SNRs) to the best of our knowledge. In the present manuscript we report our first-principles study on the geometry and stability of novel SNRs with different decorations using density functional theory within Dion's van der Waals functional (optB88-vdW)28 which has been successfully applied for many sparse systems.27,29–32 The starting materials are monosilicic acid molecules (Si(OH)4)33,34 considering the recent work by Preari and co-workers who stabilized monosilicic acid molecules in silica-supersaturated solution by polyethylene glycols (PEG)s at pH = 7.35 The building block for the SNRs is an infinite linear chain with each Si being connected to two bridging O and two terminal (OH)− groups. This was designed from the reported silica chain molecules formed during sol–gel processing.33,34,36–39 Two siloxane structures in the ribbons were taken into consideration. One type of SNRs contains silica connected by four bridging O to form squares.27 The other one contains hexagonal rings. The dangling Si bonds were terminated by (OH)− groups. Therefore, each Si atom is tetrahedrally coordinated by four O atoms satisfying its sp3 bonding. Furthermore, bilayer SNRs were also built in such a way that two hexagonal SNR sheets are positioned on top of each other and each Si is connected to three O in its plane and one O between the sheets, based on recent experimental results.5–11 The relative stability of SNRs with respect to silicic acid and water molecules is obtained according to water condensation reactions in colloidal chemistry.35–42 Local structures, chemical bonding and charge transfer in the predicted SNRs of high stability were analysed. The obtained information is not only useful for understanding the SNRs and related low-dimensional silica forms, but also helpful for experimentalists for the preparation and characterization of new silica compounds, and further for development of novel materials for e.g. catalysis, bioscience, and materials science.
Details of computational techniques
A. Formation energy of silica ribbons
The reactions of the formation of various forms of 1D and 2D siloxane from silicic acid by condensation reactions and by the release of water can be represented by the following set of reactions:33–36| 2nSi(OH)4 → [(SiO)2nOn](OH)2n + 2nH2O + 2n (−ΔH) | (1) |
B. Silica ribbon unit cells
First we build lattices of the reactants, isolated mono-silicic acid Si(OH)4 and di-silicic acid Si2O(OH)6, and water H2O molecules within cubic supercells with axis lengths of 20.0 and 16.5 Å, respectively. In this way the inter-molecular distances are longer than 14 Å, large enough to avoid inter-molecular interactions.Low-dimensional SNRs with different connections and widths are built with one-dimensional periodicity in orthorhombic cells. The starting inputs were made by arranging the distances between Si–Si atoms at around 3.0 Å, as shown in Table 1 which lists the optimized lattice parameters for the SNRs with different siloxane structures. The other two axes have dimensions greater than 20 Å to avoid inter-ribbon interactions.
| a (Å) | d Bond (Å) and angle (°) | Charges |
|---|---|---|
| Water molecule | ||
| 16.5 |
O–H: 0.971
H–O–H: 104.8 |
H+0.562O−1.12 |
| Mono-silicic acid molecule | ||
| 20.0 |
Si–O: 1.643
O–H: 0.968 |
Si+3.16(O−1.39H+0.60)4 |
| Di-silicic acid molecule with formula Si2O(OH)6 | ||
| 20 |
Si–O(Si): 1.65
Si–O(H): 1.62(×3) Si–O–Si: 145.3 |
Si: +3.16
O(Si): −1.62 O(H): −1.39 H: +0.61 |
| L a-axis (Å) | d Bond (Å) and angle (°) | Charges |
|---|---|---|
| 1D-single atom silica chain (1D-SASC) with formula SiO(OH)2 | ||
|
4.98
Z = 2 |
Si–O(Si): 1.625
Si–O(H): 1.642 O–Si–O: 111.7 Si–O–Si: 145.3 |
Si: +3.18
O(Si): −1.62 O(H): −1.39 H: +0.60 |
C. van der Waals density-functional (optB88-vdW) and computational settings
It is well-known that the standard first-principles density-functionals, such as local density approximation (LDA) and generalized gradient approximation (GGA), do not take into account van der Waals interactions.28,43 In order to have accurate and reliable results, we employed the van der Waals density functional formulated by Dion and co-workers26 for the sparse systems. The van der Waals functional was implemented in the code VASP (Vienna Ab initio Simulation Package).44,45 This approach employs the London dispersion interaction for the nonlocal electron correlation effects (optB88-vdW)26–30 whereas the exchange term is based on the exchange functional by Becke (B88).46,47The electronic wave functions were sampled on dense grids along the ab plane in the irreducible Brillouin zone (BZ) for the crystals, using the Monkhorst and Pack method48 for the total energy and electronic structure calculations of the SNRs. Only the Γ-point was used for the isolated molecules in large cubes. Structural optimizations were performed for the ribbons with fixed volume to find the periodic a-axis at the systems' energy minima, whereas all atomic coordinates in a fixed cube were relaxed for the single molecule calculations. We also used the fixed volume approach to determine the lattice parameters of the two periodic dimensions from the minimized total energies. The optimizations showed that both approaches produced results within numeric error.
As shown in our former work,27 the molecules and silica sheets in the present study contain strongly localized bonds, O 2p–Si 3s 3p and O 2p–H 1s and therefore, the calculated valence electron total energy is sensitive to the value of the cut-off energies. High cut-off energies for the electron wave-functions are therefore required. In the present work the cut-off energy of the wave-functions was set at 750 eV, and the cut-off energy of the augmentation wave-functions was at 1000 eV. Tests of k-mesh and cut-off energies showed a good energy convergence (<1 meV per atom).
Computational results
A. 1D single atom silica chain (1D-SASC) and its flexibility
First we report the structural optimizations and total energy calculations for a mono-silicic acid molecule (one silicon atom with four silanol groups attached, Fig. 1a), its first condensation derivative di-silicic acid (Fig. 1c) and a water molecule (Fig. 1b) using the optB88_vdW functional. The calculated results are shown in Tables 1 and 2. The calculated water molecule has an O–H bond length of 0.971 Å with a H–O–H angle of 104.8°, which agrees well with experimental observations (0.958 Å and 104.5°).49,50 Bader charge analysis51–53 provides a charge transfer of about 0.56 e per H to O (Table 1).27 Recently, Preari and co-workers stabilized monosilicic acid molecules in a silica-supersaturated solution by uncharged polyethylene glycols (PEGs) at pH = 7.35 They also suggested the stabilization mechanism is most likely the hydrogen bonding between the silanol groups and –CH2–CH2–O–ether moieties. In the mono-silicic acid molecule, the Si atom/ion is tetragonally coordinated (sp3 hybrid bonding). The structural optimizations produced bond lengths: 1.64 Å for Si–O and 0.97 Å for O–H (Table 1).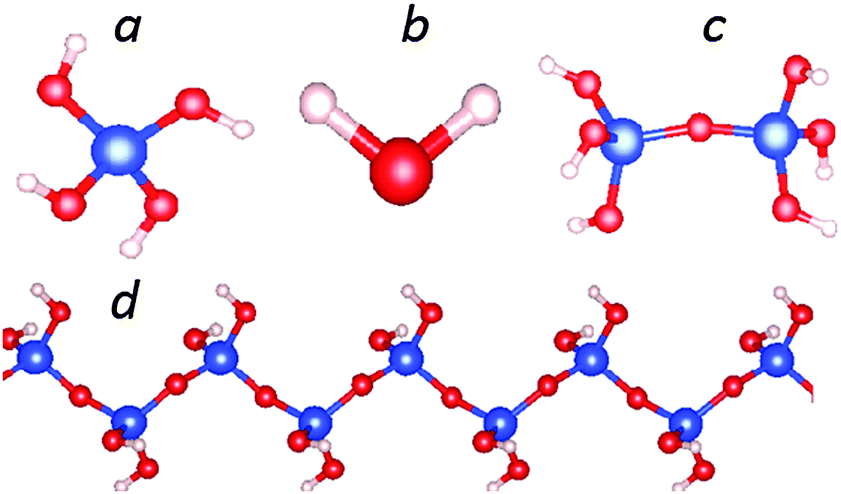 | ||
| Fig. 1 Schematic structure for an isolated silicic acid, Si(OH)4 molecule (a), a H2O molecule (b), a di-silicic acid molecule (c) and the one dimensional (1D) silica chain with chemical composition SiO(OH)2 (d). Computed results are listed in Table 1. | ||
| 1D system formulae | a (Å) | Bond length (Å) angle (°) | Bader charges | E Form (eV per Si) |
|---|---|---|---|---|
|
Di-silicic acid
Si2O(OH)6 |
-(Molecule) |
Si–O(Si): 1.65
Si–O(H): 1.62(×3) Si–O–Si: 145.3 |
Si: +3.16
O(Si): −1.62 O(H): −1.39 H: +0.60 |
−0.021 |
|
SASC
Si2O2(OH)4 |
4.980 |
Si–O(Si): 1.625
Si–O(H): 1.642 O–Si–O: 111.7 Si–O–Si: 145.3 |
Si: +3.18
O(Si): −1.62 O(H): −1.39 H: +0.60 |
−0.025 |
| SNRs with hexagonal rings (HR) | ||||
| HR_R1: Si4O5(OH)6 | 5.218 |
Si1–O(Si): 1.62
–O(H): 1.65 O–H: 0.97 |
Si: +3.19
O(Si): −1.60 O(H): −1.40 H: +0.61 |
−0.151 |
| HR_R2: Si6O8(OH)8 | 5.245 | −0.243 | ||
| HR_R3: Si8O11(OH)10 | 5.326 | −0.259 | ||
|
2D-HR sheet
Si2O3(OH)2 |
Hexagonal
a = 5.292 |
−0.336 | ||
| SNRs with squares (SQ) | ||||
|
SQ_R1
Si4O6(OH)4 |
5.500 |
Si1–O(Si): 1.62
–O(H): 1.65 O–H: 0.97 |
Si: +3.20
O(Si): −1.60 O(H): −1.40 H: +0.61 |
−0.028 |
|
SQ_R2
Si6O10(OH)4 |
5.513 | −0.058 | ||
|
SQ_R3
Si8O14(OH)4 |
5.615 | −0.060 | ||
|
2D SQ sheet
Si4O8 |
Orthorh.
a = 5.022 b = 5.545 |
−0.138 | ||
| SNRs with bilayers (BL) | ||||
|
BL_R1
Si8O14(OH)4 |
5.273 |
Si1–O(Si): 1.62
–O(H): 1.65 O–H: 0.97 |
Si: +3.20
O(Si): −1.60 O(H): −1.40 H: +0.61 |
−0.231 |
|
BL_R2
Si6O10(OH)4 |
5.271 | −0.257 | ||
|
BL_R3
Si8O14(OH)4 |
5.292 | −0.279 | ||
|
2D BL sheet
Si4O8 |
Hexagonal
a = 5.292 |
−0.332 | ||
| SNR with mixed squares and hexagonal rings | ||||
| Si8O11(OH)10 |
Orthorh.
a = 10.80 |
Si1(top) 1.63(×2) 1.64(×2)
Si1(top) 1.62(×2) 1.65(×2) |
Si: 3.18
O(Si): −1.60 O(H): −1.40 H: +0.61 |
−0.149 |
| Crystalline quartz: hexagonal lattice | ||||
| SiO2 |
a = 4.966
c = 5.475 |
Si–O: 1.63(×4) |
Si: +3.22
O: −1.61 |
−0.488 |
In Table 1, the computed results are also given of the di-silicic acid molecule (HO)3Si–O–Si(OH)3 (Fig. 1c). The calculated bond lengths are: Si–O(Si) 1.62 Å, and Si–O(H) is 1.65 Å and O–H 0.97 Å. The Si–O–Si angle is 160.8°. Therefore, the Si is in a tetragonal coordination. The calculations also showed that the formation energy of a di-silicic acid molecule is 42 meV or 21 meV per Si with respect to mono-silicic and water molecules.
Next we discuss the results of the 1D siloxane chain (1D-SASC) since we used such chains as the building block for the SNRs. The optimized geometry of a 1D-SASC is shown in Fig. 1d. The obtained chemical bond lengths and charges at different atomic sites and charge transfers in the chain are listed in Table 1. The results of structural optimization and total energy calculations are shown in Fig. 2b. The structure of the 1D-SASC is determined mainly by the Si–O bond length and the angles θ2[Si–O–Si] and θ1[(Si)O–Si–O(Si)], which are dependent on the length of the a-axis, as shown in Fig. 2.
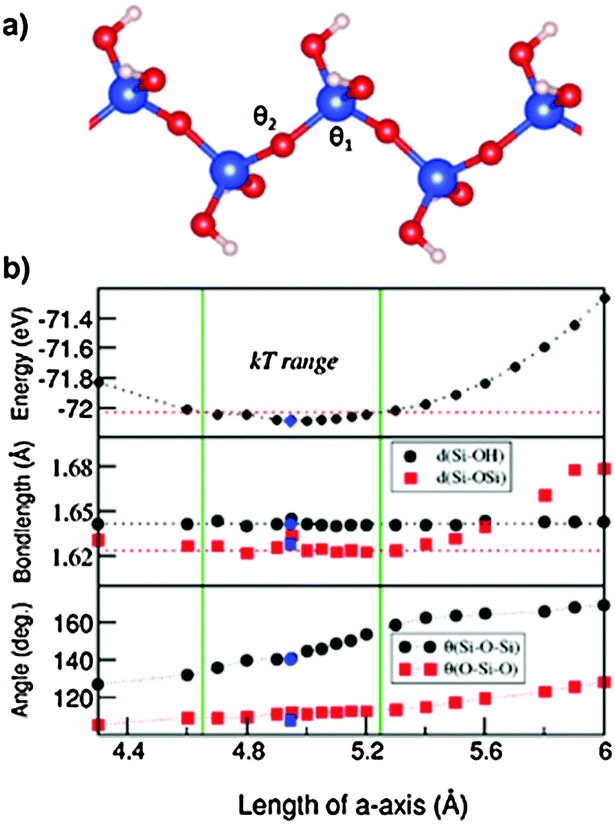 | ||
| Fig. 2 (a) The detailed schematic structure of 1D-SASC; (b) the calculated dependences of total valence-electron energy, θ1[angle (Si)O–Si–O(Si)] and θ2[angle Si–O–Si], and O(Si)–Si and O–H bond lengths of 1D-SASC (SiO(OH)2) on the length of the periodic a-axis. The dotted lines serve to guide the reader's eyes. The blue spheres and squares represent the calculated results with relaxation of both unit cells and coordinates of atoms with fixed volume. | ||
The optimized 1D-SASC structure found using the fixed lattice parameters has a length of a = 4.980 Å with total valence electron energy of E = −72.085 eV per cell, which is very close to the one found by the fixed volume approach (a = 4.945 Å and E = −72.087 eV per cell). Here we discuss the results from the fixed lattice parameters approach. The calculated structure showed the following local structural data: θ1[Si–O–Si] = 111.4° and θ2[(Si)O–Si–O(Si)] = 145.3°, and the Si–O(Si) bonds have a length of 1.624 Å which is slightly shorter than those of the two Si–O(H) bonds (1.641 Å). Therefore, the Si is in a distorted tetragonal coordination. Furthermore, the calculations showed that the bond lengths O–H (0.97 Å), Si–O(H) (1.64 Å) (Fig. 2b) and the angles H–O–Si (∼108°) and (H)O–Si–O(H) (∼111.4°) vary little variations in the length of the a-axis.
As shown in Fig. 2b, the energy curve changes slowly with the a-axis length. If we consider temperature effects (kT per Si, or about 50 meV per cell at T = 300 K, where k is the Boltzmann constant), then the flexibility of the 1D-SASC is clear: the a-axis has a range of 0.6 Å (12% of the length of a-axis), and θ1[(Si)O–Si–O(Si)] changes from 133° to 155°. Meanwhile, other structural parameters change but not significantly. The structural flexibility of the 1D-SASC originates mainly from the O–Si–O angle in the chain.
B. Optimized structures and formation energies of 1D-SNRs
Based on the 1D-SASC, there are two possibilities to connect the Si atoms via bridging O to form extensions in the second dimension of the 1D siloxane strings into ribbons: (a) one is that the siloxane bonds form hexagonal-rings with one OH cluster for each Si, (b) the other one is a square-like lattice of Si atoms (see below). Naturally, (c) a bilayer silica hexagonal ordering ribbon can also be built based on graphene-like ordering by further water condensation reaction to connect the Si atoms in two layers. The widths of the SNRs were designed from 1 to 3 units. When the widths are large enough, the SNRs can be regarded as 2D sheets which were reported previously.25 The calculated formation energies are summarized in Fig. 3 and in Table 2. In the following we discuss the systems in more detail. | ||
| Fig. 3 Calculated formation energies of the 1D silica chain and of ribbons with different decorations relative to the energies of silicic acid and water according to eqn (1). The negative values indicate favored water condensation reactions. The blue dotted line connects the relative formations of the isolated silicic acid molecule with N(Si)/[N(Si) + N(OH)] = 0.2 and the most stable form of silicon dioxide, quartz with N(Si)/[N(Si) + N(OH)] = 1.0. | ||
 | ||
| Fig. 4 Schematic structures for SNRs decorated with hexagonal rings of different widths. The ribbon contains one hexagonal ring across (c), two hexagonal rings (a) and three hexagonal rings (b). The relative stability of the SNRs is shown in Fig. 3. | ||
The formation energy of the SNR with one hexagonal ring with respect to monosilicic acid and one water molecule is about −0.15 eV per Si which is notably smaller than the formation of 1D-SASC (about 0.025 eV per Si). Similarly, SNRs with more hexagonal rings can be built in this way (Fig. 4a and b). As shown in Fig. 3 and Table 2, the calculations also showed that the SNRs become more stable with increasing number of hexagonal rings or the width of the ribbons. Naturally a 2D sheet is formed at infinite width, as shown in the insert in Fig. 3. As shown in the former simulations, for a 2D-sheet, the (OH) groups prefer to be at one side.27
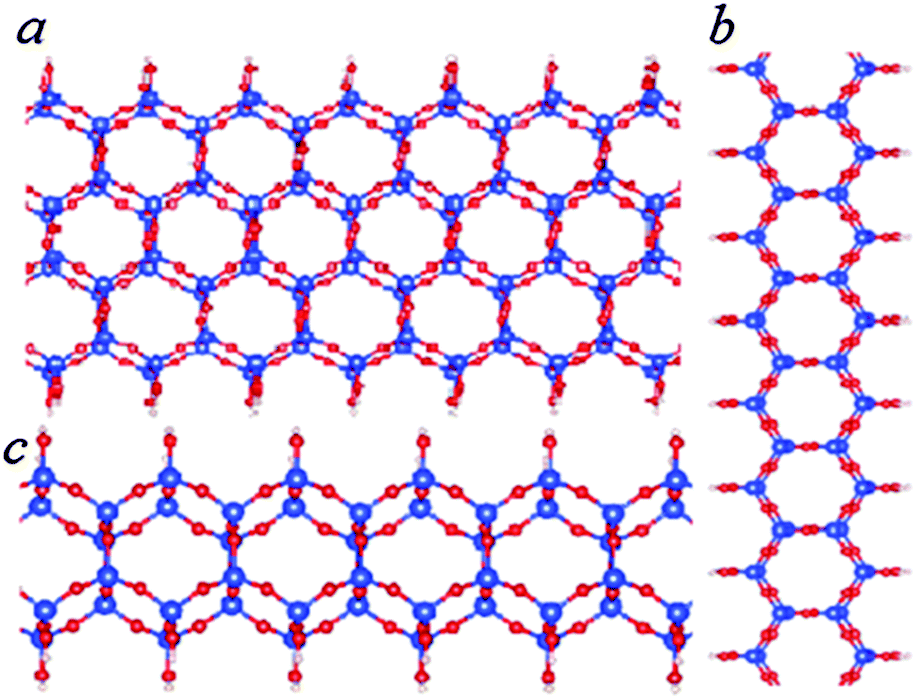 | ||
| Fig. 5 Schematic structures of bilayer SNRs with hexagonal rings. The ribbon is one hexagonal ring wide (b and c) or three hexagonal rings wide (a). The relative stability of the SNRs is shown in Fig. 3. | ||
The total energy calculations showed the formation energies for the bi-layer ribbons (Table 2 and Fig. 3). There is a trend of decreasing formation energy with increasing width of the ribbons. The most stable one is the anhydrous bi-layer sheet which has been prepared recently by vacuum evaporation deposition techniques.7–9
 | ||
| Fig. 6 Schematic structure of SNRs with squares. The ribbon is one square wide (c), two squares wide (a) or three squares wide (b). | ||
As shown in Fig. 3 and Table 2, the formation energies of the 1D ribbons with squares are negative with respect to the monisilicic and water molecules. However, the formation energies are above those of the SNRs decorated with hexagonal rings or the bilayer ribbons as shown in Fig. 3. Therefore, the SNRs with squares can only be formed and maintained under special conditions, such as an appropriate substrate.
 | ||
| Fig. 7 Schematic picture of the optimized structure of one 1D SNR decorated with both hexagonal rings and squares. | ||
As shown in Fig. 3 and Table 2, this unique 1D SNR has quite high stability. Therefore, one expects formation of such mixed siloxane structures as one of the first phases to be formed during the synthesis of low dimensional silica ribbons and sheets.
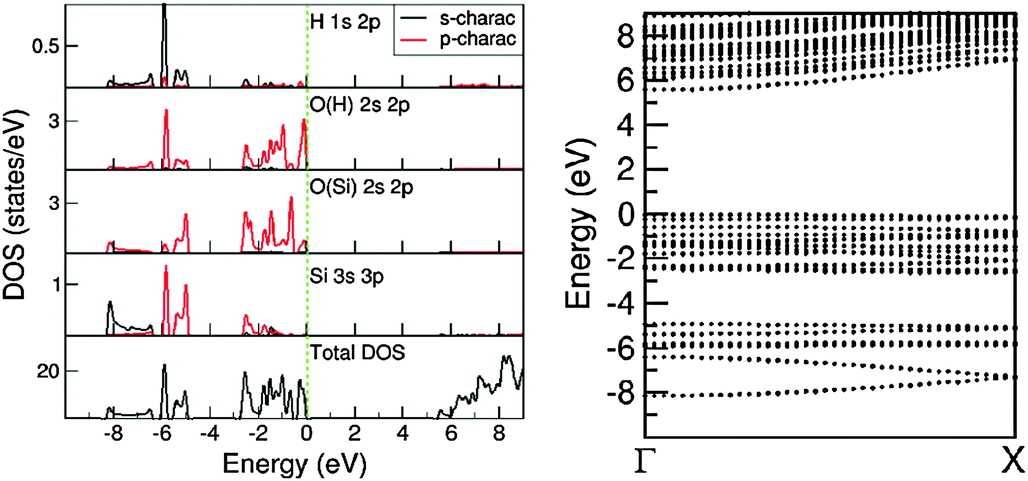 | ||
| Fig. 8 The calculated partial DOS in the atomic/ionic spheres and total density of states in the cell (left), and the dispersion curves of bands around the Fermi level (zero eV) along the periodic axis (right) for 1D-SASC chain. | ||
There are three distinct energy ranges for the electronic structure for the 1D-SASC: the s range, the p range (valence band) and the unoccupied conduction band. The O 2s states form a band from −18.98 eV to −17.15 eV with several peaks in between. The first high peak is positioned at −17.69 eV and is dominated by bonding of H 1s and O(H) 2s. The second high peak is at about −17.40 eV, being the (antibonding) states of O(Si) 2s, whereas bonding states form a band at the lower part of the s band. The O(Si) 2s states form two peaks at the lower part of the s band and the top of the s band. In the s band there are also some states from the Si atoms which can be considered as the tails of the nearby O(H) and O(Si) ions/atoms. Another notable point is that the overall shape of the H 1s 2p states is very similar to that of the O(H) 2s 2p states. That indicates a strong covalent nature between the O and H atoms even though the Bader charge analysis indicates an ionic nature.
The valence band is in the energy range of −8.30 eV to the Fermi level which is set to be zero eV. In the middle of the valence band, there is a strong peak at −5.90 eV, dominated by (antibonding) H 1s and O 2p states. The bonding/antibonding splitting of the H 1s states is about 11.8 eV, however they have no contributions to the chemical bonding of the ribbons/chains since both orbitals are occupied. Other states in the valence band are dominated by O 2p states of which the lower part is admixing with Si 3s 3p states. The calculations also showed that the top of the valence band is dominated by the O(H) 2p states. There is an energy gap of about 5.43 eV. The partial densities of states in the atomic/ionic spheres are considerably lower, whereas the total DOS at the lower part of the conduction band is quite high. That means that a large part of the density of states at the lower part of the conduction band are outside of the atomic/ionic spheres. As shown in Fig. 8, most of the bands have small dispersions, except the band at the lower s band and the ones at the lower part of the valence band. These bands correspond to O(Si) 2s and O 2p(Si)/Si 3s states, respectively, responsible for the strong interactions along the Si–O(Si)–Si chain and constitute the driving force for the formation of the 1D-SASC chains and SNR ribbons.
Fig. 9 shows the calculated total DOS for several selected systems: the 1D-SASC with schematic structure in Fig. 1d, the SNR decorated with two hexagonal rings (Fig. 4c), (c) the bi-layered SNR with three hexagonal rings (Fig. 5a) and (d) the SNR with two squares (Fig. 6a).
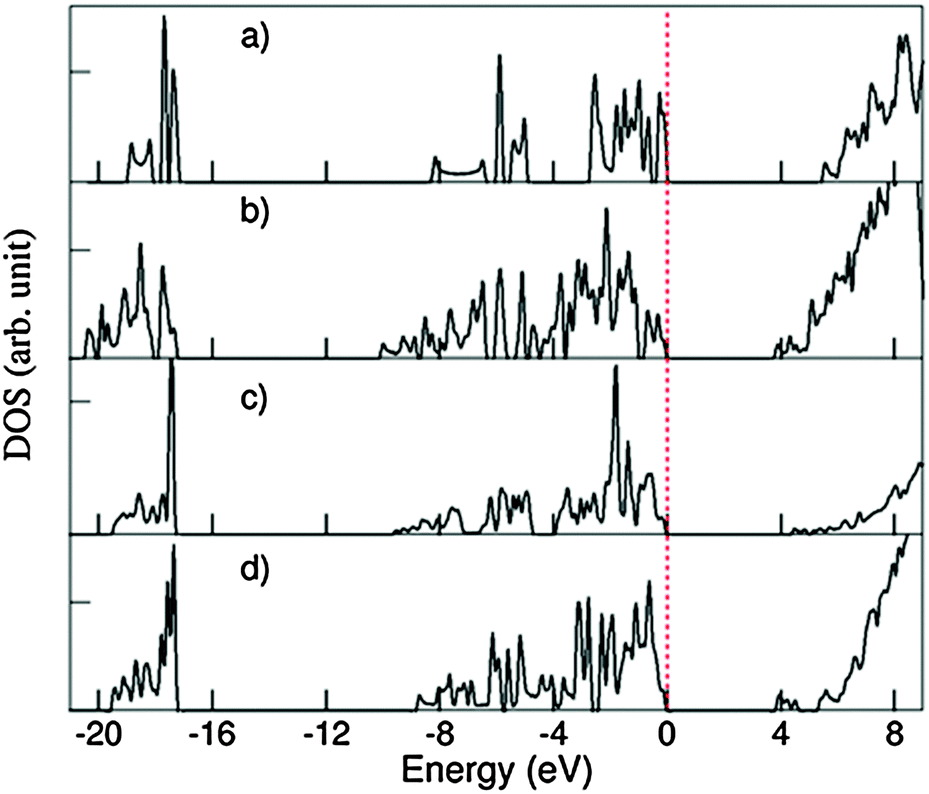 | ||
| Fig. 9 Density of states (DOS) curves for (a) 1D-SASC (Fig. 2a), (b) SNR decorated with two hexagonal rings (Fig. 4a), (c) bi-layered SNR with three hexagonal rings (Fig. 5a) and (d) SNR with two squares (Fig. 6a). | ||
As previously mentioned, there are similarities between the electronic structures of the 1D-SASC and the SNRs: (i) the lowest band is dominated by O 2s with/without bonding H 1s state, (ii) the valence bands are dominated by O 2p admixing with Si 3s 3p states and with/without antibonding H 1s state; and (iii) the conduction bands which are dominated by antibonding O 2p, Si 3s, 3p states. The calculated energy gaps vary strongly: 5.43 eV for the 1D-SASC, 3.75 eV for the SNR decorated with two hexagonal rings (Fig. 4a), 4.40 eV for the bi-layered SNR with three hexagonal rings (Fig. 5a) and 3.80 eV for the SNR with two squares (Fig. 6a). Using the same method and settings, the energy gap of quartz (SiO2) was calculated at 6.2 eV, which is smaller than the experimentally observed value of 9.3 eV.54 This not unusual since the DFT approximations generally underestimate the energy gap of insulators.43,55 Therefore, the actual energy gaps of these SNRs are expected to be larger than the values reported above. The wide band gaps of the SNRs show their highly insulating nature, making them interesting candidates as nano-scale insulating materials in nano-electronics, if they can be made sufficiently defect-free.
Summary and wide expectations
We performed first-principles simulations for various silica nano-ribbons as well as for a single atom chain built up from siloxane bonds that can be considered as an early stage product in ‘silica’ sheet or ribbon formation. The calculations showed a high stability of the SNRs. The SNRs with structured siloxane bonds in the form of hexagonal rings (HR) have similar stability as double anhydrous SNRs, both are more stable than those with square-like arrangements of the siloxane bonds. Experimentally, the double layer anhydrous SNRs were prepared by vacuum evaporation deposition techniques. The high stability of the SNRs with single hexagonal rings indicates possibilities for synthesis by means of colloidal methods as well. Furthermore, the calculations also showed that the SNRs with siloxane bond structured composed of mixed hexagonal rings and squares also have high stability. This indicates a high likelihood of formation of such mixed rings-and-squares ribbons are entropically favourable at elevated temperatures. Furthermore, an important factor is the pH value of the solution. The net concentration of H+ in acidic solutions is expect to induce formation of anhydrous silica SNRs, considering the reaction (OH)− + H+ ↔ H2O. The factors which influence the formation of the SNRs will be addressed in future work. We summarize our results/conclusions as follows.1. The present results can be helpful for understanding the growth mechanism of the various 1D and 2D forms of silica. The silica nanochains and nanoribbons might be useful as building blocks for larger silica frameworks.
2. The present results also suggest the mechanisms behind multi-string ribbons or bilayers, which may be useful 1D–2D scaffolds to arrange other elements, or functional groups such as, transition metal or noble metal atoms/ions, to form catalysts at the nano- or even at the atomic scale.
3. Unique structures and flexibility of the chains and ribbons provide possibilities to form bundles wherein other elements or molecules can be incorporated, if the synthesis can be achieved with sufficiently low levels of defects.
4. The high energy gaps indicate possibilities for 1D silica to be used as resistors for nano-electronics.
Acknowledgements
MvH acknowledges a VIDI grant from the Dutch Science Foundation NWO.References
- C. J. Brinker and G. W. Scherer, Sol-gel science: the physics and chemistry of sol-gel processing, Academic Press, Boston, 2013 Search PubMed.
- The Colloid Chemistry of Silica, ed. H. E. Bergna, ACS Washington, 1994 Search PubMed.
- G. R. Bhimanapati, Z. Lin, V. Meunier, Y. Jung, J. Cha, S. Das, D. Xiao, Y. Son, M. S. Strano, V. R. Cooper, L. B. Liang, S. G. Louie, E. Ringe, W. Zhou, S. S. Kim, R. R. Naik, B. G. Sumpter, H. Terrones, F. N. Xia, Y. L. Wang, J. Zhu, D. Akinwande, N. Alem, J. A. Schuller, R. E. Schaak, M. Terrones and J. A. Robinson, ACS Nano, 2015, 9, 11509–11539 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- A. Grinthal, S. H. Kang, A. K. Epstein, M. Aizenberg, M. Khan and J. Aizenberg, Nano Today, 2011, 7, 35–52 CrossRef.
- P. Y. Huang, S. Kurasch, A. Srivastava, V. Skakalova, J. Kotakoski, A. V. Krasheninnikov, R. Hovden, Q. Y. Mao, J. C. Meyer, J. Smet, D. A. Muller and U. Kaiser, Nano Lett., 2012, 12, 1081–1086 CrossRef CAS PubMed.
- P. Y. Huang, S. Kurasch, J. S. Alden, A. Shekhawart, A. A. Alemi, P. L. McEuen, J. P. Sethna, U. Kaiser and D. A. Muller, Science, 2013, 342, 224–227 CrossRef CAS PubMed.
- T. Björkman, S. Kurasch, O. Lehtinen, J. Katakaski, O. V. Yzxyev, A. Srivastawa, V. Skakalova, J. H. Smel, U. Kaiser and A. V. Krasheninnikov, Sci. Rep., 2013, 3, 3482 Search PubMed (7 pages).
- T. Schoeder, M. Adelt, B. Richter, M. Naschitzki, M. Baumer and H. J. Freund, Surf. Rev. Lett., 2000, 7, 7–14 CrossRef.
- J. Weissenrieder, S. Kaya, J.-L. Lu, H.-J. Gao, S. Shaikhutdinov, H. J. Freund, M. Sierka, T. Todorova and J. Sauer, Phys. Rev. Lett., 2005, 95, 076103 CrossRef CAS PubMed.
- D. Löffler, J. Uhlrich, M. Baron, B. Yang and X. Yu, Phys. Rev. Lett., 2010, 105, 146194 CrossRef PubMed.
- L. Lichtenstein, C. Bűvhner, B. Yang, S. Shaikhutdinov, M. Heyde, R. Wlodarczyk, J. Sauer and H. J. Freund, Angew. Chem., Int. Ed., 2012, 51, 404–407 CrossRef CAS PubMed.
- G. Wulf, Process of preparing silicic acid in a two-dimensional structure, US Pat., 3345132 A, 1967, Application Germany, June 3, 1964 W 36914, 8 Claims. (Cl. 23-182).
- J. Aizenberg, J. C. Weaver, M. S. Thannawala, V. C. Sundar, E. D. E. Morse and P. Fratzi, Science, 2005, 309, 275–278 CrossRef CAS PubMed.
- R. Ciriminna, M. Sciotino, G. Alonzo, A. de Schrijver and M. Pagliaro, Chem. Rev., 2011, 111, 765–770 CrossRef CAS PubMed.
- N. Baccile, F. Babonneau, B. Thomas and T. Coradin, J. Mater. Chem., 2009, 45, 8537–8559 RSC.
- L. F. Giraido, B. L. Löpez, L. Pérez, S. Urrego, L. Sierra and M. Mssa, Macromol. Symp., 2007, 258, 129–141 CrossRef.
- M. W. Zhao, R. Q. Zhang, Y. Y. Xia and S.-T. Lee, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 195412 CrossRef.
- S. T. Bromley, Nano Lett., 2004, 4, 1427–1432 CrossRef CAS.
- N. H. de Leeuw, Z. M. Du, J. Li, S. Yip and T. Zhu, Nano Lett., 2003, 3, 1347–1352 CrossRef CAS.
- M. W. Zhao, J. Z. G. Zhu, Y. Y. Xia and M. Lu, J. Phys. Chem. C, 2007, 111, 1234–1238 CAS.
- R. J. Bondi, S. H. Lee and G. S. Hwang, ACS Nano, 2011, 5, 1713–1723 CrossRef CAS PubMed.
- M. J. Walter, A. R. Thomson, W. Wang, O. T. Lod, J. Roos, S. C. MacMahon, M. A. Baron, E. Melekhova, A. K. Kleppe and S. C. Kohn, Geology, 2015, 418, 16–29 CAS.
- C. M. Fang and R. Ahuja, Phys. Earth Planet. Inter., 2006, 157, 1–7 CrossRef CAS.
- F. Corsetti and A. Mostofi, J. Phys.: Condens. Matter, 2014, 26, 055002 CrossRef PubMed.
- A. Pasquarello, M. S. Hybertsen and R. Car, Nature, 1998, 396, 58–60 CrossRef CAS.
- C. M. Fang, A. van Blaaderen and M. A. van Huis, J. Phys. Chem. C, 2015, 119, 14343–14350 CAS.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- J. Klimeš, D. R. Bowler and A. Michaelides, J. Phys.: Condens. Matter, 2010, 22, 022201 CrossRef PubMed.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- F. Tran and J. Hutter, J. Chem. Phys., 2013, 138, 204103 CrossRef PubMed (10 pages).
- C. M. Fang, W.-F. Li, R. S. Koster, J. Klimeš and M. A. Van Huis, Phys. Chem. Chem. Phys., 2015, 17, 365–375 RSC.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- L. L. Hench and J. K. West, Chem. Rev., 1990, 90, 33–72 CrossRef CAS.
- M. Preari, K. Spinder, J. Lazic and K. D. Demadis, J. Am. Chem. Soc., 2014, 136, 4236–4239 CrossRef CAS PubMed.
- W. H. Zachsriasen, J. Am. Chem. Soc., 1932, 54, 3841–3851 CrossRef.
- C. J. Brinker, J. Non-Cryst. Solids, 1988, 100, 31–50 CrossRef CAS.
- B. Dunn and J. I. Zink, Chem. Mater., 1997, 9, 2280–2291 CrossRef CAS.
- A. M. Buckley and M. Greenblatt, J. Chem. Educ., 1994, 71, 599–602 CrossRef CAS.
- A. van Blaaderen, R. Ruel and P. Wiltzius, Nature, 1997, 385, 321–324 CrossRef CAS.
- A. Kuijk, A. van Blaaderen and A. Imhof, J. Am. Chem. Soc., 2011, 133, 2346–2349 CrossRef CAS PubMed.
- J. J. Penninkhof, C. Graf, T. van Dillen, A. M. Vredenberg, A. van Blaaderen and A. Polman, Adv. Mater., 2005, 17, 1484–1488 CrossRef CAS.
- J. Neugebauer and T. Hickel, Density functional theory in materials science, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 438–448 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- J. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- J. Paier, R. Hirschl, M. Marsman and G. Kresse, J. Chem. Phys., 2005, 122, 234102 CrossRef PubMed (13 pages).
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- L. R. Painter, R. N. Hamm, E. T. Arakawa and R. D. Birkhoff, Phys. Rev. Lett., 1968, 21, 282–284 CrossRef CAS.
- R. E. Verrall and W. A. Senior, J. Chem. Phys., 1969, 50, 2746–2750 CrossRef CAS.
- R. F. W. Bader, J. Phys. Chem. A, 1998, 102, 7314–7323 CrossRef CAS.
- R. F. W. Bader and M. A. Austen, J. Chem. Phys., 1997, 107, 4271–4285 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- Z. A. Weinberg, G. W. Rubloff and E. Bassous, Phys. Rev. B: Condens. Matter Mater. Phys., 1979, 19, 3107–3117 CrossRef CAS.
- R. O. Jones, Rev. Mod. Phys., 2015, 87, 897–923 CrossRef.
| This journal is © the Owner Societies 2016 |