
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The surface structure matters: thermal stability of phthalic acid anchored to atomically-defined cobalt oxide films
Tao
Xu
a,
Matthias
Schwarz
a,
Kristin
Werner
a,
Susanne
Mohr
a,
Max
Amende
a and
Jörg
Libuda
*ab
aLehrstuhl für Physikalische Chemie II, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 3, D-91058 Erlangen, Germany. E-mail: joerg.libuda@fau.de; Fax: +49 9131 8527308
bErlangen Catalysis Resource Center and Interdisciplinary Center Interface Controlled Processes, Friedrich-Alexander-Universität Erlangen-Nürnberg, 91058 Erlangen, Germany
First published on 22nd March 2016
Abstract
We have investigated the influence of the structure of oxide surfaces on the thermal stability of anchored phthalic acid (PA) thin films. Specifically, we have performed temperature programmed infrared reflection absorption spectroscopy (TP-IRAS) of PA films deposited by physical vapor deposition (PVD) in ultra-high vacuum (UVH) onto three well-ordered surfaces: Co3O4(111), CoO(111) and CoO(100), all grown on Ir(100). Restructuring and desorption of PA were monitored in situ by TP-IRAS. Upon annealing of PA multilayers, co-adsorbed phthalic anhydride (PAA) desorbs at 200 K and a structural transition to a flat-lying adsorption geometry occurs at 250 K, before the PA multilayer desorbs at 300 K. At temperatures up to 400 K co-adsorbed mono-carboxylates partially desorb and partially convert to bis-carboxylates. Pronounced structure dependencies are observed regarding the thermal stability of the anchored bis-carboxylate monolayers. From Co3O4(111) the anchored PA desorbs over a wide range of temperatures centered at around 540 K. Weaker binding is observed for CoO(111) with desorption temperatures centered around 490 K. The strongest binding occurs on CoO(100), where the anchored PA films are found to be perfectly stable up to 510 K, before desorption starts and centers at around 580 K. The differences in binding strength are rationalized based on the density and the accessibility of the surface Co2+ ions. The findings show that the atomic structure of the oxide surface plays an important role in the stability of organic hybrid interfaces.
1. Introduction
Organic thin films on oxide surfaces play a key role in hybrid materials for molecular electronics1,2 or photovoltaics.3–5 To design stable and functional interfaces, the organic molecules often need to be immobilized on the oxide surface. Anchoring groups like carboxylic acid, phosphoric acid or hydroxyl are commonly used to covalently bind the functional organic units to the semiconducting or insulating oxide surface. The binding mechanism, the kinetics and the energetics of the immobilized organic films are essential factors that control the growth and structure formation of the film and, thereby, its electronic and physico-chemical properties.6–12Despite the wide-spread practical applications, very little is known about the binding sites that the anchor groups are attached to at the atomic level. The same holds for the influence of the binding sites on the stability, geometry, energetics and the formation kinetics of the organic film. The reasons for this lack of knowledge are the intrinsic complexity of such organic/oxide interfaces and the experimental challenges to study such systems on atomically controlled surfaces.
To address this challenge we follow a surface science approach under ultrahigh vacuum (UHV) conditions. Clean and atomically well-defined surfaces can be readily obtained for a variety of oxides, sometimes with different crystallographic orientations. Such surfaces can be prepared either as bulk single crystals or in the form of ordered thin films supported on metal single crystals.13–16 The latter approach prevents charging when working with photoelectron spectroscopy or with scanning tunneling microscopy (STM) and, in addition, allows varying the stoichiometry and surface structure in some cases. Based on the systematic work of Heinz, Hammer and coworkers (see their review17 and references therein), we have prepared Co3O4(111), CoO(111) and CoO(100) thin films on Ir(100). Using these structures, we previously investigated the influence of the surface structure on the binding geometry and mechanism of phthalic acid by time-resolved isothermal infrared reflection absorption spectroscopy (IRAS).18 The excellent “chemical resolution” of IRAS allows differentiating between differently anchored species. Taking advantage of the metal surface selection rule (MSSR),19 which is also valid for thin oxide films on metal surfaces,20 we simultaneously obtain information on the molecular orientation. We have found that the binding mechanisms are very different on the three cobalt oxide surfaces.18 This finding would suggest that the different binding geometries could also lead to different bonding strength and stability. In this work, we explore the thermal stability of the different surface bound carboxylates on Co3O4(111), CoO(111) and CoO(100) by temperature-programmed in situ spectroscopy. Indeed it is found that thermal behavior and stability of the anchored films is very different on the three surfaces.
2. Experimental
All measurements were conducted in an UHV system (base pressure of 1.0 × 10−10 mbar) which was described in detail elsewhere.21 The UHV system consists of a preparation chamber for sample cleaning and preparation and a measurement chamber for the IRAS experiments. The latter is equipped with a FTIR spectrometer (Bruker VERTEX 80v) which is connected via differentially pumped KBr windows.Preparation of Co3O4(111)/Ir(100)
A Co3O4(111) thin film is prepared via reactive deposition of Co atoms in O2 atmosphere based on the method described in literature22 with little modifications of parameters like O2 partial pressure, annealing temperature and the amount of Co. The detailed procedure is as follows: first the Ir(100) single crystal (MaTeck) is cleaned by cycles of Ar+ sputtering (1.8 keV, room temperature, 1 hour; Linde, 6.0) and annealing (1100 °C, 3 min) until a clear LEED pattern (room temperature) of the Ir(100)-(5 × 1) reconstructed surface can be seen. Secondly, the Ir(100)-(5 × 1) surface is heated to 1000 °C in 5 × 10−8 mbar O2 (Linde, 5.0) for 3 min and cooled down in O2 atmosphere to room temperature. This leads to an Ir(100)-(2 × 1)O reconstructed surface which shows a clear (2 × 1) pattern in LEED. Starting with the Ir(100)-(2 × 1)O, Co is evaporated onto the surface (cooled to temperatures below 0 °C) using a commercial electron beam evaporator (Focus EFM3, 2 mm Co rod, Alfa Aesar 99,995%) in an atmosphere of 1.0 × 10−6 mbar O2 for 18 min. The evaporation rate of Co was determined to be 2 Å min−1 by means of a quartz micro-balance. After the growth, the film was annealed in O2 (1.0 × 10−6 mbar) to 250 °C for 2 min and then in UHV to 430 °C for 5 min. The film was checked qualitatively by comparing the LEED I–V curves with literature.22Preparation of CoO(111)/Ir(100)
The CoO(111) film is obtained from a Co3O4(111) thin film which is prepared via the procedure described above. Simple heating of the Co3O4(111) film in UHV leads to loss of oxygen and transformation to CoO(111).23 In our experiments, Co3O4(111) thin films (Co amount equivalent of 36 Å Co metal) were heated to 620 °C for 5 min. The quality of the CoO(111) thin film was checked by LEED.23Preparation of CoO(100)/Co/Ir(100)
To obtain the CoO(100) thin film, a metastable Ir(100)-(1 × 1) reconstructed surface is prepared in the first step. To this end, we start with a clean Ir(100)-(5 × 1) surface and prepare the Ir(100)-(2 × 1)O reconstructed surface following the procedure described above for the Co3O4(111). Then, the sample is heated to 275 °C in 1 × 10−7 mbar H2 (Linde, 5.3) for 1 min and then heated in UHV to 275 °C for 1 min before cooling down to room temperature. This procedure yields a Ir(100)-(1 × 1) surface characterized by a sharp (1 × 1) pattern in LEED. Subsequently, metallic Co is deposited onto the surface (50 °C) at a rate of 2 Å min−1 for 6 min. After preparation of the Co buffer layer, the sample is cooled down to temperatures below −50 °C: then Co is deposited reactively (2 Å min−1) in 4 × 10−7 mbar O2 for 3 min. Subsequently the film is annealed to 100 °C for 1 min resulting in an ordered CoO(100) structure. For thicker and better ordered CoO(100) films, a second reactive deposition of Co in O2 for 18 min is conducted while keeping the sample at low temperature (<−50 °C). After that, the CoO(100) film is annealed to 600 °C for 10 min. The as-prepared CoO(100) thin film shows a sharp (1 × 1) pattern in LEED.Organic thin film deposition
PA (Sigma-Aldrich, 99.9%) was dosed from a home-built Knudsen source that can be separated from the UHV chamber by a gate valve. The evaporator was baked out overnight before first usage. PA was placed into a glass reservoir with an orifice. For degassing, the glass reservoir with the PA was heated to 350 K for at least 30 min while being pumped through a separate pumping line. Subsequently, the gate valve to the chamber was opened. For deposition, the evaporator was placed in front of the sample and the sample shutter was opened. The evaporation temperature was adjusted to ensure a constant deposition rate during the time scale of the dosing experiment. The deposition rate is estimated to be around 0.03 ML per min on Co3O4(111) and CoO(111) and 0.08 ML per min on CoO(100), respectively, as determined from low temperature adsorption experiments where we can distinguish monolayer from multilayer.18Temperature-programmed IRAS (TP-IRAS)
Two types of TP-IRAS experiments were conducted for the PA films on the three cobalt oxides. In a first set of experiments we investigated the thermal stability of multilayer PA films deposited at 133 K. The TP-IRAS experiments were performed up to a temperature of 363 K. The second type of experiments aimed at the thermal stability of surface-bound monolayers formed at room temperature (300 K). Within this experiment, the monolayer films were heated from 300 to 620 K (570 K for the CoO(111)). A heating rate of 2 K min−1 was applied in all heating ramps. During the heating IRAS spectra were taken with a resolution of 4 cm−1 at a rate of 30 s per spectrum.IRAS scaling
IRAS spectrum is practically defined as normalized reflectivity which is obtained by:where R is the reflectance from the adsorbate-covered surface and R0 is the reflectance of the clean surface.19,24 Thus, IRAS peak intensity is determined by the ratio of R and R0. With increasing temperature, the reflectivity of the metal substrate is known to decrease.25,26 As a result, R would be continuously reduced so that sensitivity of IRAS is compromised. The overall effect would be a decrease of adsorbed molecular IRAS signals, even if no desorption is taking place. To compensate the loss of R at high temperatures, the spectra were scaled by a factor ai, with
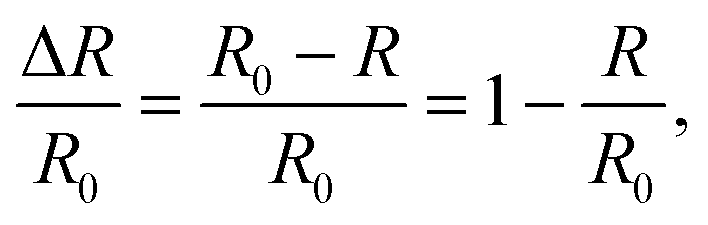
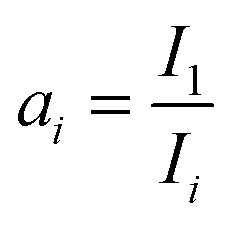 and Ii being the intensity at 2000 cm−1 in the ith spectrum. We chose the intensity at 2000 cm−1 for referencing as there are no relevant bands in this region. The scaling procedure was only applied for TP-IRAS spectra above 300 K as the normalized reflectivity loss is neglectable below 300 K.
and Ii being the intensity at 2000 cm−1 in the ith spectrum. We chose the intensity at 2000 cm−1 for referencing as there are no relevant bands in this region. The scaling procedure was only applied for TP-IRAS spectra above 300 K as the normalized reflectivity loss is neglectable below 300 K.
3. Results and discussion
3.1. Multilayer films of phthalic acid on Co3O4(111), CoO(111) and CoO(100)
To study the thermal behavior of PA multilayers, the three surfaces were first cooled to 133 K and, subsequently, PA was continuously deposited for 60 min to accumulate multilayer films on each surface. Although the deposition rate of PA was slightly different for each deposition, within each deposition process the deposition rate was reasonably constant (for details about the dosing of PA we refer to ref. 18 and 27). By comparing the IRAS spectra at saturation of the monolayer to the final spectrum of each series, the PA multilayer films on Co3O4(111) and CoO(111) were estimated to be 6 ML thick, while on CoO(100) the PA multilayer was only 2 ML thick. The TP-IRAS spectra (133 to 363 K) of the PA multilayer on Co3O4(111), CoO(111) and CoO(100) are shown in Fig. 1 as contour color plots.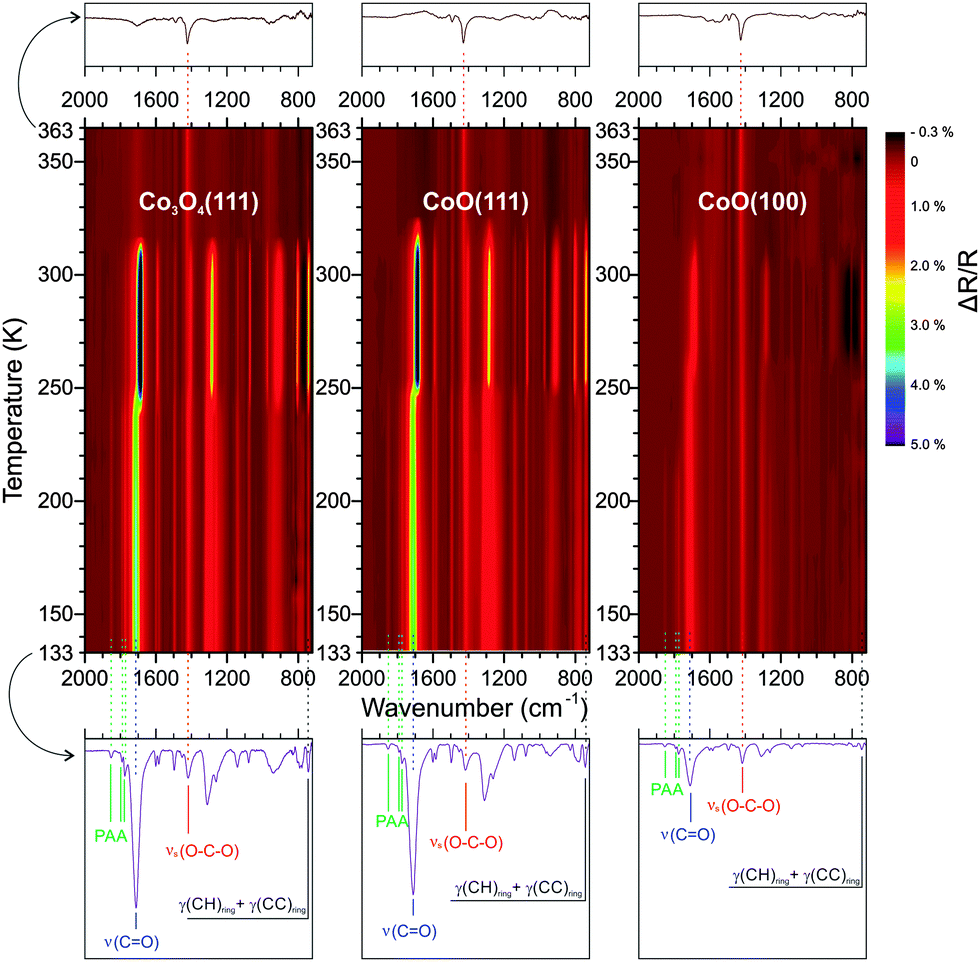 | ||
Fig. 1 TP-IRAS of PA multilayers on three cobalt oxide surfaces. Characteristic peaks are marked using the following color code: PAA = green; ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, carboxylic acid) = blue; νs(OCO, carboxylate) = red; γ(CH) = grey. O, carboxylic acid) = blue; νs(OCO, carboxylate) = red; γ(CH) = grey. | ||
At 133 K, the spectra on the three surfaces are similar to each other and also resemble the bulk transmission spectrum.28 Prominent peaks are observed at 743, 1259, 1312, 1419, 1494, 1548, 1583, 1710, 1775, 1790 and 1853 cm−1. The assignments of peaks based on literature and our previous work18 are summarized in Table 1. Specifically, the peak at 743 cm−1 can be attributed to the out-of-plane CH deformation. The 1710 cm−1 peak corresponds to the characteristic CO stretching mode in the carboxyl group. The 1419 and 1583 cm−1 peaks can be assigned to symmetric and asymmetric OCO stretching of the surface-bound carboxylate. Noteworthy, the peaks at 1775, 1790 and 1853 cm−1 cannot be assigned to any vibrational mode of PA, but correspond to phthalic anhydride (PAA).28–30 It was previously found that a small fraction of PAA is formed during PVD of PA under UHV conditions.27 Upon heating, the spectra undergo shifts of the peak positions and variations of the peak intensities. Some general features can be identified. First, the PAA peaks decrease dramatically at around 200 K while the intensity of the other peaks remains largely unchanged. Secondly, a shift of spectral positions and a change of peak intensities takes place at around 250 K. Finally, a drastic decrease of the most prominent peaks occurs at around 310 K. At the highest temperature in this experiment (363 K), the most prominent peak is the OCO symmetric stretch of the surface-bound carboxylate at 1419 cm−1.
| Peak position (cm−1) | Assignment29,43–48 | Comment | ||
|---|---|---|---|---|
| CoO(100) | CoO(111) | Co3O4(111) | ||
| 743 | 743 | 743 | γ(CH)ring + γ(CC)ring | PA, PAA |
| 1260 | 1262 | 1259 | ν s(COC) | PAA |
| 1309 | 1312 | 1312 | ν(CO) + ν(CC) + δ(OH) | PA |
| 1417 | 1416 | 1419 | ν sym(OCO) | Carboxylate |
| 1497 | 1495 | 1494 | ν(C–Cring) + νCCring + ν(CH) | PA, PAA |
| 1552 | 1550 | 1548 | ν asym(OCO) | Carboxylate |
| 1585 | 1583 | 1583 | ν(CC)ring | PA, PAA |
| 1602 | 1601 | 1603 | ν(CC)ring | PA, PAA |
| 1712 | 1712 | 1710 |
ν(CO) |
PA |
| 1776 | 1776 | 1775 |
ν
asym(CO) |
PAA |
| 1793 | 1793 | 1790 | δ(CH) + δ(CC)ring | PAA |
| 1853 | 1855 | 1853 |
ν
sym(CO) |
PAA |
To follow the changes quantitatively, we integrated the peak area of the out-of-plane deformation mode γ(CH) at 743 cm−1, the symmetric carboxylate stretching mode νs(OCO) at 1419 cm−1 and the CO stretching mode of the free acid νs(CO) at 1710 cm−1 for all three surfaces and plotted them as a function of the surface temperature. The resulting data is shown in Fig. 2a–c. Three integrated PAA peak areas for the bands at 1775, 1785 and 1850 cm−1 are shown separately in Fig. 2d and e. In line with the three major spectral changes describe above, we can divide the full temperature range into four regimes which are indicated in Fig. 2a–f.
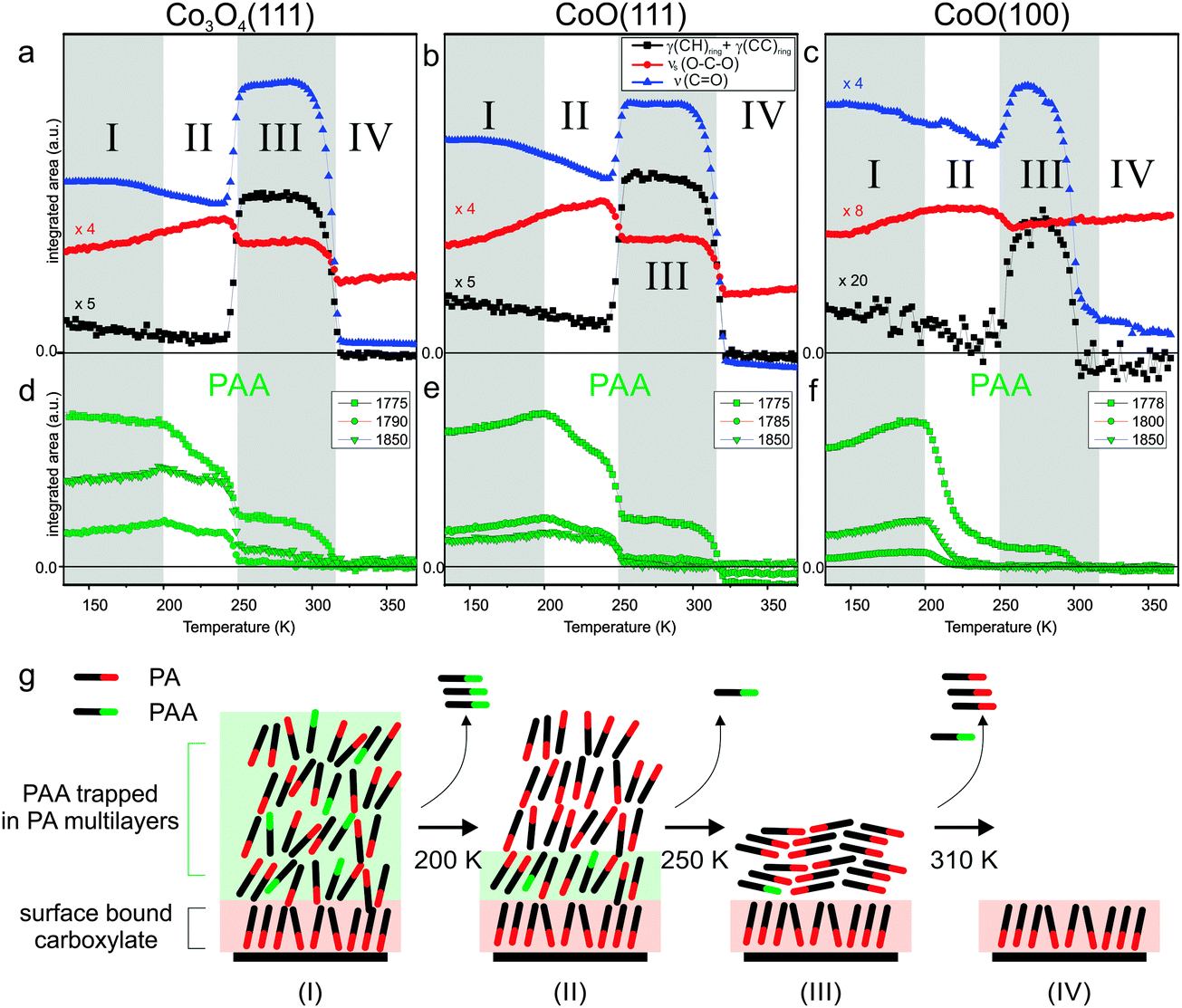 | ||
| Fig. 2 (a–f) Integrated peak areas for PA and PAA on the three cobalt oxide surfaces; (g) schematic representation the molecular orientation in PA multilayer films as a function of substrate temperature. | ||
In the low temperature regime from 133 to 200 K there is very little change in intensity for the PAA bands. Most likely, this minor change is due to temperature induced ordering to some extent. For the PA, we observe an increase of the symmetric OCO stretch at 1419 cm−1 and a decrease of γ(CH) out-of-plane peak at 743 cm−1. In general the relatively low intensity of the γ(CH) band indicates that most of the PA molecules remain in an upright-standing geometry (note that the γ(CH) mode is polarized perpendicularly to the molecular plane and thus shows highest intensity in IRAS at small adsorbate tilting angles towards the surface). The decrease in intensity with increasing temperature indicates an even better aligned upright-standing adsorption geometry of PA, probably due to an increased fraction of PA molecules bonding to the surface. At 200 K a dramatic decrease of the PAA peaks can be observed on all three surfaces indicating that a large amount of PAA is desorbing. This observation fits to our previous studies of multilayer PAA desorption from MgO(100) which is found to occur at around 220 K.31 This is also consistent with the standard sublimation enthalpy (ΔsubH°) of PAA (88.7 ± 2.3 kJ mol−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 32) which is lower than that of PA (129.8 ± 0.6 kJ mol−133). The fact that on the CoO(100) the PAA peaks are very small can be explained by the much thinner PA film (2 ML) in this case (the remaining intensity at 1776 cm−1 is an artifact due to the broad PA CO stretching peak at 1710 cm−1 which overlaps in frequency with the PAA modes). We assume that desorption of PAA occurs preferentially from the topmost PA layers. This explains that the thinner films lose most PAA at 200 K, whereas in the thicker films the PAA remains trapped in deeper PA layers at temperatures higher than 200 K.
32) which is lower than that of PA (129.8 ± 0.6 kJ mol−133). The fact that on the CoO(100) the PAA peaks are very small can be explained by the much thinner PA film (2 ML) in this case (the remaining intensity at 1776 cm−1 is an artifact due to the broad PA CO stretching peak at 1710 cm−1 which overlaps in frequency with the PAA modes). We assume that desorption of PAA occurs preferentially from the topmost PA layers. This explains that the thinner films lose most PAA at 200 K, whereas in the thicker films the PAA remains trapped in deeper PA layers at temperatures higher than 200 K.
While in the second temperature regime from 200 to 250 K the PAA bands decrease rather slowly, a dramatic decrease is observed around 250 K. At the same time we observe an increase of the ν(CO) band at 1710 cm−1 and of the out-of-plane γ(CH) band at 743 cm−1, together with a narrowing of the peak width. These observations indicate that a more homogeneous structure is formed in which the PA adopts a largely flat-lying orientation. Similar recrystallization phenomena were reported for other organic thin films,34–36 including our previous studies of PA on MgO(100).27 Interestingly, the orientation of the bulk PA film appears to have an effect on the surface-bound phthalate, for which the intensity of the νs(OCO) peak is observed to decrease. A possible explanation is that also the surface-anchored layer adopts a more tilted orientation. Simultaneously to the crystallization transition, the PAA features decrease dramatically. We assign this desorption of PAA to an enhanced mobility of the pre-trapped PAA during the restructuring process of the PA film.
In the third temperature period from 250 to 310 K the band intensities remain largely constant, before they suddenly decrease at around 310 K. The latter effect indicates desorption of the PA multilayer. In the temperature regime above 310 K only the surface-bound monolayer remains on the surfaces. The dominating band in this regime is the symmetric OCO stretching mode of the carboxylate at 1420 cm−1. We will discuss the thermal behavior of the anchored monolayers in the next section. Fig. 2g shows a schematic summary of the molecular orientation in PA multilayer films with increasing temperature.
3.2. Anchored monolayers of phthalic acid on Co3O4(111)
To investigate the thermal stability of surface-bound carboxylate species on Co3O4(111), we performed TP-IRAS experiments between 310 K and 620 K. Details on the procedures are given in the Experimental section. The resulting data is shown in Fig. 3a as contour color plots where changes of the peak intensity are represented by a color gradient. In contrast to the TP-IRAS experiments for the PA multilayers (Section 3.1) for which the clean surface at low temperature was used as reference, here we use as a reference the IRAS spectrum at the highest temperature when all PA has desorbed.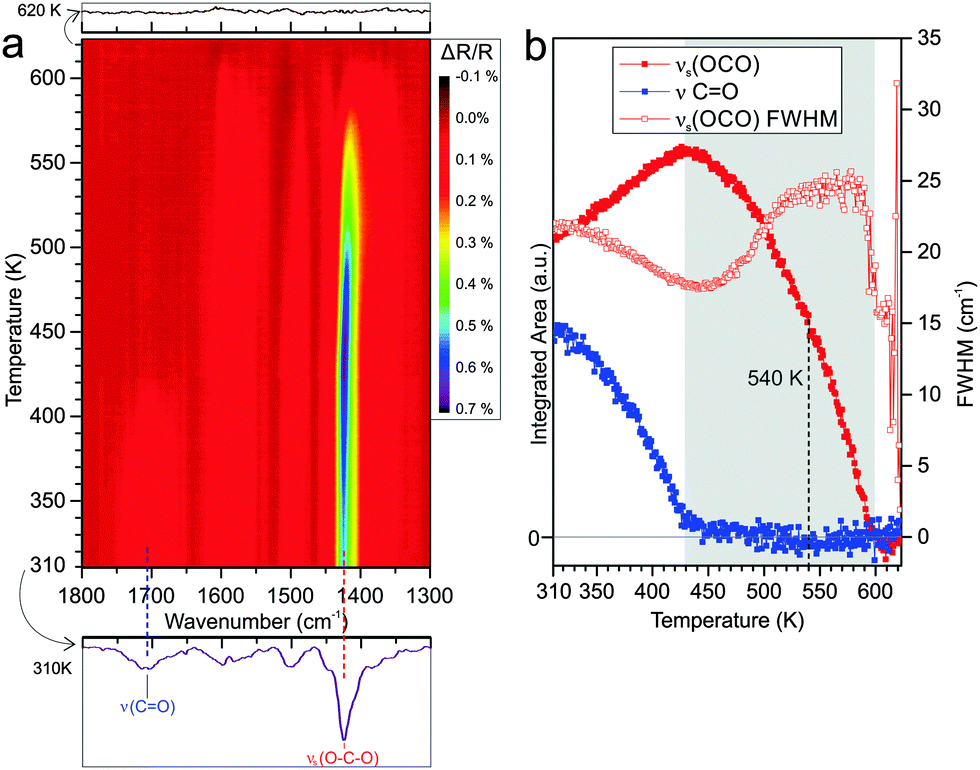 | ||
| Fig. 3 (a) Contour plot of the TP-IRAS spectra recorded after exposure of Co3O4(111) to PA at 300 K. (b) Integrated peak area of the symmetric stretching band of the carboxylate νs(OCO) (1425 cm−1) and the CO stretching band of the free acid ν(CO) (1706 cm−1) as a function of the surface temperature. The change of the FWHM of the νs(OCO) band is also shown (hollow red rectangles). | ||
As can be seen from Fig. 3a, the most prominent peak is the OCO symmetric stretching band of the surface-bound carboxylate at 1425 cm−1. The CO stretching band of the free carboxylate is observed as well at 1706 cm−1 but has lower intensity than the OCO peak. The other bands at 1494 and 1548 cm−1 are assigned to aromatic ring vibrations and to the asymmetric OCO stretching mode, respectively (see Table 1). As discussed in our previous work, the spectral features indicate that PA is anchored to the surface in form of a chelating bis-carboxylate and co-adsorbed with a chelating mono-carboxylate at larger exposure.18
With increasing temperature, the principal spectral features remain the same, indicating that no new infrared-active surface species are formed. However, the peak intensities and peak widths change dramatically. To follow these changes quantitatively, the main bands are scaled for temperature-dependent changes in the reflectivity (see Experimental section) and the peak areas are integrated. The resulting data is shown in Fig. 3b. Specifically we focus on the peak area and peak width of the bound carboxylate mode νs(OCO) at 1425 cm−1 and of the free acid mode ν(CO) at 1706 cm−1. We observe that thermally induced changes can be distinguished and divided into two temperature regimes. In the first regime from 310 to 430 K the free acid CO band decreases in intensity until it completely vanishes at 430 K while the carboxylate OCO band increases in intensity and reaches a maximum at 430 K. At 300 K PA binds to the surface as a chelating bis-carboxylate on presence of a small amount of chelating mono-carboxylate.18 The decreasing intensity of the free acid band in the lower temperature regime indicates conversion of mono-carboxylate to bis-carboxylate. The much weaker increase of the carboxylate band suggests, however, that the process is accompanied by desorption of a large fraction of the mono-carboxylate, which would, thereby, generate free adsorption sites for the formation of the bis-carboxylate. Meanwhile, the decrease of the full width at half maximum (FWHM) of the carboxylate band indicates a more homogeneous chemical environment of the surface-bound species with increasing temperature, i.e. a thermally induced ordering of the adsorbate layer.
In the second temperature regime from 430 to around 600 K, we observe a continuous decrease of the carboxylate peak until the band vanishes completely at 600 K. The FWHM of the OCO peak increases continuously in this regime, indicating a decrease of ordering in the partially desorbed layer. The reason behind the broad range of desorption temperatures is not clear yet and will be further discussed below. As a semi-quantitative measure for the desorption activation energy we determine the temperature at which 50% of the maximal OCO band intensity is lost. This point is reached at a temperature of 540 K (±10 K). Above 600 K, no PA induced bands can be identified anymore, indicating complete desorption of the carboxylate.
3.3. Anchored monolayers of phthalic acid on CoO(111)
Using the same experimental procedure and conditions as described in Section 3.2 for Co3O4(111), we have performed a TP-IRAS experiment for PA on CoO(111). The results are displayed in Fig. 4. After adsorption at 300 K, we observe a spectrum that is dominated by the symmetric OCO band at 1430 cm−1 together with a weak feature originating from the CO vibration of the free acid (indicating the presence of a small fraction of mono-carboxylate). In a previous study we showed that the distorted bis-carboxylate is formed at 300 K, but not at low adsorption temperatures. Based on this observation we suggested that on the oxygen-terminated CoO(111), thermally-induced restructuring of the Co2+ surface ions is required to facilitate formation of the bis-carboxylate species.18
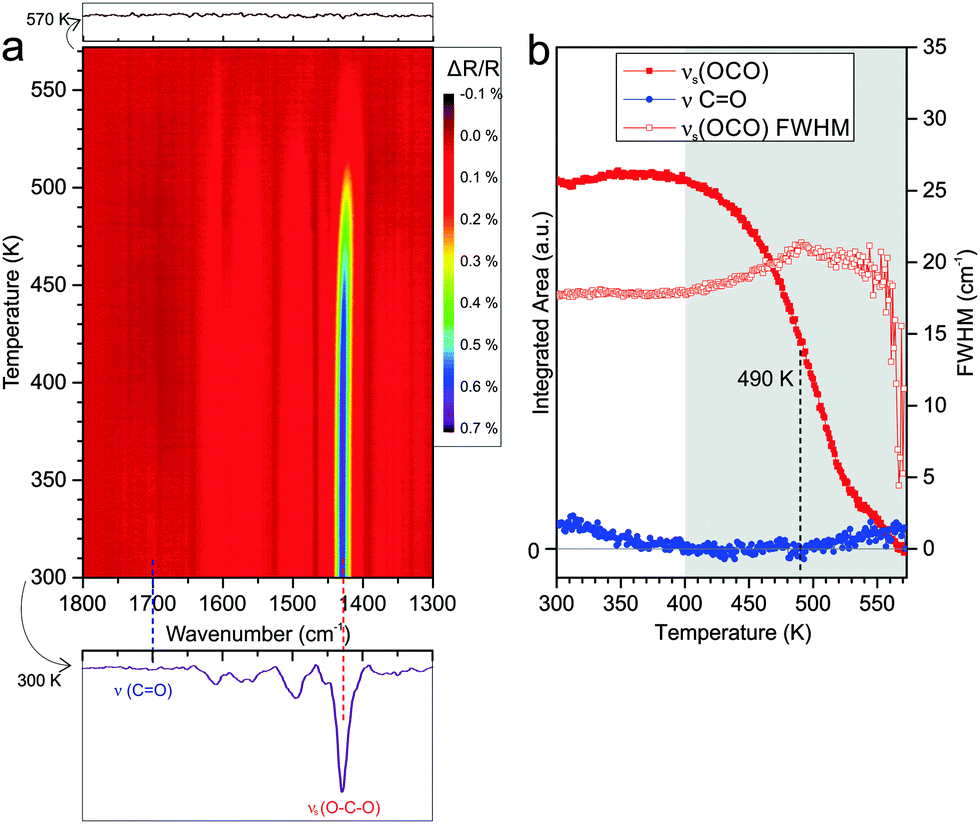 | ||
| Fig. 4 (a) Contour plot of the TP-IRAS spectra recorded after exposure of CoO(111) to PA at 300 K. (b) Integrated peak area of the symmetric stretching band of the carboxylate νs(OCO) (1430 cm−1) and the CO stretching band of the free acid ν(CO) (1700 cm−1) as a function of the surface temperature. The change of the FWHM of the νs(OCO) band is also shown (hollow red rectangles). | ||
Similar as for Co3O4(111), the TP-IRAS shows no indication for thermally induced formation of other IR-active species upon annealing. We subdivide the experiment into two temperature regimes. Within the first regime from 300 to 400 K the carboxylate OCO peak does neither change in intensity nor in width. Traces of co-adsorbed mono-carboxylates desorb and are converted up to a temperature of 350 K, as indicated by the decrease in intensity of the CO vibrational band. We conclude that the bis-carboxylate layer on CoO(111) is stable up to 400 K. No indication for restructuring and or reordering of the monolayer is observed. In the second temperature regime from 400 to 570 K we observe a continuous decrease of the OCO band, implying continuous desorption of the carboxylate species. Similar to the experiment on Co3O4(111), the temperature interval in which desorption occurs is rather broad and complete desorption is observed at temperatures above 570 K. The temperature at which half of the OCO peak intensity is lost is found to be reached at around 490 K (±10 K), which is 50 K lower than the value observed for Co3O4(111). During desorption we observe an increase of the FWHM of the carboxylate mode, indicating a less homogeneous environment of the species remaining on the surface.
3.4. Anchored monolayers of phthalic acid on CoO(100)
Finally we performed an equivalent TP-IRAS experiment for PA adsorbed at 300 K on CoO(100). In Fig. 5a the corresponding data is displayed. After adsorption at 300 K, we observe a prominent band corresponding to the OCO carboxylate mode at 1429 cm−1 together with a weak CO vibration band originating from free carboxylic acid groups at 1700 cm−1. In a previous study we showed that PA forms a bridging bis-carboxylate at 300 K, with some tendency to form additional co-adsorbed mono-carboxylate species at higher exposure.18
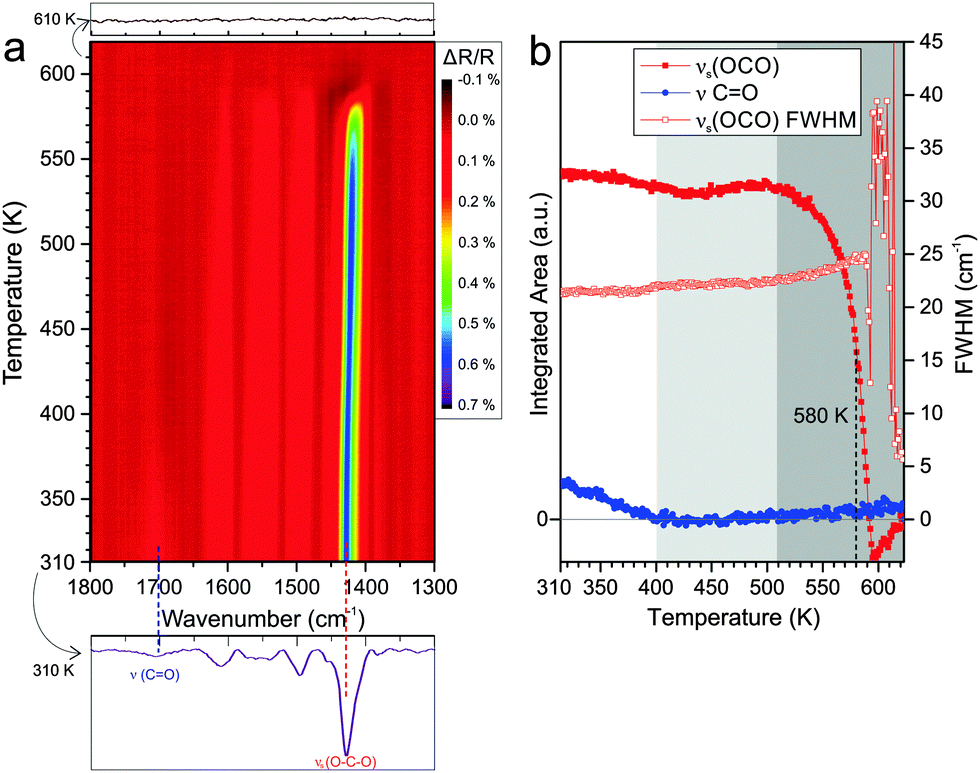 | ||
| Fig. 5 (a) Contour plot of the TP-IRAS spectra recorded after exposure of CoO(100) to PA at 300 K. (b) Integrated peak area of the symmetric stretching band of the carboxylate νs(OCO) (1429 cm−1) and the CO stretching band of the free acid ν(CO) (1700 cm−1) as a function of the surface temperature. The change of the FWHM of the νs(OCO) band is also shown (hollow red rectangles). | ||
In the TP-IRAS spectra we again observe changes in intensity and peak width, but no indication for the appearance of new bands. Similar as for the experiments on the other two surfaces, we analyze the peak intensity and peak width quantitatively. The resulting data is shown in Fig. 5b. We subdivide the temperature dependence into three regimes. In the first regime from 310 to 400 K, the CO stretching band of the free acid decreases and, finally, vanishes completely. This effect is neither associated with a change in the band intensity nor in the band width of the OCO carboxylate band. We associate this effect with partial desorption of mono-carboxylate while partial mono-carboxylate is transformed to bis-carboxylate, refilling the vacant sites thus the overall OCO band intensity is preserved. The fact that the peak width does not change at all indicates that there is very little change in the local environment of the adsorbed PA. This observation may indicate that the bis-carboxylate adsorbs on the CoO(100) surface with very little distortion due to intermolecular interactions. In the second temperature regime from 400 to 510 K the bis-carboxylate film is perfectly stable and does not show any indication for desorption or restructuring. In the third temperature regime from 510 to 600 K we observe desorption of the surface carboxylate. Similar as on the other surfaces, the formation of an inhomogeneous layer after partial desorption leads to broadening of the carboxylate band, but the effect is much weaker on CoO(100) than on the other two surfaces. This is in line with the hypothesis that the intermolecular interaction within the anchored PA layer on CoO(100) is weaker than in the other two cases. The temperature at which desorption leads to loss of 50% of the intensity of the carboxylate OCO band is found to be reached at 580 K (±10 K). This value is the highest observed on the three cobalt oxide surfaces studied.
3.5. Discussion: influence of the surface structure on the structure and thermal stability of phthalic acid thin films
The experiments described in Section 3.1 show that there is little influence of the interfacial structure on the orientation and structure of PA multilayer films. However, the molecular orientation of the anchored monolayer and the presence of contaminations such as PAA play an important role during thin film growth. Major differences between the three surfaces are witnessed regarding the thermal stability of anchored PA monolayers. The main findings are summarized in Fig. 6.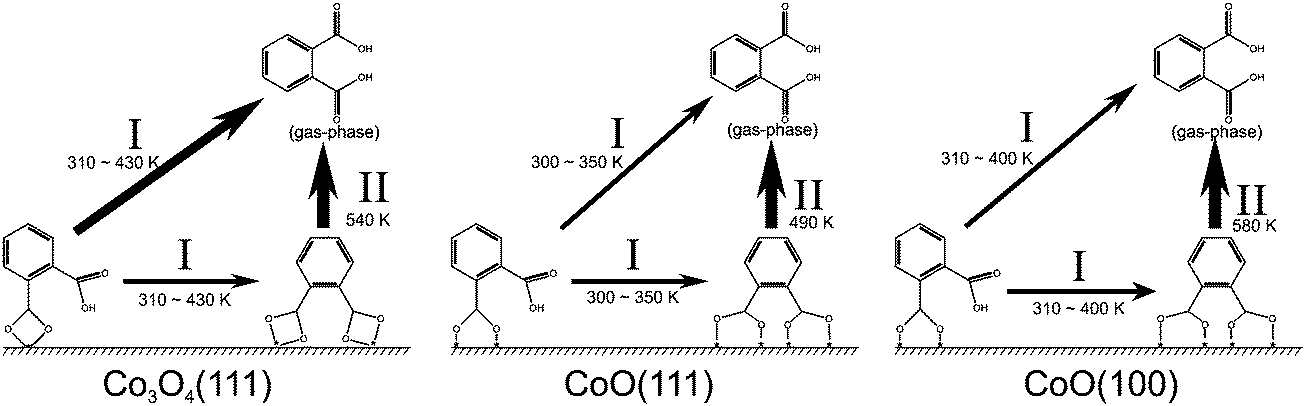 | ||
| Fig. 6 Schematic representation of surface bound carboxylates and the thermally induced changes as a function of the substrate temperature. | ||
The differences appear in relation to two characteristic stability parameters: (i) the desorption temperature Tdes, which is related to the activation barrier for desorption. This parameter is experimentally characterized by the temperature at which the carboxylate band decreases to 50% of its maximal intensity. The desorption temperature decreases in the order PA/CoO(100) (Tdes = 580 K ± 10 K) > PA/Co3O4(111) (Tdes = 540 K ± 10 K) > PA/CoO(111) (Tdes = 490 K ± 10 K). (ii) The second parameter is the width of the desorption region ΔTdes. This value characterizes the coverage dependence of the desorption activation energy. ΔTdes is found to be much larger for PA/Co3O4(111) (ΔTdes = 170 K ± 30 K) and PA/CoO(111) (ΔTdes = 170 K ± 30 K) than for PA/CoO(100) (ΔTdes = 90 K ± 20 K).
The different desorption temperatures Tdes – or different stabilities of the bis-carboxylate species – can be rationalized on the basis of the distribution and accessibility of the Co2+ ions on the surface. On CoO(100), a nearly perfectly bridging bis-carboxylate is formed at 300 K.18 The bridging adsorption geometry is typically the most stable one, as reflected by the preferential formation of bridging species for other carboxylic acids and oxides (e.g. formic acid on ZnO(10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0),37 rutile TiO2(110),38 anatase TiO2(110)39 and on CeO2(111)40 or benzoic acid on rutile TiO2(110)41 and on MgO(100)27). In order to realize bridging adsorption for the two carboxylate groups of a single PA molecule, a sufficiently high density-small Co–Co distance- of surface cobalt ions is required. On CoO(100), the distance between the surface Co2+ ions is only 3.0 Å17 and, therefore, the distance is small enough to allow formation of a bridging bis-carboxylate. On Co3O4(111), however, the distance between the surface Co2+ ions is 5.7 Å,17 which is too large for the carboxylate to form bridge between two adjacent Co2+ ions. Therefore, a chelating bis-carboxylate must be formed. The lowering of the desorption temperature by 40 K for PA on Co3O4(111) is attributed to the energetic penalty for the formation of the chelating carboxylate. On CoO(111) we are facing a special situation, as the surface is polar and undergoes reconstruction from a rocksalt to a wurtzite structure in the topmost surface layer.23,42 The distance between the Co2+ ions on CoO(111) is similar to that on CoO(100), but the surface is oxygen-terminated and the Co2+ ions are less accessible to adsorbates.17 The limited accessibility is reflected by the fact that at low temperature no bis-carboxylate is formed. Previously, we suggested that formation of the bis-carboxylate at 300 K requires restructuring of the surface.18 Thus, energetically a less stable bis-carboxylate is formed as reflected by the substantially lower desorption temperature on this surface.
0),37 rutile TiO2(110),38 anatase TiO2(110)39 and on CeO2(111)40 or benzoic acid on rutile TiO2(110)41 and on MgO(100)27). In order to realize bridging adsorption for the two carboxylate groups of a single PA molecule, a sufficiently high density-small Co–Co distance- of surface cobalt ions is required. On CoO(100), the distance between the surface Co2+ ions is only 3.0 Å17 and, therefore, the distance is small enough to allow formation of a bridging bis-carboxylate. On Co3O4(111), however, the distance between the surface Co2+ ions is 5.7 Å,17 which is too large for the carboxylate to form bridge between two adjacent Co2+ ions. Therefore, a chelating bis-carboxylate must be formed. The lowering of the desorption temperature by 40 K for PA on Co3O4(111) is attributed to the energetic penalty for the formation of the chelating carboxylate. On CoO(111) we are facing a special situation, as the surface is polar and undergoes reconstruction from a rocksalt to a wurtzite structure in the topmost surface layer.23,42 The distance between the Co2+ ions on CoO(111) is similar to that on CoO(100), but the surface is oxygen-terminated and the Co2+ ions are less accessible to adsorbates.17 The limited accessibility is reflected by the fact that at low temperature no bis-carboxylate is formed. Previously, we suggested that formation of the bis-carboxylate at 300 K requires restructuring of the surface.18 Thus, energetically a less stable bis-carboxylate is formed as reflected by the substantially lower desorption temperature on this surface.
The effects that lead to the differences in the width of the desorption region ΔTdes are more difficult to rationalize. We envisage that two contributions may affect ΔTdes. First, we have to consider intermolecular interactions originating from the bulky phenyl groups. These interactions may enforce the carboxylate units to adopt a less favorable geometry at high coverage, thus decreasing the adsorption energy. Theoretical calculations will be required to verify this hypothesis. The second possible effect is related to OH groups formed by deprotonation of the carboxylic acid. First experiments indicate that on Co3O4(111) hydroxyl groups of different nature and concentration are stable over a very broad range of temperatures up to above 500 K. It is likely that the presence of these OH groups greatly influences the binding of the phthalate species to the cobalt oxide surface.
4. Conclusion
We have studied the temperature-dependent behavior and thermal stability of phthalic acid (PA) monolayer and multilayer films on three different cobalt surfaces, Co3O4(111), CoO(111) and CoO(100). The cobalt oxides were prepared in UHV in the form of thin films on Ir(100). Using temperature-programmed infrared reflection absorption spectroscopy (TP-IRAS) we followed the restructuring and desorption processes in situ. The main findings are summarized as follows:(1) Upon deposition of the PA multilayers by PVD at 133 K, a monolayer of surface-anchored carboxylates is formed first. This anchored monolayer has an influence on the orientation of the multilayer, but little effect of the cobalt oxide structure itself on the stability or thermal behavior of the PA multilayer films is observed. A small fraction of phthalic anhydride (PAA) is formed during PVD and is co-deposited. At 200 K, the PAA desorbs from the surface of the multilayer films and at 250 K the PA multilayer films themselves undergo a structural transition during which they adopt a largely flat-lying geometry. The PA multilayer desorbs at 310 K.
(2) On Co3O4(111) PA adsorbs at 300 K in form of a chelating bis-carboxylate together with co-adsorbed mono-carboxylate species. The surface-bound mono-carboxylate partially converts to bis-carboxylate and partially desorbs in the temperature regime between 310 and 430 K, accompanied by an ordering of the adsorbate layer. In the temperature range between 430 and 600 K the bis-carboxylate desorbs from the Co3O4(111) surface.
(3) On CoO(111) PA adsorbs at 300 K in form of a bridging bis-carboxylate co-adsorbed with a very small amount of mono-carboxylate. The mono-carboxylate desorbs and converts to bis-carboxylate at temperatures up to 350 K. Between 400 to 570 K the bis-carboxylate desorbs from the CoO(111) surface.
(4) On CoO(100) PA adsorbs at 300 K in form of a bridging bis-carboxylate co-adsorbed with a small amount of mono-carboxylate. The mono-carboxylate desorbs and converts to bis-carboxylate in the temperature interval from 310 to 400 K. The bis-carboxylate film is perfectly stable in the temperature range from 400 to 510 K. Between 510 to 600 K the bis-carboxylate desorbs from the CoO(100) surface.
(5) Two structure-dependent observables are identified for the surface-anchored PA films: the mean desorption temperatures (Tdes) for the bis-carboxylates is found to increase in the order: CoO(111) (Tdes = 490 K ± 10 K) < Co3O4(111) (Tdes = 540 K ± 10 K) < CoO(100) (Tdes = 580 K ± 10 K). These differences are related to the density and accessibility of the Co2+ surface ions in the three cobalt oxide structures. In addition we observe that the width of the temperature region for desorption is much larger on Co3O4(111) and CoO(111) (ΔTdes = 170 K ± 30 K) than for PA/CoO(100) (ΔTdes = 90 K ± 20 K).
These observations show that the atomic structure of the surface plays an important role for the anchoring of organic films to oxide materials. Both the ordering and the thermal stability critically depend on the arrangement and coordination environment of the surface cations. Therefore a high level of structural control over the oxide surface is an essential precondition for a controlled synthesis of organic-oxide interfaces.
Acknowledgements
This project was financially supported by the Deutsche Forschungsgemeinschaft (DFG) within the Research Unit FOR 1878 “funCOS – Functional Molecular Structures on Complex Oxide Surfaces”. Additional support is acknowledged from the Excellence Cluster “Engineering of Advanced Materials” in the framework of the excellence initiative. The authors further acknowledge additional support by the Deutsche Forschungsgemeinschaft (DFG) within the DACH Project “COMCAT”, the European Commission (“chipCAT”, Grant Agreement no. 310191), by the COST Action CM1104 “Reducible oxide chemistry, structure and functions”. T. X. gratefully acknowledges support by a PhD grant of the China Scholarship Council (CSC).References
- H. Klauk, U. Zschieschang, J. Pflaum and M. Halik, Nature, 2007, 445, 745–748 CrossRef CAS PubMed.
- E. C. P. Smits, S. G. J. Mathijssen, P. A. van Hal, S. Setayesh, T. C. T. Geuns, K. A. H. A. Mutsaers, E. Cantatore, H. J. Wondergem, O. Werzer, R. Resel, M. Kemerink, S. Kirchmeyer, A. M. Muzafarov, S. A. Ponomarenko, B. de Boer, P. W. M. Blom and D. M. de Leeuw, Nature, 2008, 455, 956–959 CrossRef CAS.
- J. S. Kim, J. H. Park, J. H. Lee, J. Jo, D. Y. Kim and K. Cho, Appl. Phys. Lett., 2007, 91, 112111 CrossRef.
- K.-J. Jiang, K. Manseki, Y.-H. Yu, N. Masaki, K. Suzuki, Y.-l. Song and S. Yanagida, Adv. Funct. Mater., 2009, 19, 2481–2485 CrossRef CAS.
- O. K. Varghese, M. Paulose and C. A. Grimes, Nat. Nanotechnol., 2009, 4, 592–597 CrossRef CAS PubMed.
- Y. Joseph, M. Wühn, A. Niklewski, W. Ranke, W. Weiss, C. Wöll and R. Schlögl, Phys. Chem. Chem. Phys., 2000, 2, 5314–5319 RSC.
- S. C. Li, J. G. Wang, P. Jacobson, X. Q. Gong, A. Selloni and U. Diebold, J. Am. Chem. Soc., 2009, 131, 980–984 CrossRef CAS PubMed.
- P. Rahe, M. Nimmrich, A. Nefedov, M. Naboka, C. Woll and A. Kuhnle, J. Phys. Chem. C, 2009, 113, 17471–17478 CAS.
- J. S. Prauzner-Bechcicki, S. Godlewski, A. Tekiel, P. Cyganik, J. Budzioch and M. Szymonski, J. Phys. Chem. C, 2009, 113, 9309–9315 CAS.
- L. M. Liu, S. C. Li, H. Z. Cheng, U. Diebold and A. Selloni, J. Am. Chem. Soc., 2011, 133, 7816–7823 CrossRef CAS PubMed.
- P. Rahe, M. Kittelmann, J. L. Neff, M. Nimmrich, M. Reichling, P. Maass and A. Kühnle, Adv. Mater., 2013, 25, 3948–3956 CrossRef CAS PubMed.
- S. Godlewski and M. Szymonski, Int. J. Mol. Sci., 2013, 14, 2946–2966 CrossRef CAS PubMed.
- J. Libuda, F. Winkelmann, M. Bäumer, H. J. Freund, T. Bertrams, H. Neddermeyer and K. Müller, Surf. Sci., 1994, 318, 61–73 CrossRef CAS.
- H.-J. Freund, H. Kuhlenbeck and V. Staemmler, Rep. Prog. Phys., 1996, 59, 283 CrossRef CAS.
- H.-J. Freund and G. Pacchioni, Chem. Soc. Rev., 2008, 37, 2224–2242 RSC.
- M. Sobota, I. Nikiforidis, W. Hieringer, N. Paape, M. Happel, H. P. Steinruck, A. Gorling, P. Wasserscheid, M. Laurin and J. Libuda, Langmuir, 2010, 26, 7199–7207 CrossRef CAS PubMed.
- K. Heinz and L. Hammer, J. Phys.: Condens. Matter, 2013, 25, 173001 CrossRef CAS PubMed.
- T. Xu, M. Schwarz, K. Werner, S. Mohr, M. Amende and J. Libuda, Chem. – Eur. J., 2016 DOI:10.1002/chem.201504810.
- F. Hoffmann, Surf. Sci. Rep., 1983, 3, 107 CrossRef CAS.
- J. Libuda and H. J. Freund, Surf. Sci. Rep., 2005, 57, 157–298 CrossRef CAS.
- A. Desikusumastuti, T. Staudt, M. Happel, M. Laurin and J. Libuda, J. Catal., 2008, 260, 315–328 CrossRef CAS.
- W. Meyer, K. Biedermann, M. Gubo, L. Hammer and K. Heinz, J. Phys.: Condens. Matter, 2008, 20, 265011 CrossRef CAS PubMed.
- W. Meyer, K. Biedermann, M. Gubo, L. Hammer and K. Heinz, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 4 CrossRef.
- V. P. Tolstoy, I. V. Chernyshova and V. A. Skryshevsky, Handbook of infrared spectroscopy of ultrathin films, Wiley-Interscience, Hoboken, N.J., 2003 Search PubMed.
- P. W. Chan, Y. W. Chan and H. S. Ng, Phys. Lett. A, 1977, 61, 151–153 CrossRef.
- K. Ujihara, J. Appl. Phys., 1972, 43, 2376 CrossRef CAS.
- T. Xu, S. Mohr, M. Amende, M. Laurin, T. Dopper, A. Gorling and J. Libuda, J. Phys. Chem. C, 2015, 119, 26968–26979 CAS.
- National Institute of Standards and Technology (U.S.), in NIST standard reference database 69, National Institute of Standards and Technology, Washington, D.C.
- S. Haq, R. C. Bainbridge, B. G. Frederick and N. V. Richardson, J. Phys. Chem. B, 1998, 102, 8807–8815 CrossRef CAS.
- B. C. Smith, Infrared spectral interpretation: a systematic approach, CRC Press, Boca Raton, 1999 Search PubMed.
- S. Mohr, T. Xu, T. Dopper, M. Laurin, A. Gorling and J. Libuda, Langmuir, 2015, 31, 7806–7814 CrossRef CAS PubMed.
- D. A. Crooks and F. M. Feetham, J. Chem. Soc., 1946, 899–901 RSC.
- R. Sabbah and L. Perez, Can. J. Chem., 1999, 77, 1508–1513 CAS.
- C. Naselli, J. P. Rabe, J. F. Rabolt and J. D. Swalen, Thin Solid Films, 1985, 134, 173–178 CrossRef CAS.
- S. R. Cohen, R. Naaman and J. Sagiv, J. Phys. Chem., 1986, 90, 3054–3056 CrossRef CAS.
- H. P. Steinruck, J. Libuda, P. Wasserscheid, T. Cremer, C. Kolbeck, M. Laurin, F. Maier, M. Sobota, P. S. Schulz and M. Stark, Adv. Mater., 2011, 23, 2571–2587 CrossRef PubMed.
- M. Buchholz, Q. Li, H. Noei, A. Nefedov, Y. M. Wang, M. Muhler, K. Fink and C. Woll, Top. Catal., 2015, 58, 174–183 CrossRef CAS.
- B. E. Hayden, A. King and M. A. Newton, J. Phys. Chem. B, 1999, 103, 203–208 CrossRef CAS.
- M. Xu, H. Noei, M. Buchholz, M. Muhler, C. Wöll and Y. Wang, Catal. Today, 2012, 182, 12–15 CrossRef CAS.
- W. O. Gordon, Y. Xu, D. R. Mullins and S. H. Overbury, Phys. Chem. Chem. Phys., 2009, 11, 11171–11183 RSC.
- M. Buchholz, M. Xu, H. Noei, P. Weidler, A. Nefedov, K. Fink, Y. Wang and C. Wöll, Surf. Sci., 2016, 643, 117–123 CrossRef CAS.
- C. Ebensperger, M. Gubo, W. Meyer, L. Hammer and K. Heinz, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 235405 CrossRef.
- L. Colombo, V. Volovšek and M. Lepostollec, J. Raman Spectrosc., 1984, 15, 252–256 CrossRef CAS.
- B. G. Frederick, F. M. Leibsle, S. Haq and N. V. Richardson, Surf. Rev. Lett., 1996, 03, 1523–1546 CrossRef CAS.
- B. G. Frederick, M. R. Ashton, N. V. Richardson and T. S. Jones, Surf. Sci., 1993, 292, 33–46 CrossRef CAS.
- K. D. Dobson and A. J. McQuillan, Spectrochim. Acta, Part A, 2000, 56, 557–565 CrossRef CAS.
- J. van den Brand, O. Blajiev, P. C. J. Beentjes, H. Terryn and J. H. W. de Wit, Langmuir, 2004, 20, 6308–6317 CrossRef CAS PubMed.
- J. F. Arenas and J. I. Marcos, Spectrochim. Acta, Part A, 1980, 36, 1075–1081 CrossRef.
| This journal is © the Owner Societies 2016 |