
Metalorganocatalysis: cooperating transition-metal catalysis and organocatalysis through a covalent bond
Xiu-Qin
Dong
*a,
Qingyang
Zhao
b,
Pan
Li
ac,
Caiyou
Chen
a and
Xumu
Zhang
*a
aCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan, Hubei 430072, P. R. China. E-mail: xumu@whu.edu.cn; xiuqindong@whu.edu.cn
bCollege of Chemistry & Materials Science, Northwest University, Xuefu Road, Xi'an 710127, P. R. China
cCollege of Chemical Engineering, Zhejiang University of Technology, Hangzhou, Zhejiang 310014, P. R. China
First published on 6th August 2015
Abstract
Asymmetric catalysis has grown rapidly and made considerable progress in the last few decades, but there still remain significantly unachievable reactions through either organocatalysis or transition-metal catalysis alone. The concept of combination of transition-metal catalysis with organocatalysis emerged as a powerful strategy for developing asymmetric catalysis, and has attracted great attention. In order to avoid the incompatibility existing in catalysts, substrates, intermediates and solvents through combining transition-metal catalysis and organocatalysis, it is urgently necessary to develop a new catalytic strategy to resolve these problems. Therefore, we are devoted to designing a series of novel bifunctional catalysts based upon the synergistic activation strategy via cooperating transition metal-catalysis and organocatalysis through a covalent bond forming a bifunctional molecule. In this review, this momentous strategy is illustrated with several recent outstanding examples and prospective promising applications, with the aim of elaborating the synthetic utilities and potentialities of this concept as a powerful tool in organic synthesis.
 Xiu-Qin Dong | Xiu-Qin Dong was born in 1985. She received her B.S. degree from Huanggang Normal College in 2007, and her Ph.D. degree from Wuhan University under the guidance of Professor Chun-Jiang Wang in 2012. After postdoctoral research (2012–2014) at the Hong Kong University of Science and Technology in Prof. Jianwei Sun's group, she joined Wuhan University as an Associate Professor in 2015. Her research interests mainly focus on asymmetric metalorganocatalysis, hydroformylation, hydrogenation and the development of new synthetic methods for biologically active compounds. |
 Qingyang Zhao | Qingyang Zhao was born in 1985. He received his B.Sc. degree from Lanzhou University (P. R. China) in 2007 and then started his graduate studies under the guidance of Professor Rui Wang at the same university. After two-year joint Ph.D. research (2011–2013) with Prof. Xumu Zhang at Rutgers University (U.S.A.), he obtained his Ph.D. degree in 2014. Then he joined Northwest University (P.R. China) as a lecturer. His research interests focus on cooperative catalysis (metal catalysis and organocatalysis), ligand design and their implementation for the synthesis of bioactive molecules, especially the chiral molecules. |
 Pan Li | Pan Li was born in 1990. He obtained his B.S. degree (2012) at Zhejiang University of Technology. He started his Ph.D. under the guidance of Professor Xinquan Hu at the same university. As a visiting student of Xumu Zhang's group, his research work mainly focused on metalorganocatalysis and asymmetric hydrogenation. |
 Caiyou Chen | Caiyou Chen was born in Hubei Province, China in 1989. He obtained his B.S. degree (2012) from Wuhan University. He joined Prof. Xumu Zhang's group during his fourth year of undergraduate study and started his Ph.D. study in September 2012. His research work focuses on hydroformylation, asymmetric hydrogenation and nickel catalyzed copolymerization reactions. |
 Xumu Zhang | Xumu Zhang received his Ph.D. degree from Stanford University under the guidance of Professor James P. Collman in 1992. After two-year postdoctoral work at Stanford University, he joined the department of chemistry at The Penn State University as an Assistant Professor. In 1999, he was promoted as an Associate Professor. In 2003–2007, he was appointed as a full professor. In 2007, he moved to The State University of New Jersey as a distinguished Professor. From 2011 to the present, he has been appointed as a Professor (Thousand Talent Program) in Wuhan University. His research interests include the development of chiral phosphine ligands for asymmetric reactions and ligands for hydroformylation, hydrogenation of esters, and copolymerization reactions. |
1. Introduction
Asymmetric catalysis has been regarded as one of the most fascinating approaches for construction of functionalized optically active compounds and plays an important role in organic synthetic chemistry.1 Asymmetric catalysis including enzyme catalysis, transition-metal catalysis and organocatalysis has obvious advantages over chiral pool synthesis and chiral auxiliary approaches (Scheme 1). Transition-metal catalysis and organocatalysis have been well established to allow lots of different organic reactions serving as powerful tools in modern organic synthesis.1 Since the initiative studies in the 1960s,2 asymmetric transition-metal catalysis has dominated the center of asymmetric synthesis for a long period and has achieved great success, mainly owing to development of privileged chiral ligands.1 Asymmetric transition-metal catalysis has allowed the reactions through coordination between metal–ligand complexes and substrates. On the other hand, organocatalysis has grown explosively to be recognized as one of the most promising research areas in current organic chemistry by the breaking work in the 1990s and early 2000s.3 It can promote various organic transformations through distinct activation modes between catalysts and substrates,4–12 such as, by forming enamine intermediates,6 imine intermediates,7 hydrogen-bonds11 and ion-pair complexes.13 In general, transition-metal catalysis and organocatalysis have their unique advantages. Transition-metal catalysis has enabled a broader scope of chemical transformations to participate in organic synthesis, while organocatalysis has permitted a diverse range of functional groups to perform asymmetric reactions. However, there are still a lot of challenging transformations which can't be realized by either of these catalytic systems alone.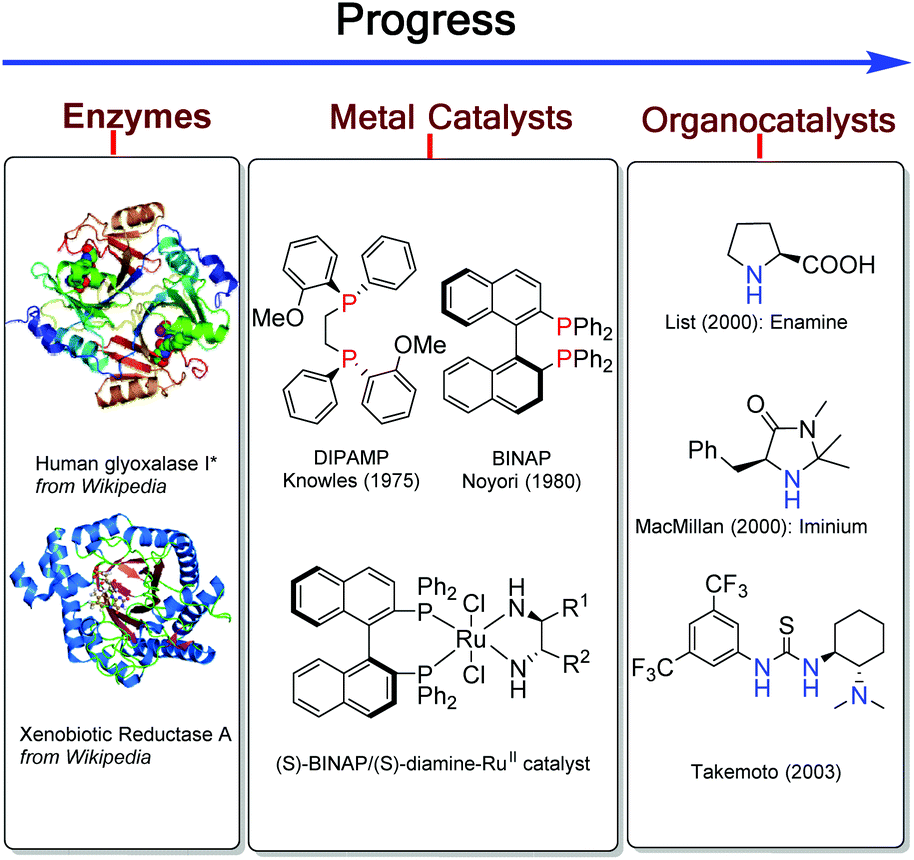 | ||
| Scheme 1 Progress of asymmetric catalysis. | ||
In recent years, the combination of transition-metal catalysis and organocatalysis as a promising strategy has been introduced to develop challenging and unprecedented transformations.14 Some effective strategies such as cooperative catalysis,15 synergistic catalysis,16 sequential/relay catalysis17 and supramolecular catalysis,18 have been established. However, these strategies were hindered by the incompatibility of catalysts, substrates, intermediates and solvents. For example, the potential self-quenching of a Lewis basic amine and a Lewis acidic transition-metal may lead to catalyst inactivation.19 Therefore, it is urgent to develop a new resolution. A different and special strategy, wherein both activation modes of transition-metal catalysis and organocatalysis are incorporated into one single bifunctional molecule, could avoid these tough problems and catalyze the reactions smoothly through multiple activation modes (Scheme 2). In this review, we summarize some recent remarkable research results and illustrate their future promising wide applications in organic synthesis as a novel methodology.
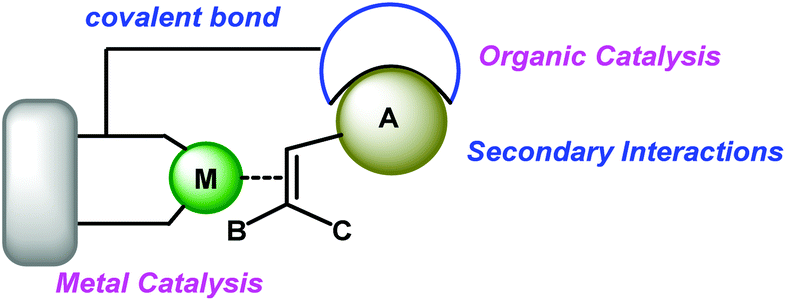 | ||
| Scheme 2 Proposed activation modes of metalorganocatalysis. | ||
2. Recent advances
To the best of our knowledge, there are only a few reported examples of this powerful strategy of combining transition-metal catalysis with organocatalysis through a covalent bond.20–26 Most of them were applied in asymmetric carbon–carbon bond-forming reactions, such as aldol reaction, hydrocyanation of aldehyde reaction, inverse-electron-demand hetero-Diels–Alder reaction, nitro-aldol reaction, and hydrogenation reactions with high activities and selectivities. Early in 1986, Hayashi and co-workers have reported asymmetric aldol reaction of isocyanoacetate with aldehydes catalyzed by a chiral bifunctional ferrocenylphosphine–gold(I) complex, producing optically active 5-alkyl-2-oxazoline-4-carboxylates with excellent selectivities (Scheme 3).20 The corresponding products are useful synthetic intermediates to access optically active 8-hydroxyamino acids and their derivatives. They proposed that the high efficiency of the bifunctional ferrocenylphosphine ligand was mainly ascribed to the dialkylamino group, which was at the end of the side chain. This secondary interaction participated in the formation of enolate of isocyanoacetate coordinated with gold.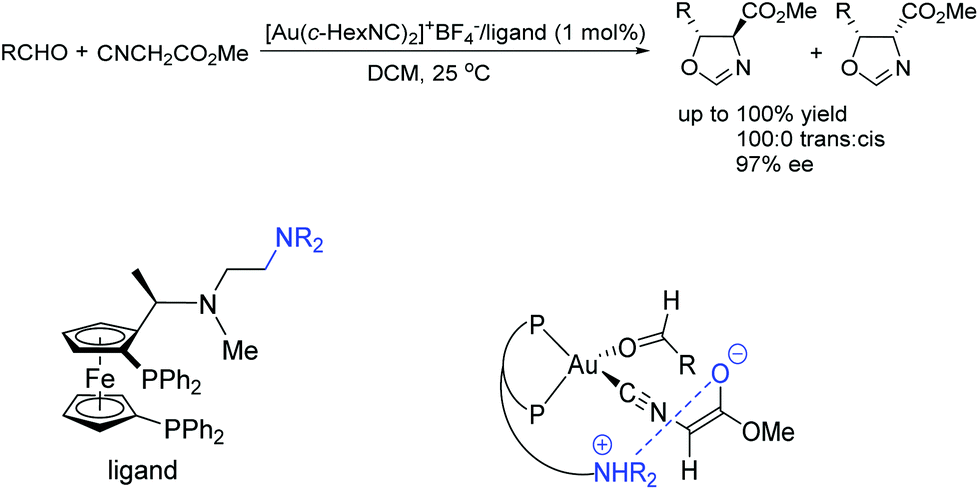 | ||
| Scheme 3 Asymmetric aldol reaction of isocyanoacetate with aldehyde. | ||
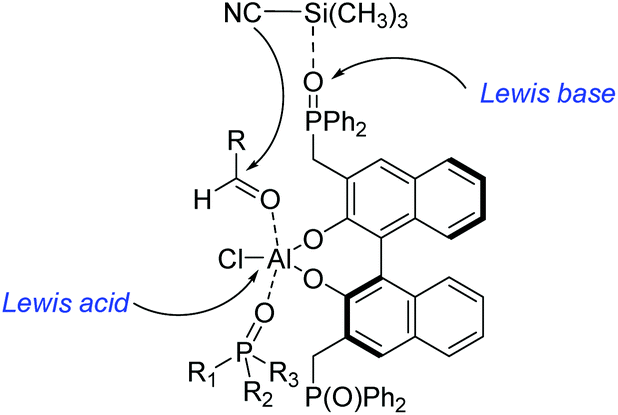
Unfortunately, this new catalytic strategy didn't attract people's attention for a long time. In 1999, this concept was proceeded by Shibasaki's group through a new type of chiral monometallic and bifunctional phosphinoyl-containing catalyst.21 It was successfully applied into the asymmetric hydrocyanation of aldehydes, which was able to coordinate both nucleophilic and electrophilic substrates through dual Lewis acid/Lewis base activation (Scheme 4). It was helpful to drastically improve the efficiency of asymmetric reactions with respect to the enantioselectivity and conversion rate. In this reaction system, the external additive phosphine oxide should coordinate with aluminum, thus converting the tetragonal Al(III) into a pentavalent center metal. As shown in Scheme 5, this geometry would allow the aldehyde to position itself at the apical side and close to the phosphine oxide of the ligand, and the trimethylsilylcyanide interacts with the Lewis base part of the chiral ligand. Both the aldehyde and trimethylsilylcyanide are coordinated to the bifunctional catalytic complex through dual Lewis acid/Lewis base activation. Subsequently, the attack of the cyanide to the aldehyde results in excellent enantioselectivities.
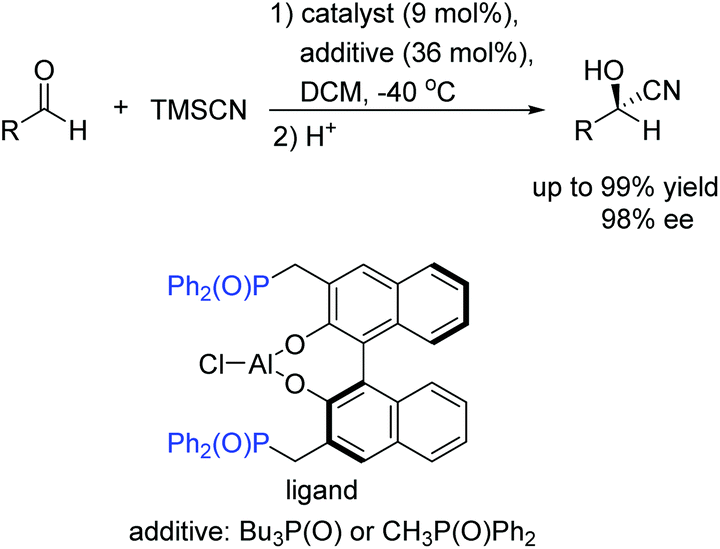 | ||
| Scheme 4 Asymmetric hydrocyanation of aldehydes. | ||
 | ||
| Scheme 5 Mechanism of Lewis acid/Lewis base catalysis. | ||
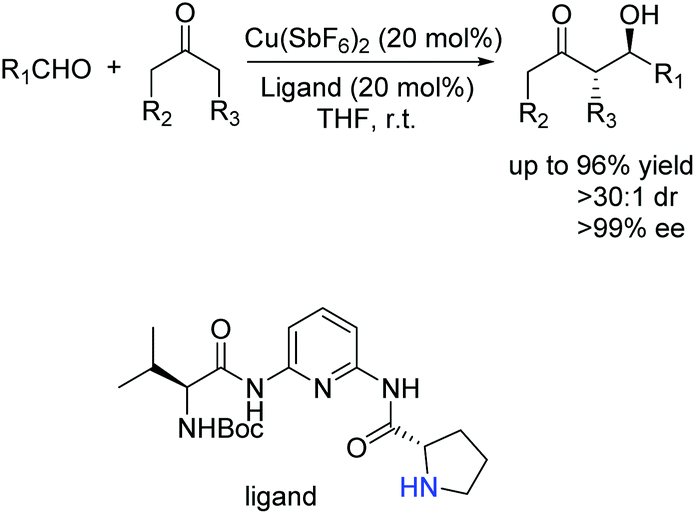
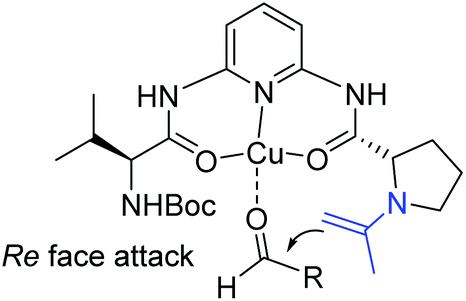
Ten years later, Wang and co-workers developed a novel class of primary amine-based enamine-metal Lewis acid cooperative bifunctional catalysts, and successfully applied them into asymmetric direct aldol reactions with good to excellent stereoselectivities (Scheme 6).22 It is worth noting that the coexistence of the secondary amine and metal Lewis acid is critical for the reaction. They speculated that the metal coordinated with chelating ligands to form a rigid chiral structure and acted as a Lewis acid to activate the aldehyde, and the secondary amine reacted with ketone to form an enamine. Then the enamine attacked the activated aldehyde from the Re-face to produce the desired products (Scheme 7).
 | ||
| Scheme 6 Asymmetric direct aldol reaction. | ||
 | ||
| Scheme 7 Proposed reaction transition state. | ||
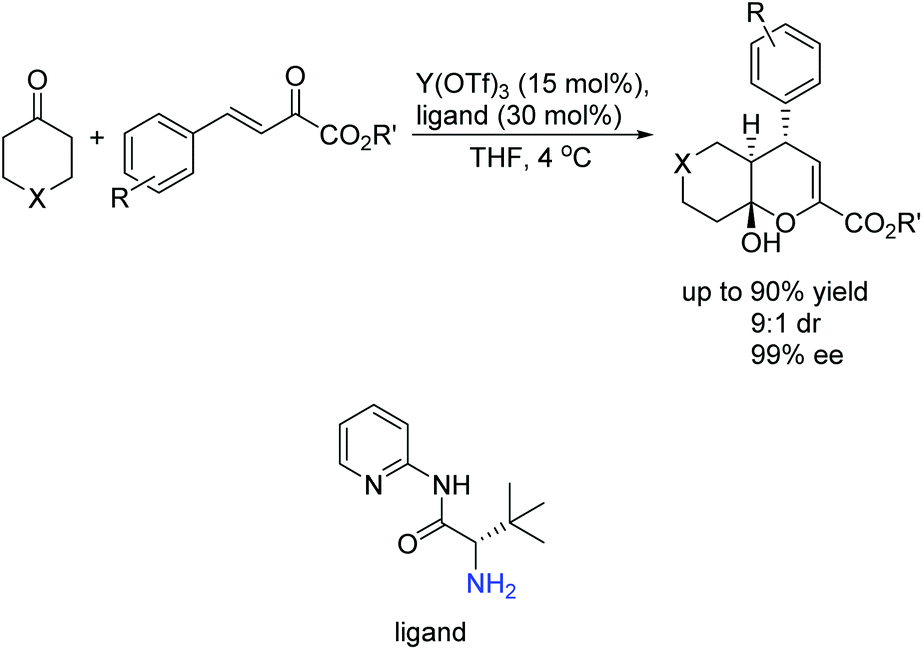
Subsequently, Wang's group successfully introduced the novel concept of enamine/metal Lewis acid bifunctional catalysis into challenging asymmetric inverse-electron-demand hetero-Diels–Alder reaction, providing chiral dihydropyran derivatives bearing stereochemically well-controlled quaternary carbon center products with high chemo- and enantioselectivities (Scheme 8).23
 | ||
| Scheme 8 Asymmetric inverse-electron-demand hetero-Diels–Alder reaction. | ||
The strong activation of the activated enone through chelation with the metal and intramolecular nature of the bifunctional catalyst was helpful to obtain high activities and stereoselectivities. As shown in Scheme 9, the proposed transition state matched well with the relative stereochemistry bearing an endo-selective mode of the reaction. In addition, the sterically hindered R (tert-butyl) group shielded the Re face of the enamine, thus the activated enone tended to attack from the Si face, contributing for the absolute configuration of the final products.
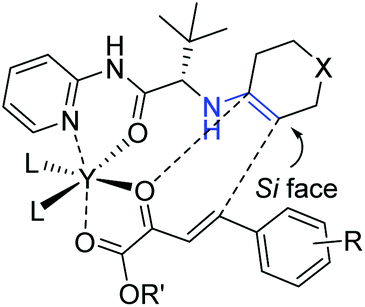 | ||
| Scheme 9 Proposed reaction transition state. | ||
And then a new type of cooperative catalyst featuring urea H-bonds and a cobalt center has been developed for anti-selective asymmetric nitro-aldol reactions. In 2012, Hong's group has developed high anti-selective asymmetric nitro-aldol reactions catalyzed by a cooperative catalyst bearing bisurea H-bonding and salen-Co(III) (Scheme 10).24 The urea–cobalt bifunctional catalytic system successfully extended the substrate scope of anti-selective nitro-aldol reactions to previously unexplored aldehydes, and the synthetic utility is demonstrated by the concise asymmetric synthesis of (1R, 2S)-methoxamine hydrochloride. The urea H-bonds played an important role in the impressive improvement of reaction activities and selectivities.
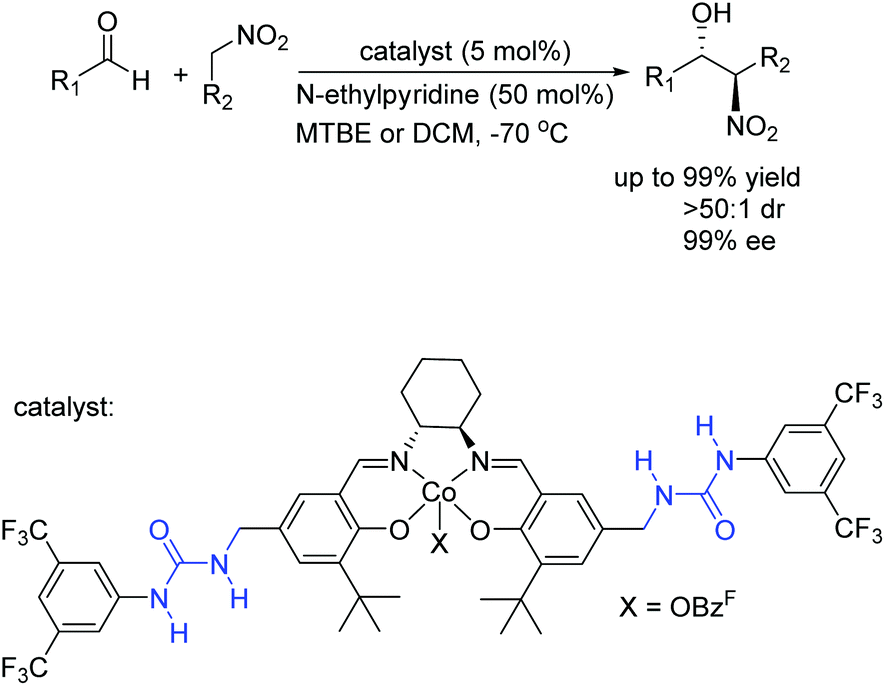 | ||
| Scheme 10 Asymmetric nitro-aldol reaction. | ||
In 2013, our group developed a series of novel chiral bisphosphine-thiourea ligands based on the concept of combining transition-metal catalysis with organocatalysis into a bifunctional molecule. We have successfully applied them into Rh-catalyzed asymmetric hydrogenation of various challenging β,β-disubstituted nitroalkenes with excellent enantioselectivities (Scheme 11).25 The thiourea backbone played a remarkable role in this catalytic system through forming hydrogen bonding with the nitro group to activate the substrate. As a result, the sensitive nitro group was reserved without hydrogenation.
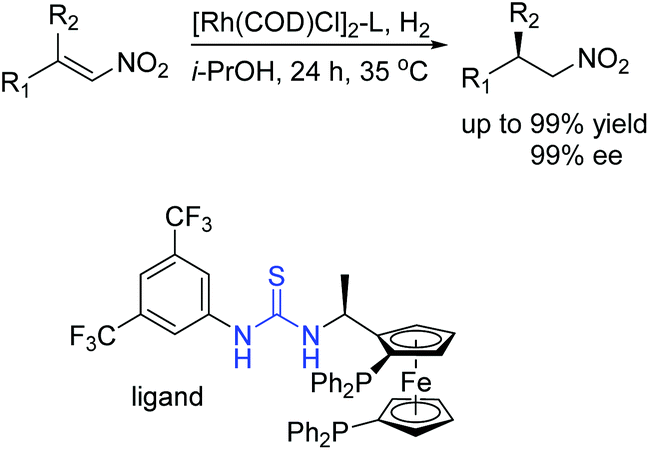 | ||
| Scheme 11 Asymmetric hydrogenation of challenging β,β-disubstituted nitroalkenes. | ||
It is generally known that the thiourea unit was widely used as a hydrogen bonding donor in organocatalysis.26 In addition, some research work took advantage of its anion binding of ion-pairing intermediates.27 Inspired by the strategies developed in organocatalysis28–30 and our previous research work about the Rh–bisphosphine–thiourea catalytic system for asymmetric hydrogenation of β,β-disubstituted nitroalkenes,25 our group made efforts to broaden the anion-binding interaction into the transition-metal-catalyzed asymmetric hydrogenation. We envisioned that the thiourea block could interact with a counterion in the catalytic system (Scheme 12).
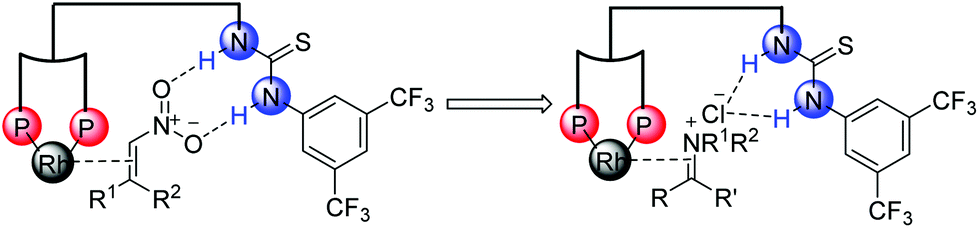 | ||
| Scheme 12 Activation model of thiourea block. | ||
Then we successfully developed asymmetric hydrogenation of unprotected NH imines catalyzed by rhodium/bis(phosphine)-thiourea for the first time (Scheme 13).31 The corresponding chiral amine products were obtained in high yields and enantioselectivities. We proposed that the anion-binding interaction between the thiourea and chloride counterion played a crucial role in the catalytic system based on the control experiments and 1H NMR studies.
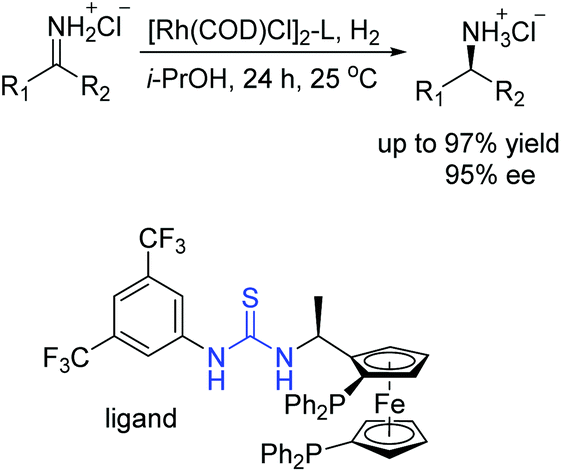 | ||
| Scheme 13 Asymmetric hydrogenation of unprotected NH imines. | ||
3. Catalyst design and applications
A catalyst is the core of chemical transformations, so it is greatly important to design appropriate catalysts. Inspired by the previous excellent research results, it is extremely critical to develop novel bifunctional catalysts through the synergistic activation strategy via cooperating transition-metal catalysis and organocatalysis with a covalent bond. The metal unit and organic unit work cooperatively to promote many types of unprecedented transformations. Generally, metal catalysts can offer high activities in hydrogenation and other carbon–carbon bond forming reactions, and the organic motif can achieve excellent selectivities through secondary interactions providing an excellent directing induction.Chiral metal catalysts bearing monophosphine, biphosphine, amino alcohol, oxazoline, or salen ligands etc. are powerful catalysts for a variety of asymmetric reactions. The great catalytic potential of these catalysts can be enhanced by introducing organic motifs to provide secondary interactions between substrates and organic motifs. Until now many types of organocatalysts have been developed, such as chiral amines, thioureas, squaramides, and phosphoric acids. Combining these kinds of organic motifs with transition-metal catalysts will produce a tremendous amount of metalorganocatalysts forming a catalytic library (Scheme 14).
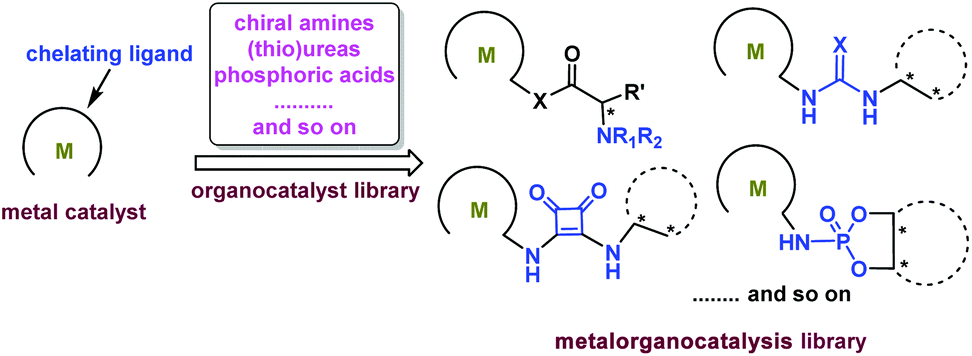 | ||
| Scheme 14 Proposed metalorganocatalysis library. | ||
We believe that this new catalytic concept will enable unprecedented transformations with excellent activities and selectivities, and be efficient to resolve some challenging problems. These new catalytic systems will be applied to develop highly enantioselective asymmetric reactions, such as hydrogenation reactions, asymmetric allylic alkylation (AAA), Michael addition, the aldol reaction, Mannich reaction, Henry reaction etc. In future, the novel bifunctional catalysts derived from the synergistic activation strategy via cooperating transition-metal catalysis and organocatalysis with a covalent bond will occupy a special place in organic chemistry and it is extremely necessary for them to be explored.
4. Conclusion
The new research direction of combining metal catalysis with organocatalysis has emerged as a powerful tool for challenging and unprecedented transformations. Up to now, some effective strategies have been developed, such as cooperative catalysis,15 synergistic catalysis,16 sequential/relay catalysis17 and supramolecular catalysis.18 However, there always exists incompatibility of catalysts, substrates, intermediates and solvents. A different strategy is needed to avoid these problems and promote the transformations through multiple activation modes. Several groups made use of single bifunctional molecules combining activation modes of metal catalysis and organocatalysis to efficiently resolve these problems. In this review, we summarize some recent remarkable examples and propose a library of bifunctional catalysts combining activation modes of transition-metal catalysis and organocatalysis through a covalent bond. In the near future, they will be widely applied to asymmetric reactions, such as asymmetric allylic alkylation (AAA), Michael addition, the aldol reaction, Mannich reaction and Henry reaction. We believe that this momentous strategy will become a powerful tool in organic synthetic chemistry.Acknowledgements
We thank the grant from Wuhan University (203273463, 203410100064), and “111” Project of the Ministry of Education of China for financial support and the National Natural Science Foundation of China (Grant No. 21372179, 21432007). Dr Qingyang Zhao thanks the grant from Northwest University (338050061).Notes and references
- (a) Comprehensive Asymmetric Catalysis, ed. E. N. Jacobson, A. Pfaltz and H. Yamamoto, Springer, Heidelberg, 1999 Search PubMed; (b) Asymmetric Catalysis on Industrial Scale, ed. H. U. Blaser and H.-J. Federsel, Wiley, Weinheim, Germany, 2010 Search PubMed; (c) Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Groger and O. May, Wiley, Weinheim, Germany, 2012 Search PubMed; (d) Q.-L. Zhou, Privileged Ligands and Catalysts, Wiley, Weinheim, Germany, 2011 Search PubMed.
- (a) H. Nozaki, S. Moriuti, H. Takaya and R. Noyori, Tetrahedron Lett., 1966, 7, 5239–5244 CrossRef; (b) H. Nozaki, H. Takaya, S. Moriuti and R. Noyori, Tetrahedron, 1968, 24, 3655–3669 CrossRef CAS; (c) W. S. Knowles and M. J. Sabacky, Chem. Commun., 1968, 1445–1446 RSC; (d) L. Horner, H. Siegel and H. Büthe, Angew. Chem., Int. Ed. Engl., 1968, 7, 942–942 CrossRef CAS PubMed.
- For a review on organocatalysis, see: S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178–2189 RSC.
- For a general review on organocatalysis by N-heterocyclic carbene, see: N. Marion, S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 26, 2988–3000 CrossRef PubMed.
- For a review on phase transfer catalysis: T. Ooi and K. Maruoka, Angew. Chem., Int. Ed., 2007, 46, 4222–4266 CrossRef CAS PubMed.
- For a recent review on enamine catalysis, see: S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS PubMed.
- For a recent review on iminium ion catalysis, see: A. ErkkilV, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416–5570 CrossRef PubMed.
- For a seminal report on SOMO catalysis, see: T. D. Beeson, A. Mastracchio, J. Hong, K. Ashton and D. W. C. MacMillan, Science, 2007, 316, 582–585 CrossRef CAS PubMed.
- For a recent review on aminocatalysis, see: P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138–6171 CrossRef CAS PubMed.
- For a review on Brønsted base catalysis, see: C. Palomo, M. Oiarbide and R. López, Chem. Soc. Rev., 2009, 38, 632–653 RSC.
- For selected reviews on Brønsted acid catalysis: (a) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS PubMed; (b) T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed; (c) M. Terada, Curr. Org. Chem., 2011, 15, 2227–2256 CrossRef CAS; (d) Z. Shao and F. Peng, Curr. Org. Chem., 2011, 15, 4144–4160 CrossRef.
- For a review on dienamine catalysis, see: D. B. Ramachary and Y. V. Reddy, Eur. J. Org. Chem., 2012, 865–887 CrossRef CAS PubMed.
- For a review on Lewis base catalysis, see: S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560–1638 CrossRef CAS PubMed.
- For an account on metal–organic cooperative catalysis, see: (a) Y. J. Park, J.-W. Park and C.-H. Jun, Acc. Chem. Res., 2008, 41, 222–234 CrossRef CAS PubMed; (b) Z. Shao and H. Zhang, Chem. Soc. Rev., 2009, 38, 2745–2755 RSC; (c) C. Zhong and X. Shi, Eur. J. Org. Chem., 2010, 2999–3025 CrossRef CAS PubMed; (d) C. C. J. Loh and D. Enders, Chem. – Eur. J., 2012, 18, 10212–10225 CrossRef CAS PubMed.
- Recent examples of cooperative catalysis: (a) L. Stegbauer, F. Sladojevich and D. J. Dixon, Chem. Sci., 2012, 3, 942–958 RSC; (b) W. Tang, S. Johnston, J. A. Iggo, N. G. Berry, M. Phelan and L. Lian, Angew. Chem., Int. Ed., 2012, 52, 1668–1672 CrossRef PubMed.
- Recent examples of synergistic catalysis: (a) A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633–658 RSC; (b) M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593–1596 CrossRef CAS PubMed.
- Recent example of sequential/relay catalysis: (a) F. Shi and L.-Z. Gong, Angew. Chem., Int. Ed., 2012, 51, 11423–11425 CrossRef CAS PubMed; (b) N. T. Patil, V. S. Shinde and B. Gajula, Org. Biomol. Chem., 2012, 10, 211–224 RSC; (c) D.-F. Chen, Z.-Y. Han, X.-L. Zhou and L.-Z. Gong, Acc. Chem. Res., 2014, 47, 2365–2377 CrossRef CAS PubMed.
- Recent example of self-assembling supramolecular catalysis: (a) J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 CrossRef CAS PubMed; (b) T. Šmejkal and B. Breit, Angew. Chem., Int. Ed., 2008, 47, 311–315 CrossRef PubMed.
- J. Zhou, Chem. – Asian J., 2010, 5, 422–434 CrossRef CAS PubMed.
- Y. Ito, M. Sawamura and T. Hayashi, J. Am. Chem. Soc., 1986, 108, 6405–6406 CrossRef CAS.
- Y. Hamashima, D. Sawada, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 1999, 121, 2641–2642 CrossRef CAS.
- Z. Xu, P. Daka, I. Budik, H. Wang, F. Bai and H. X. Zhang, Eur. J. Org. Chem., 2009, 4581–4585 CrossRef CAS PubMed.
- Z. Xu, L. Liu, K. Wheeler and H. Wang, Angew. Chem., Int. Ed., 2011, 50, 3484–3488 CrossRef CAS PubMed.
- K. Lang, J. Park and S. Hong, Angew. Chem., Int. Ed., 2012, 51, 1620–1624 CrossRef CAS PubMed.
- Q. Zhao, S. Li, K. Huang, R. Wang and X. Zhang, Org. Lett., 2013, 15, 4014–4017 CrossRef CAS PubMed.
- For reviews: (a) P. M. Pihko, Hydrogen Bonding in Organic Synthesis, Wiley-VCH, Weinheim, 2009 Search PubMed; (b) Z. Zhang and P. R. Schreiner, Chem. Soc. Rev., 2009, 38, 1187–1198 RSC; (c) R. R. Knowles and E. N. Jacobsen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20678–20685 CrossRef CAS PubMed.
- For reviews: (a) R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603–614 CrossRef CAS PubMed; (b) K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534–561 CrossRef CAS PubMed; (c) M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518–533 CrossRef CAS PubMed.
- (a) M. S. Taylor and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 10558–10559 CrossRef CAS PubMed; (b) I. T. Raheem, P. S. Thiara, E. A. Peterson and E. N. Jacobsen, J. Am. Chem. Soc., 2007, 129, 13404–13405 CrossRef CAS PubMed; (c) S. J. Zuend and E. N. Jacobsen, J. Am. Chem. Soc., 2009, 131, 15358–15374 CrossRef CAS PubMed; (d) R. R. Knowles, S. Lin and E. N. Jacobsen, J. Am. Chem. Soc., 2010, 132, 5030–5032 CrossRef CAS PubMed; (e) H. Xu, S. J. Zuend, M. G. Woll, Y. Tao and E. N. Jacobsen, Science, 2010, 327, 986–990 CrossRef CAS PubMed; (f) J. A. Birrell, J.-N. Desrosiers and E. N. Jacobsen, J. Am. Chem. Soc., 2011, 133, 13872–13875 CrossRef CAS PubMed; (g) N. Z. Burns, M. R. Witten and E. N. Jacobsen, J. Am. Chem. Soc., 2011, 133, 14578–14581 CrossRef CAS PubMed; (h) S. Lin and E. N. Jacobsen, Nat. Chem., 2012, 4, 817–824 CrossRef CAS PubMed; (i) M. P. Lalonde, M. A. McGowan, N. S. Rajapaksa and E. N. Jacobsen, J. Am. Chem. Soc., 2013, 135, 1891–1894 CrossRef CAS PubMed; (j) G. Bergonzini, C. S. Schindler, C.-J. Wallentin, E. N. Jacobsen and C. R. Stephenson, Chem. Sci., 2014, 5, 112–116 RSC.
- (a) C. K. De, E. G. Klauber and D. Seidel, J. Am. Chem. Soc., 2009, 131, 17060–17061 CrossRef CAS PubMed; (b) E. G. Klauber, C. K. De, T. K. Shah and D. Seidel, J. Am. Chem. Soc., 2010, 132, 13624–13626 CrossRef CAS PubMed; (c) C. K. De and D. Seidel, J. Am. Chem. Soc., 2011, 133, 14538–14541 CrossRef CAS PubMed; (d) C. Min, N. Mittal, D. X. Sun and D. Seidel, Angew. Chem., Int. Ed., 2013, 52, 14084–14088 CrossRef CAS PubMed.
- (a) Y. S. Lee, M. M. Alam and R. S. Keri, Chem. – Asian J., 2013, 8, 2906–2919 CrossRef CAS PubMed; (b) A. G. Schafer, J. M. Wieting, T. J. Fisher and A. E. Mattson, Angew. Chem., Int. Ed., 2013, 52, 11321–11324 CrossRef CAS PubMed.
- Q. Zhao, J. Wen, R. Tan, K. Huang, P. Metola, R. Wang, E. V. Anslyn and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 8467–8470 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2015 |