
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
AuRu/AC as an effective catalyst for hydrogenation reactions
Alberto
Villa
a,
Carine E.
Chan-Thaw
a,
Sebastiano
Campisi
a,
Claudia L.
Bianchi
a,
Di
Wang
b,
Paul G.
Kotula
c,
Christian
Kübel
b and
Laura
Prati
*a
aDipartimento di Chimica, Università degli Studi di Milano, via Golgi 19, 20133 Milano, Italy. E-mail: Laura.Prati@unimi.it
bInstitute of Nanotechnology and Karlsruhe Nano Micro Facility, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
cMaterials Characterization Department, Sandia National Laboratories, Albuquerque, NM 87185-0886, USA
First published on 23rd March 2015
Abstract
AuRu bimetallic catalysts have been prepared by sequential deposition of Au on Ru or vice versa obtaining different nanostructures: when Ru has been deposited on Au, a Aucore–Rushell has been observed, whereas the deposition of Au on Ru leads to a bimetallic phase with Ru enrichment on the surface. In the latter case, the unexpected Ru enrichment could be attributed to the weak adhesion of Ru on the carbon support, thus allowing Ru particles to diffuse on Au particles. Both structures result very active in catalysing the liquid phase hydrogenolysis of glycerol and levulinic acid but the activity, the selectivity and the stability depend on the structure of the bimetallic nanoparticles. Ru@Au/AC core–shell structure mostly behaved as the monometallic Ru, whereas the presence of bimetallic AuRu phase in Au@Ru/AC provides a great beneficial effect on both activity and stability.
1. Introduction
In the last decade, metal nanoparticles have been extensively investigated because of their unique properties and their potential applications in different fields, such as catalysis, electronics, optics, imaging, and biology.1In particular, gold nanoparticles have been recognized to play an important role in the field of catalysis since, twenty years ago, Haruta and Hutchings disclosed the peculiarity of the activity of this metal in CO oxidation and ethylene hydrochlorination.2
In addition, gold has shown more recently an attractive behaviour in the liquid phase oxidation of alcohols when O2 is used as the oxidant being more resistant to deactivation compared to classical Pd or Pt catalysts.3 Moreover, it has been revealed that when gold is used as modifier of a second more active metal, the bimetallic systems take advantage from the gold presence resulting not only generally more active but also more resistant to deactivation.4 This latter aspect is of great importance for the exploitation of the heterogeneous processes to the industrial scale, especially, in liquid phase reactions.3
More recently Au has been investigated as catalyst for the liquid phase hydrogenation of various molecules, such as cinnamaldehyde, citral and glycerol.5 The lower catalytic activity of Au in hydrogen mediated reactions relative to conventional transition metals (Pd, Pt, Ru) is generally attributed to the less effective activation/dissociation of dihydrogen.6 However, supported gold has shown promising results in terms of selectivity and stability.7 As well as for oxidative processes, also in hydrogenations gold based bimetallic systems normally show a great enhancement in their activity generally addressed to a synergistic effect between the two metals. For example, the addition of Au to Pd, or Ru results in a great improvement in the catalytic activity and selectivity in the hydrogenation of cinnamaldehyde and glycerol.5
In this paper, we will focus on the impact of AuRu nanoparticle structure in the hydrogenation of two important compounds deriving from biomass, namely glycerol and levulinic acid. Hydrogenolysis of glycerol can produce 1,2- and 1,3-propandiols, lactic acid as well as different alcohols which are all important chemicals.8 From levulinic acid the main product is gamma valerolactone (GVL) that can be transformed in pentanediol or pentanoic acid, both useful additives in biofuels.9
Several studies reported on Ru as the most effective catalyst for the cited reactions, showing good activity and selectivity despite deactivation phenomena have been envisaged.8,9 We recently reported the beneficial effect of gold addition to Ru catalysts in terms of activity, selectivity and stability in the oxidation of glycerol and aliphatic alcohols.10 Therefore, we thought that a possible solution to the high deactivation of Ru catalysts in the glycerol or levulinic acid hydrogenolysis could be the addition of Au. It has been highlighted that the synergistic effect between Au and Ru strongly depends on the structure of the bimetallic system. In the case of liquid phase oxidation, the addition of Au to Ru is beneficial only when alloyed AuRu nanoparticles were formed.10a Thus, to investigate the effect of AuRu structure on the catalytic activity in the hydrogenation of levulinic acid and glycerol, two different AuRu/AC bimetallic catalysts were prepared by successive deposition of Au on Ru or Ru on Au. Their catalytic performances revealed that a beneficial effect can be obtained only when a bimetallic phase is present.
2. Experimental
2.1 Catalyst preparation
Activated carbon (AC) was obtained from Camel (X40S, surface area 1100 m2 g−1, pore volume 1.5 mL g−1). NaAuCl4·2H2O and RuCl3 were obtained from Aldrich (99.99% purity), NaBH4 of purity >96% from Fluka and polyvinylalcohol (PVA) (Mw = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–23000, 87–89% hydrolyzed) from Aldrich were used. Gaseous oxygen and hydrogen from SIAD were 99.99% pure.
000–23000, 87–89% hydrolyzed) from Aldrich were used. Gaseous oxygen and hydrogen from SIAD were 99.99% pure.
Monometallic 1% Ru/AC has been prepared by incipient wetness impregnation. Solid RuCl3 (equivalent to Ru 10 mg) was dissolved in water (1.5 mL) and added to 1 g of support (final ruthenium loading of 1 wt%). The catalyst is then dried at 80 °C for 2 h and reduced in H2 at 200 °C for 2 h.
Monometallic 1% Au/AC has been prepared by impregnating the activated carbon with preformed Au nanoparticles. Solid NaAuCl4·2H2O (0.051 mmol) and PVA (2 wt%) solution (Au/PVA 1/1 wt%) were added to 100 mL of H2O. After 3 min, NaBH4 (0.1 M) solution (Au/NaBH4 1/4 mol mol−1) was added to the yellow solution under vigorous magnetic stirring. The ruby red Au(0) sol was immediately formed. Within a few minutes of sol generation, the colloid (acidified at pH 2, by sulphuric acid) was immobilised by adding the support under vigorous stirring. The amount of support was calculated as having a total final metal loading of 1 wt%. After 2 h the slurry was filtered, the catalyst washed thoroughly with distilled water (neutral mother liquors) and dried at 80 °C for 4 h.
Bimetallic systems were prepared following two steps procedure using two different techniques.
In the first preparation, RuCl3 was added under stirring to a slurry of 0.73 wt% Au/AC prepared as reported above and reduced with H2 at 200 °C after filtration. The catalyst was labelled as Ru@Au/AC. The total metal loading was 1 wt% and the molar ratio Au/Ru 6:4 (Table 1).
| Catalyst | Au (wt%) | Ru (wt%) | Au:Ru (mol mol−1) |
Particle d (nm) |
|---|---|---|---|---|
| Au/AC | 1 | — | — | 3.3 |
| Ru/AC | — | 1 | — | 2.1 |
| Ru@Au/AC | 0.73 | 0.27 | 6:4 |
4.1 |
| Au@Ru/AC | 0.73 | 0.27 | 6:4 |
4.3 |
In the second preparation, 0.27 wt% Ru/AC was added under stirring to a PVA stabilized AuNPs suspension, prepared following the procedure reported above. The catalyst was labelled as Au@Ru/AC. The total metal loading was 1 wt% and the molar ratio Au/Ru 6:4 (Table 1).
2.2 Characterisation
The catalysts were examined in an image aberration corrected FEI Titan 80-300 electron microscope with conventional EDX detector as well as in a probe aberration corrected Titan 80-200 with in column Super-X EDX detector. The microscope was operated at an accelerating voltage of 300 kV in the image corrected Titan and 200 kV in the probe corrected Titan, respectively. Spectrum imaging with probe corrected STEM and Super-X EDX detector offers high spatial resolution and the opportunity to resolve the components by multivariate analysis before changing the structure significantly during electron beam irradiation.The metal content was checked by ICP analysis on a Jobin Yvon JY24.
2.3 Catalytic tests
Glycerol hydrogenolysis was performed at 150 °C, using a stainless steel reactor (50 mL capacity), equipped with heater, mechanical stirrer, gas supply system and thermometer. The glycerol solution (30 mL; 0.3 M) was added into the reactor and the desired amount of catalyst (glycerol/metal ratio = 1000, mol mol−1) was suspended in the solution. The autoclave was purged three times with nitrogen before charging 7 bar of H2. The mixture was heated to the reaction temperature, 150 °C, and mechanically stirred (1250 rpm).Levulinic acid (LA) hydrogenation was performed at 70 °C, using a stainless steel reactor (50 mL capacity), equipped with heater, mechanical stirrer, gas supply system and thermometer. The LA solution (30 mL; 0.3 M) was added into the reactor and the desired amount of catalyst (LA/metal ratio = 1000 mol mol−1) was suspended in the solution. The autoclave was then purged three times with nitrogen before charging 7 bar of H2. The mixture was heated to the reaction temperature, 70 °C, and mechanically stirred (1250 rpm).
Recycling test for levulinic acid hydrogenation: each run was carried out under the same conditions (LA = 0.3 M; metal/LA = 1/1000 mol mol−1. 70 °C, 7 bar H2). The catalyst was recycled in the subsequent run after filtration without any further treatment.
Products analysis: the reaction mixture, after separation from the catalysts by filtration, was analysed using high performance liquid chromatography (HPLC). Samples were removed periodically (0.5 mL) under stirring and analysed by high-performance liquid chromatography (HPLC) using a column (Alltech OA-10308, 300 mm × 7.8 mm) with UV and refractive index (RI) detection in order to analyse the product mixtures. H3PO4 0.1 wt% solution was used as the eluent. The identification of the possible products was done by comparison with the original samples.
3. Results and discussion
3.1 Catalyst characterization
Two different AuRu bimetallic catalysts, Ru@Au/AC and Au@Ru/AC were prepared by successive deposition of the two metals. For Ru@Au/AC, active carbon was impregnated with Ru(III) salt added to the slurry of Au/AC, and reduced with H2 at 80 °C. On the contrary Au@Ru/AC was prepared by immobilising PVA stabilized Au nanoparticles onto preformed Ru/AC. In both cases the Au:Ru molar ratio was Au:Ru 6:4 (Table 1) and the total metal loading 1 wt%.
Monometallic Au/AC was prepared by preformed Au nanoparticles using PVA as protective agent, whereas Ru/AC was prepared by incipient wetness. In both cases the metal loading was the same as for bimetallics (1 wt%). The catalysts were examined in an aberration corrected FEI Titan 80-300 electron microscope with conventional EDX detector as well as in a probe aberration corrected Titan 80-200 with in column Super-X EDX detector.
All the catalysts are composed by small nanoparticles with an average diameter of 3.3 and 2.1 for Au/AC and Ru/AC, respectively. In the case of AuRu slightly bigger particles of 4.1–4.3 nm are present (Table 1).
Ru@Au/AC showed a good metal dispersion on the support. Spectrum imaging with probe corrected STEM and Super-X EDX detector offers high spatial resolution and the opportunity to resolve the components by multivariate analysis before changing the structure significantly during electron beam irradiation. Two main components were revolved containing separately Au and Ru for the metallic particles. The superimposed component map is shown in Fig. 1a, revealing an Au core (in red)–Ru shell (in green) structure. The corresponding EDX spectra of these two components are shown in Fig. 1b and there is no alloyed Au–Ru. The high resolution HAADF STEM image in Fig. 1c shows two Au particles with Ru clusters (lower intensity) situated on their surface. Furthermore, small Ru clusters were observed separately in some areas on AC. In the case of Au@Ru/AC, when Ru/AC was used as seeds for nucleating Au, we obtained a more inhomogeneous distribution, with the presence of small particles principally composed of Ru and larger bimetallic particles, as seen in Fig. 2a.
 | ||
| Fig. 1 (a) Component maps and (b) component EDX spectra from multivariate analysis of STEM-EDX spectrum imaging for Ru@Au/AC catalyst. Red and green correspond to Au and Ru containing components, respectively; (c) HAADF-STEM image showing Ru clusters decorating on the surface of Au particles. | ||
 | ||
| Fig. 2 (a) HAADF STEM image of segregated small Ru particles together with big bimetallic particles for the Au@Ru/AC catalyst; (b) component maps and (c) component EDX spectra from multivariate analysis of STEM-EDX spectrum imaging for Au@Ru/AC. Red and green correspond to Au and Ru containing components, respectively. | ||
Interestingly, multivariate analysis on the Au@Ru bimetallic particles, as shown e.g. in Fig. 2b, reveal also an Au core and Ru shell structure. The unexpected core–shell sequence could be attributed to weak adhesion of Ru to the carbon support therefore the ultra-small Ru particles may migrate and relocate around the PVA protected Au particles. Moreover, diffusion of Ru onto Au particles could also take place during drying the catalyst at 80 °C. The EDX spectra of the two components are displayed in Fig. 2c.
3.2 Hydrogenation reactions
Ru has been frequently used for the base free hydrogenolysis of glycerol and levulinic acid, showing good activity in both reaction, but not a good resistance to deactivation. On the contrary few studies reported the use of monometallic gold in these reactions, highlighting a low activity even when harsh conditions were chosen.5c,8dThe catalysts were first tested in the glycerol hydrogenolysis. In Scheme 1, the different pathways for this reaction are shown. Many products interesting from an industrial point of view can be obtained, in particular 1,2 propanediol (1,2-PD), and 1,3 propanediol (1,3-PD) and ethylene glycol (EG). Studies reported using Ru as catalysts showed that 1,2-PD and EG are the main products. 1,2-PD is commercially produced from propylene oxide and it is widely used as monomer for polyester resins, as antifreeze agent and in cosmetics and food industry.11 EG is largely produced via hydration of ethylene oxide and it is mainly used as antifreezing agent.12
 | ||
| Scheme 1 Pathways of glycerol hydrogenolysis. | ||
Glycerol hydrogenolysis has been performed under relative milder reaction conditions, 150 °C and 7 bar of dihydrogen, compared to the ones normally used in the literature, to make the process more appealing for a possible industrial upscaling (Table 2).
| Catalysta | Time (h) | Conv. (%) | Selectivity (%) | Carbon balance | ||
|---|---|---|---|---|---|---|
| EG | 1,2-PD | 1-PrOH | ||||
| a Reaction condition: glycerol = 0.3 M, pH2 = 7 bar, T = 150 °C, metal/glycerol = 1/1000 mol mol−1. EG = ethylene glycol, 1,2-PD = 1,2 propanediol, 1-PrOH = 1-propanol. | ||||||
| Au/AC | 8 | 2 | 23 | 75 | 2 | 100 |
| 16 | 5 | 25 | 71 | 2 | 98 | |
| Ru/AC | 8 | 23 | 29 | 56 | 3 | 88 |
| 16 | 38 | 24 | 58 | 5 | 87 | |
| Ru@Au/AC | 8 | 15 | 24 | 64 | 5 | 92 |
| 16 | 35 | 22 | 63 | 4 | 89 | |
| Au@Ru/AC | 8 | 33 | 17 | 73 | 3 | 96 |
| 16 | 60 | 17 | 70 | 2 | 94 | |
As expected, Au/AC showed a low activity in absence of a base, reaching only 5% of conversion after 16 h. On the contrary, Ru/AC reached a conversion of 23 and 38% after 8 h and 16 h, respectively, with a selectivity of 56% to 1,2-PD, 29% to EG and 5% of 1-PrOH at 16 h (Table 2). No other expected detectable products in the liquid phase (such as 1,3-propandiol, 2-propanol, methanol or ethanol) have been revealed. The carbon balance around only 90% observed in all the reactions was then ascribed to the formation of CH4 and CO2 (Scheme 1) not detectable analysing the liquid phase. Ru@Au/C consisting in a Rushell–Aucore structure showed a slightly lower activity compared to the monometallic Ru/AC, with a conversion of 35% after 16 h (Table 1). However, the presence of Au seems to have a beneficial effect in term of selectivity to 1,2-PD (63% instead of 58%) decreasing the amount of EG formed (Table 2).
Different results have been obtained in the case of Au@Ru/AC where a different structure of bimetallic particles is present. This catalyst showed an almost doubled activity with respect of Ru/AC or Ru@Au/AC reaching 60% of conversion after 16 h (Table 2). Moreover, a substantial enhancement of the selectivity to 1,2-PD was obtained by limiting the formation of degradation products such as EG or C1 products (mass balance of 95%).
The same catalysts have been also tested in the hydrogenolysis of levulinic acid, which is performed under milder conditions than the previous reaction (70 °C instead 150 °C). Levulinic acid is an important platform molecule deriving from the acid catalysed dehydration of cellulose to hydroxymethylfurfural (HMF) followed by the rehydration of this latter one to levulinic acid. The hydrogenation of levulinic acid (Scheme 2) results in the formation of γ-valerolactone (GVL) an important intermediate for the production of different biofuels. Under higher pressure and temperature than the ones used in this study, GVL can be fully converted to 1,4 pentanediol and pentanoic acid, important precursors for the production of valeric biofuels.
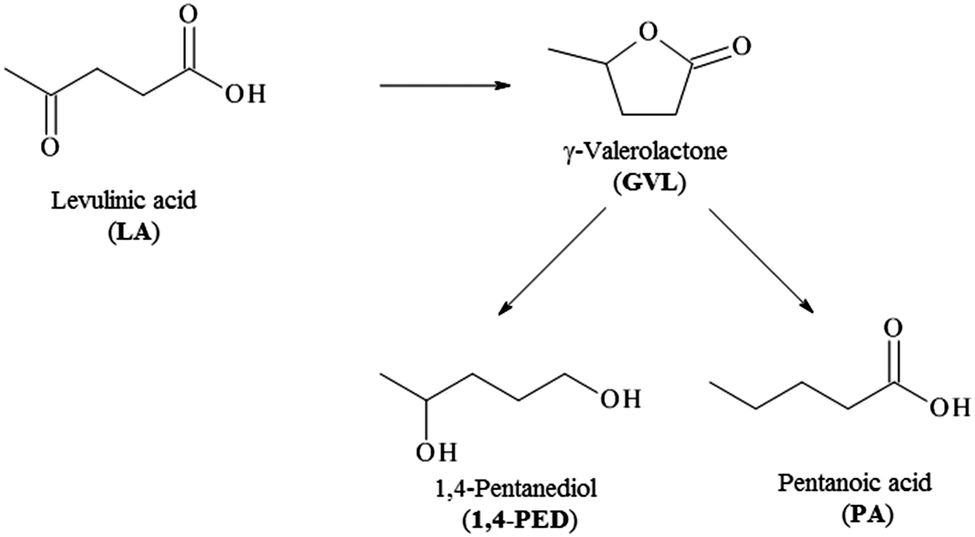 | ||
| Scheme 2 Pathways of levulinic acid hydrogenolysis. | ||
Table 3 reports the results we obtained under the following conditions (Levulinic acid = 0.3 M; pH2 = 7 bar; T = 70 °C; metal/LA = 1/1000 mol mol−1). All the catalysts showed almost the same trend evidenced in the glycerol hydrogenolysis. Ru/AC showed a pretty good activity with a conversion of 65% after 6 h, whereas Au/AC as expected was not very active reaching a conversion of only 10% at the same reaction time. Ru@Au/C was substantially as active as the monometallic Ru with a conversion of 58% after 6 h, whereas Au@Ru/AC increased the activity of more than 30% respect to Ru/AC with a conversion of 87% at 6 h.
All the catalysts showed a selectivity >97% to GVL, with a very small amount of pentanoic acid (1–2%) as by-product. This result is in agreement with the literature, where very high selectivity to GVL were obtained using Ru catalyst at low temperature and hydrogen pressure.9
To explore the usefulness of the Au addition to Ru in terms of catalyst durability, that actually limits the use of Ru catalyst in large-scale production, we investigated the recyclability of the catalyst. Recycling experiments carried out just by filtering the catalyst and adding fresh solution of levulinic acid revealed that Au@Ru/AC not only is the most active but also the most resistant catalyst compared to Ru/AC and Ru@Au/AC (Fig. 3). In fact the activity of these two latter progressively declines during the 6 runs, whereas Au@Ru/AC showed almost the same conversion. The selectivity remains stable for all the catalysts. ICP analysis resulted in a loss of 5 wt% of total Ru for Ru/AC and Ru@Au/AC whereas no metal was found in the solution in the case of Au@Ru/AC system.
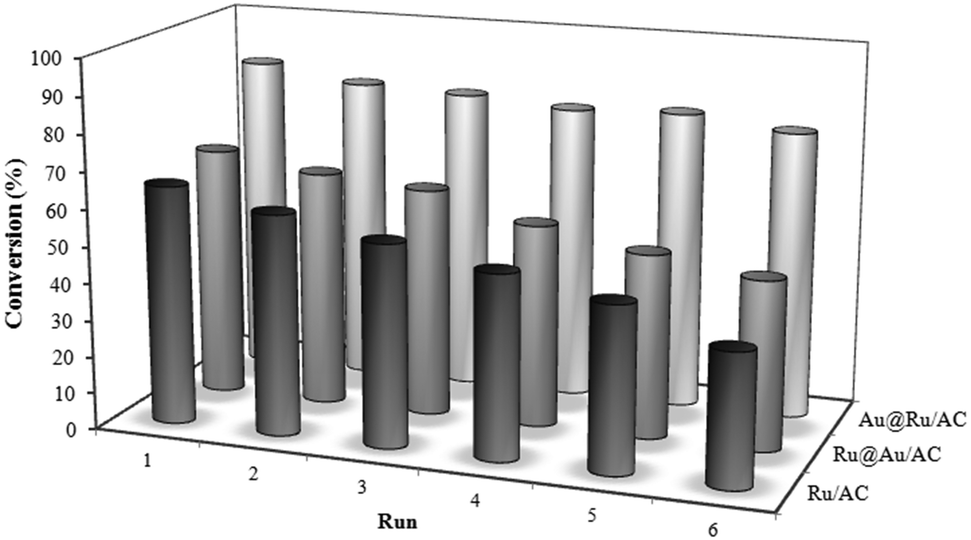 | ||
| Fig. 3 Recycling tests for the levulinic acid hydrogenolysis. | ||
Considering the two different reactions, we can then generally conclude that the structure of AuRu catalysts significantly influenced the catalytic activity. Ru@Au/AC, where a Rushell–Aucore structure was evidenced by TEM studies, showed a slight lower activity compared to Ru/AC that could be explained, according to the literature, with a geometric dilution (ensemble effect).13 To support this statement we performed an additional test of levulinic hydrogenation using 0.27% Ru/AC (the same loading as in the bimetallic system) in the same S/M ratio as in the test carried out with monometallic Ru/AC (1000 mol mol−1). At 6 h the conversion resulted of only 51% compared to 65% obtained with 1% Ru/AC but similar to conversion obtained with Ru@Au/AC (Table 3). On the contrary, using Au@Ru/AC, a synergistic effect was envisaged in terms of both activity and durability.
The different behaviour of Au@Ru and Ru@Au on carbon catalysts should be therefore ascribed to a different role of Au on Ru properties in the two different structures. Unfortunately, we cannot obtain any reliable information by XPS on possible electronic effects as Ru-signals partially overlap with C-ones thus not allowing any definite answer. We thus formulated some hypotheses based on TEM results. We showed that Au@Ru underwent diffusion of Au on Ru particle probably forming alloyed domains whereas in Ru@Au a real core–shell structure is obtained. Moreover, we also demonstrated that diluting Ru on the AC surface (lowering the loading) a less active catalyst is obtained, thus supporting the geometric effect of Au. A similar geometric effect was expected also for Au@Ru, however in this case a real more active catalyst is obtained. Therefore, we could conclude that in this latter case the alloyed domains should be responsible of the synergistic effect observed and that the main role of Au should be an electronic one.
Conclusions
AuRu bimetallic catalysts have been prepared by successive deposition of Au on Ru or in alternative Ru on Au on activated carbon as the support. TEM investigation evidenced the formation of bimetallic nanoparticles with similar size (4 nm) but different structure. When Ru was deposited on Au, a Rucore–Aushell structure was formed, whereas the deposition of Au on Ru resulted in the formation of Au–Ru bimetallic phase with a partial surface Ru enrichment.These catalysts were tested in the glycerol and levulinic acid hydrogenolysis and compared to the monometallic counterparts. Au/AC, as expected, showed a very low activity whereas Ru/AC successfully converted both the substrates. AuRu core–shell structure showed a slightly lower conversion compared to Ru/AC whereas AuRu phase (Au@Ru/AC) resulted the most active catalyst. This latter one showed also a good durability in the levulinic acid hydrogenation, showing almost the same activity in six consecutive runs. On the contrary, Ru/AC and Ru@Au/AC constantly deactivated losing the 30% of the initial activity.
Acknowledgements
TEM characterization was carried out in KIT and sponsored by Karlsruhe Nano Micro Facility (KNMF).Notes and references
- (a) M. A. El-Sayed, Acc. Chem. Res., 2001, 34, 257 CrossRef CAS PubMed; (b) K. G. Thomas and A. V. Kamat, Acc. Chem. Res., 2003, 36, 888 CrossRef CAS PubMed; (c) M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS PubMed; (d) E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740 RSC; (e) Y. Zhang, X. Cui, F. Shi and Y. Deng, Chem. Rev., 2012, 112, 2467 CrossRef CAS PubMed; (f) K. An and G. A. Somorjai, Catal. Lett., 2015, 145, 233 CrossRef CAS PubMed.
- (a) M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405 CrossRef CAS; (b) G. J. Hutchings, J. Catal., 1985, 96, 292 CrossRef CAS.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037 CrossRef CAS PubMed.
- (a) M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 809 RSC; (b) K. Singh and Q. Xu, ChemCatChem, 2013, 5, 652 CrossRef PubMed; (c) G. J. Hutchings and C. J. Kiely, Acc. Chem. Res., 2013, 8, 1759 CrossRef PubMed; (d) S. Shan, J. Luo, L. Yang and C.-J. Zhong, Catal. Sci. Technol., 2014, 4, 3570 RSC; (e) I. N. Francesco, F. Fontaine-Vive and S. Antoniotti, ChemCatChem, 2014, 6, 278 CrossRef PubMed; (f) G. J. Hutchings, Catal. Today, 2014, 238, 69 CrossRef CAS PubMed; (g) A. Villa, D. Wang, D. S. Su and L. Prati, Catal. Sci. Technol., 2015, 5, 55 RSC.
- (a) X. Yang, D. Chen, S. Liao, H. Song, Y. Li, Z. Fu and Y. Su, J. Catal., 2012, 291, 36 CrossRef CAS PubMed; (b) R. Liu, Y. Yancun, K. Yoshida, G. Li, H. Jiang, M. Zhang, F. Zhao, S.-I. Fujita and M. Arai, J. Catal., 2010, 269, 191 CrossRef CAS PubMed; (c) E. P. Maris, W. C. Ketchie, M. Murayama and R. J. Davis, J. Catal., 2007, 251, 281 CrossRef CAS PubMed.
- (a) G. C. Bond, C. Louis and D. T. Thompson, Catalysis by Gold, Imperial College Press, London, 2006 Search PubMed; (b) R. Zanella, C. Louis, S. Giorgio and R. Touroude, J. Catal., 2004, 223, 328 CrossRef CAS PubMed.
- (a) T. Mitsudome and K. Kaneda, Green Chem., 2013, 15, 2636 RSC; (b) F. Cárdenas-Lizana and M. A. Keane, J. Mater. Sci., 2013, 48, 543 CrossRef.
- (a) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS PubMed; (b) C. H. Zhou, J. N. Beltramini, Y. X. Fan and G. Q. Lu, Chem. Soc. Rev., 2008, 37, 527 RSC; (c) B. Katryniok, H. Kimura, E. Skrzynska, J.-S. Girardon, P. Fongarland, M. Capron, R. Ducoulombier, N. Mimura, S. Paula and F. Dumeignil, Green Chem., 2011, 13, 1960 RSC; (d) Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2011, 1, 179 RSC.
- (a) T. Horváth, H. Mehdi, V. Fábos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238 RSC; (b) D. Martin Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584 RSC; (c) M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2013, 114, 1827 CrossRef PubMed; (d) M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516 RSC; (e) W. R. H. Wright and R. Palkovitz, ChemSusChem, 2012, 5, 1657 CrossRef CAS PubMed; (f) J. Q. Bond, D. Martin Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110 CrossRef CAS PubMed; (g) J. Q. Bond, D. Martin Alonso, D. Wang, R. M. West and J. A. Dumesic, J. Catal., 2011, 281, 290 CrossRef CAS PubMed; (h) J. C. Serrano-Ruiz, D. J. Braden, R. M. West and J. Dumesic, Appl. Catal., B, 2010, 100, 184 CrossRef CAS PubMed.
- (a) L. Prati, F. Porta, D. Wang and A. Villa, Catal. Sci. Technol., 2011, 1, 1624 RSC; (b) A. Villa, C. E. Chan-Thaw, M. Schiavoni, S. Campisi, D. Wang and L. Prati, Chin. J. Catal., 2014, 35, 945 CrossRef CAS.
- D. T. Johnson and K. A. Taconi, Environ. Prog., 2007, 26, 338 CrossRef CAS PubMed.
- H. Yue, Y. Zhao, X. Ma and J. Gong, Chem. Soc. Rev., 2012, 41, 4218 RSC.
- A. G. Shastri and J. Schwank, J. Catal., 1985, 95, 271 CrossRef CAS.
| This journal is © the Owner Societies 2015 |