
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversible oxygen addition on a triplet sensitizer molecule: protection from excited state depopulation†
Mikhail A.
Filatov
*ab,
Ernesta
Heinrich
a,
Dmitry
Busko
a,
Iliyana Z.
Ilieva
c,
Katharina
Landfester
a and
Stanislav
Baluschev
acd
aMax Planck Institute for Polymer Research, Ackermannweg 10, D-55128 Mainz, Germany. E-mail: filatov@mpip-mainz.mpg.de; Fax: +49 6131379100
bInstitute of Polymers, Bulgarian Academy of Sciences, Acad. G. Bonchev Str., block 103-A, BG - 1113 Sofia, Bulgaria
cOptics and Spectroscopy Department, Faculty of Physics, Sofia University “St. Kliment Ochridski”, 5 James Bourchier, 1164 Sofia, Bulgaria
dFreiburg Institute for Advanced Studies (FRIAS), Albert-Ludwigs-Universität Freiburg, Albertstraße 19, D-79104 Freiburg, Germany
First published on 28th January 2015
Abstract
We demonstrate that photoactivated oxygen addition to diphenylanthracene moities can be used as a tool for protection of porphyrin's phosphorescence against oxygen quenching. Phosphorescent palladium(II) tetrabenzoporphyrin, covalently linked to four diphenylanthracene moieties, was synthesized and studied. Upon irradiation with ambient light or red laser in solution in air, addition of oxygen and formation of the corresponding endoperoxides were observed. Heating of the irradiated samples afforded the parent porphyrin material.
Introduction
Organic chromophores able to form triplet excited states upon absorption of light (triplet sensitizers) are used in various fields, e.g. electroluminescence,1 bioimaging and molecular sensing,2 photocatalytic organic reactions,3 and triplet–triplet annihilation photon upconversion (TTA-UC).4 Organic electronic materials based on triplet sensitizers promise to make revolutionary transformations in solar energy conversion technologies by improving the light-harvesting of amorphous silicon solar cells5 and extending the infrared limit of oxygenic photosynthesis.6 However, compared to fluorophores, applications for triplet sensitizers are much less developed considering a substantially higher sensitivity of triplet states towards non-emissive deactivation processes. The most common process which leads to the loss of triplet excited state population is related to the presence of molecular oxygen in the corresponding samples. It involves a triplet energy transfer between an excited chromophore and oxygen, giving rise to singlet oxygen and the ground state of the chromophore.7An efficient protection against oxygen quenching is essential for applications which include triplet excited state formation. Two general approaches for the protection are known: (1) passive protection, based on decreasing oxygen permeability e.g. by means of encapsulation in polymer films8 or nano-9 and microcarriers,10 or incorporation into supramolecular complexes11 or dendrimers12 and (2) active protection – by applying oxygen scavenging species.13
The problem of triplet excited state quenching is exceptionally important in the case of the TTA-UC process. Both the sensitizer and emitter ensembles can transfer triplet energy to the molecular oxygen and be depopulated, leading to the aging of samples and loss of quantum efficiency. Straightforward embedding of UC-active substances into inert polymer films with high oxygen barrier properties leads to a substantial decrease of the TTA-UC efficiency, because in solid state environments the local mobility of the chromophores involved a significant decrease.14 Until recently the direct incorporation of dyes into polymers was not enough to provide the imperative requirements for effective and sustainable annihilation upconversion, that is, high local mobility and exhaustive oxygen protection of the UC-chromophores. Recently we succeeded in developing organic polyphosphate oxygen protection matrices15 and cellulose bioinspired oxygen protection films.16 However, these approaches either require a change in the sample's architecture or affect the photophysical and chemical properties of a triplet sensitizer.
Herein we report a new strategy for protection of triplet excited state depopulation by quenching, relying on chemical modification of a triplet sensitizer molecule. It is based on binding the molecular oxygen, present in a sample, to specially designed structural subunits which do not affect the photophysical properties and allow the triplet sensitizer to act in an undisturbed manner further. This protection strategy is of sacrificial character, and is time-limited depending on the integral photon flux, applied to the sample. However, the starting triplet sensitizer material can be fully regenerated afterwards through oxygen release upon moderate heating. Furthermore, protective groups do not bind oxygen in its ground (triplet) state and the corresponding material is stable towards photooxidation in the course of synthetic procedures and purification. The protection is active only against singlet oxygen, thus only when sensitizer triplet states are formed. Therefore, the presence of an additional stimulus, i.e. optical excitation of the sensitizer, triggers the protection process.
In our recent work on the synthesis of tetraanthraporphyrins17 we observed a photoactivated addition of up to four oxygen molecules per porphyrin molecule, leading to the formation of the corresponding endoperoxides. We proposed that such an addition can serve as a tool for preventing oxygen from quenching of porphyrin phosphorescence. However, in the case of tetraanthraporphyrins, the addition of oxygen leads to a drastic change in the optical properties, particularly to a blue-shift of absorption and emission bands by 200 nm due to a partial loss of conjugation in the π-system. Here we suggested to introduce anthracene subunits in meso-positions of the porphyrin in order to avoid alteration of the optical properties.
The concept is illustrated in Fig. 1 using an example of Pd-porphyrin bearing four anthracene subunits in meso-positions of the macrocycle. Pd-porphyrins are known to possess high intersystem crossing coefficient18 values and are widely used as triplet sensitizers.19 Alternatively, anthracenes are capable of reversible binding of singlet oxygen species.20 Upon selective excitation of the porphyrin chromophore, a triplet excited state is formed (step 1). In the presence of the molecular oxygen a quenching process takes place, leading to the ground state of the sensitizer and singlet oxygen (step 2). The key point is step 3 where binding of the singlet oxygen to the attached hydrocarbon via the Diels–Alder-type process takes place.21 We proposed that complete binding to all of four hydrocarbon subunits can be achieved (shown as step 4 in Scheme 1) and the corresponding (O2)4 adduct can be obtained, otherwise adducts with a variable number of bound oxygens are produced. Consumption of oxygen results in partial or complete termination of the quenching process that provides emissive relaxation of newly formed triplet states of the Pd-porphyrin core. Due to the reversibility of the oxygen addition to anthracenes, the starting material can further be regenerated by means of heating (step 5).
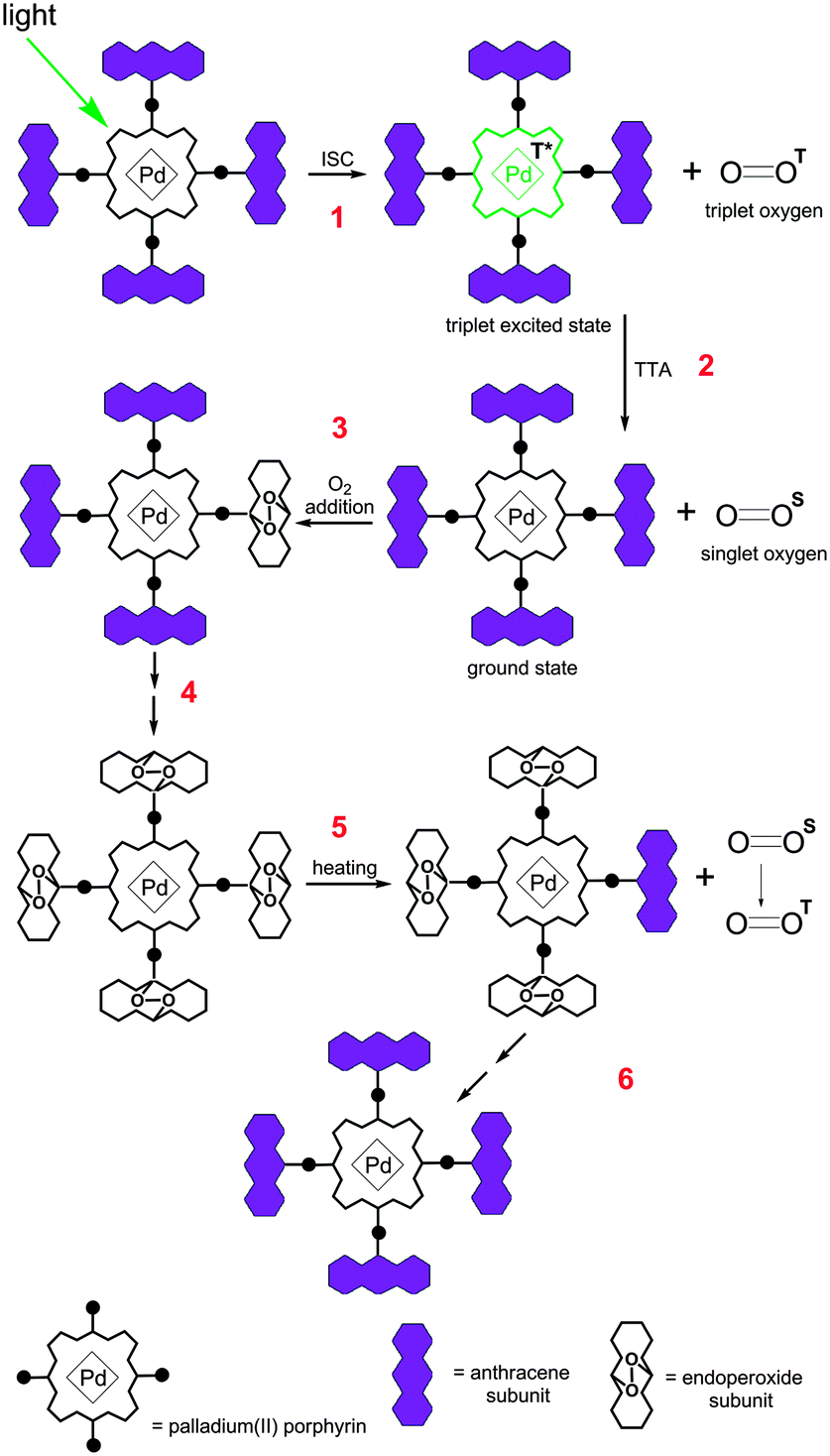 | ||
| Fig. 1 General scheme of oxygen addition to a triplet sensitizer and further thermal release. | ||
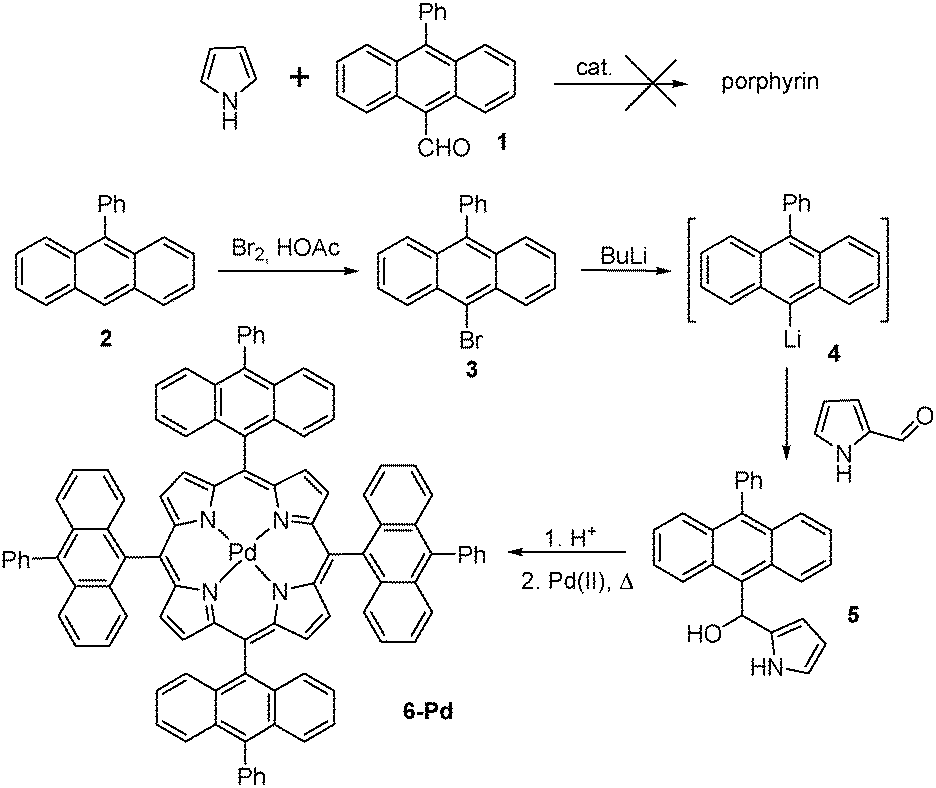 | ||
| Scheme 1 Synthesis of porphyrin 6-Pd. | ||
Although many polyaromatic hydrocarbons, e.g. naphthalenes,22 2-pyridone23 or rubrenes,24 are known to bind oxygen in a reversible manner, anthracenes are most suitable to be used for oxygen protection of the phosphorescence. First, the absorption bands of the corresponding anthracenes do not intersect with porphyrin absorption that allows monitoring of the protection by absorption spectroscopy. Second, anthracene endoperoxides decompose only upon heating above 100 °C whereas naphthalene and 2-pyridone endoperoxides release oxygen even at room temperature. Finally, anthracene provides a scaffold that allows straightforward generation of suitable substitution patterns.
The introduction of such type of triplet sensitizer into the arsenal of materials used in the relevant areas of research would open new opportunities for the control of the corresponding photophysical processes. Particularly, it provides an alternative way to oxygen protection of phosphorescent samples or to develop new molecular “singlet oxygen reservoirs” for oxygen storage and thermal release. In this paper we report the synthesis and properties of phosphorescent palladium(II) porphyrin, bearing substituted anthracenes in meso-positions, which is capable of photoactivated oxygen addition. Anthracene groups preserve the phosphorescence properties of porphyrin even in the oxygen rich environment. Furthermore, thermal-mediated reversibility of the oxygen binding was demonstrated.
Results and discussion
Synthesis
Our attempts towards the synthesis of the target molecule were firstly focused on β-unsubstituted porphyrin with anthracene groups directly attached in meso-positions.According to literature data, the conventional method of porphyrin chemistry – Lewis acid catalyzed condensation of pyrrole with the corresponding 10-phenyl-anthracene-9-carboxaldehyde 1 – results in very low preparative yields.25 An alternative method based on pyrrole-carbinol tetramerization is being commonly applied for such type of substrates.26 Following this approach, the starting 9-phenylanthracene 2 was brominated with Br2 to give 9-bromo-10-phenylanthracene 3. It was then subjected to the reaction with n-butyllithium to give the corresponding organolithium compound 4 which was further introduced into the reaction with pyrrole-2-carboxaldehyde (Scheme 1). The obtained pyrrole-carbinol 5 was used for acid-catalyzed tetramerization and delivered the corresponding porphyrin 6 with reasonable yield (15%). Subsequent insertion of palladium was achieved by reflux in benzonitrile. Unfortunately the obtained product 6-Pd was found to possess too low solubility for adequate characterization and study of oxygen addition.
Generally, for large conjugated aromatic systems, solubility is ensured by introduction of suitable side chains into the aromatic moiety, e.g. alkyl and alkoxy groups.27 However, the introduction of electron-donating groups on anthracenes is known to have a pronounced effect on the rates of oxygen binding and decomposition of the endoperoxide. In some cases it results in the irreversibility of the oxygen addition.20 Thus in order to optimize the sensitizer structure we decided to modify the porphyrin core instead of the anthracene subunit. It was shown that β-substitution of the porphyrin macrocycle, especially along with introduction of meso-aryl substituents leads to a strong distortion of the macrocycle due to steric repulsion.28 This in turn results in an improved solubility. Following this approach we decided to introduce two additional structural features: (1) a phenylene bridge between porphyrin and anthracene subunits and (2) annelated cyclohexane rings at β-positions of the porphyrin macrocycle. Such a modification also allows for shifting of absorption and emission bands into the red or far red region, the latter by means of aromatization of annelated rings.29
9-Bromo-10-phenylanthracene 3 was subjected to Suzuki-coupling with formylboronic acid giving the corresponding aldehyde 8. Its condensation with 4,5,6,7-tetrahydrosioindole 9 provided the corresponding porphyrin 10 (Scheme 2), which was then metallated with bis(benzonitrile)palladium(II) in boiling benzonitrile. Further aromatization of porphyrin 10-Pd into the corresponding tetrabenzoporphyrin 11-Pd was achieved by reflux with an excess of DDQ in toluene. Complete aromatization of all four annelated rings was achieved without significant by-product formation. Porphyrin 11-Pd was found to be well-soluble in the common solvent (chlorinated hydrocarbons, THF, toluene) and was unambiguously characterized by NMR and mass spectroscopy (see ESI†).
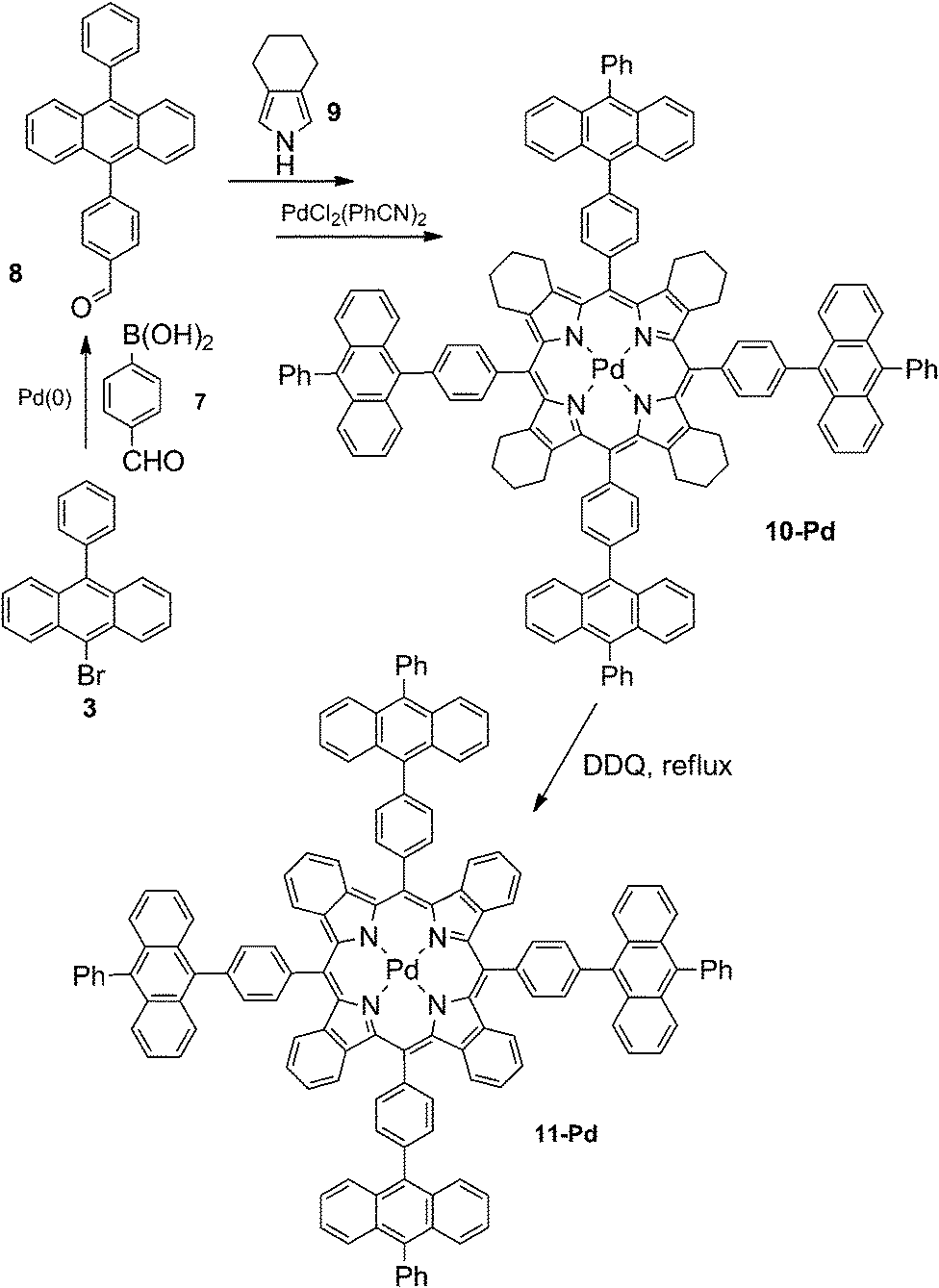 | ||
| Scheme 2 Synthesis of porphyrins 10-Pd and 11-Pd. | ||
Oxygen addition and release
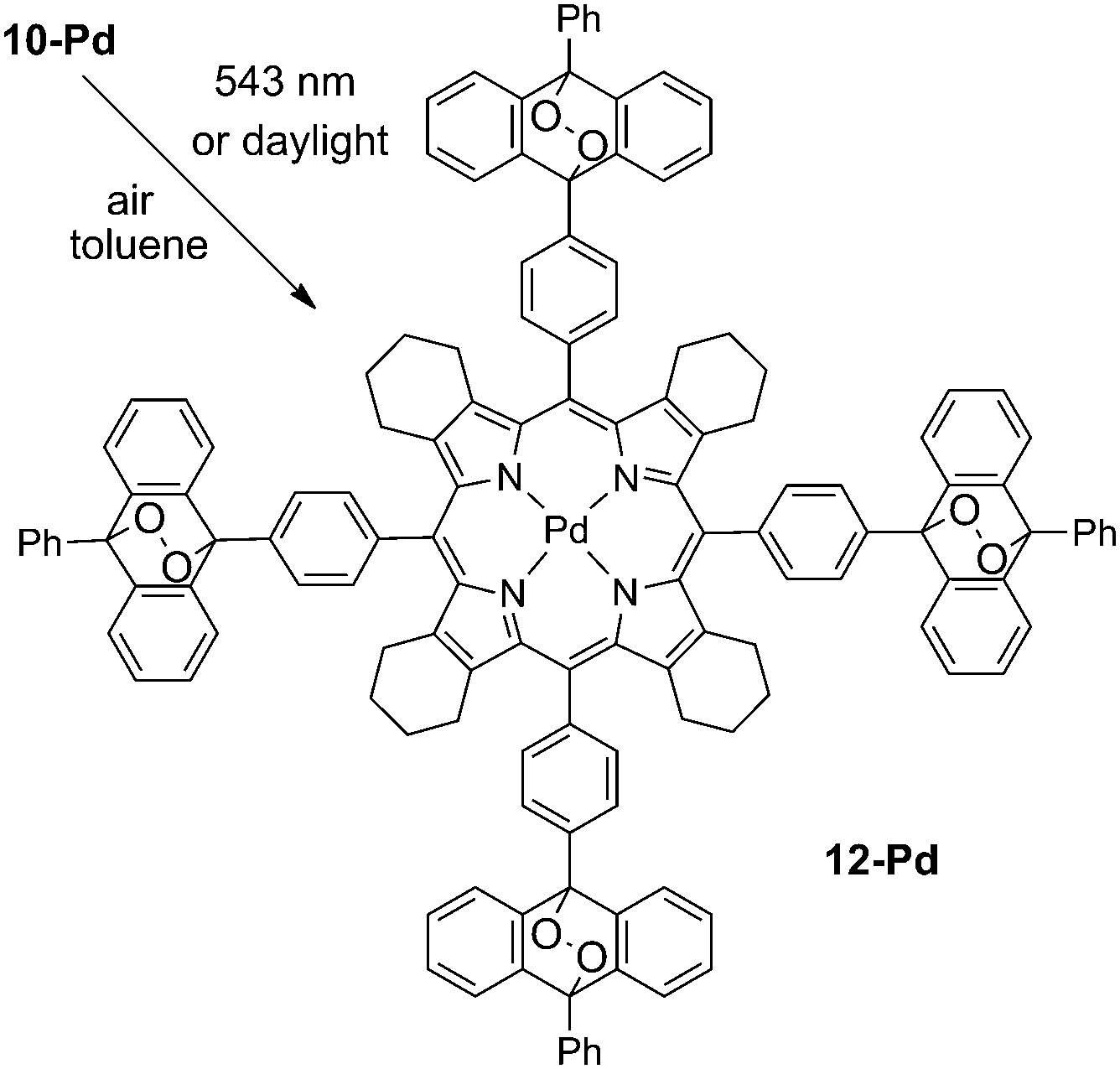
As is shown in Fig. 2, porphyrin 10-Pd possesses characteristic absorption in the region of 300–400 nm, which correspond to the anthracene subunits. When a solution of 10-Pd was kept under daylight for several days or irradiated with the green line of the HeNe laser (λ = 543 nm, broad beam), disappearance of the anthracene absorption was observed (Fig. 2, red line) giving instead a shapeless background absorption. On the other hand, spectral features corresponding to the porphyrin macrocycle – Soret (430 nm) and Q-bands (540 and 570 nm) – remained unchanged. Light irradiation of the same solution prepared and sealed in a glove-box (<1 ppm oxygen) showed no change in absorption.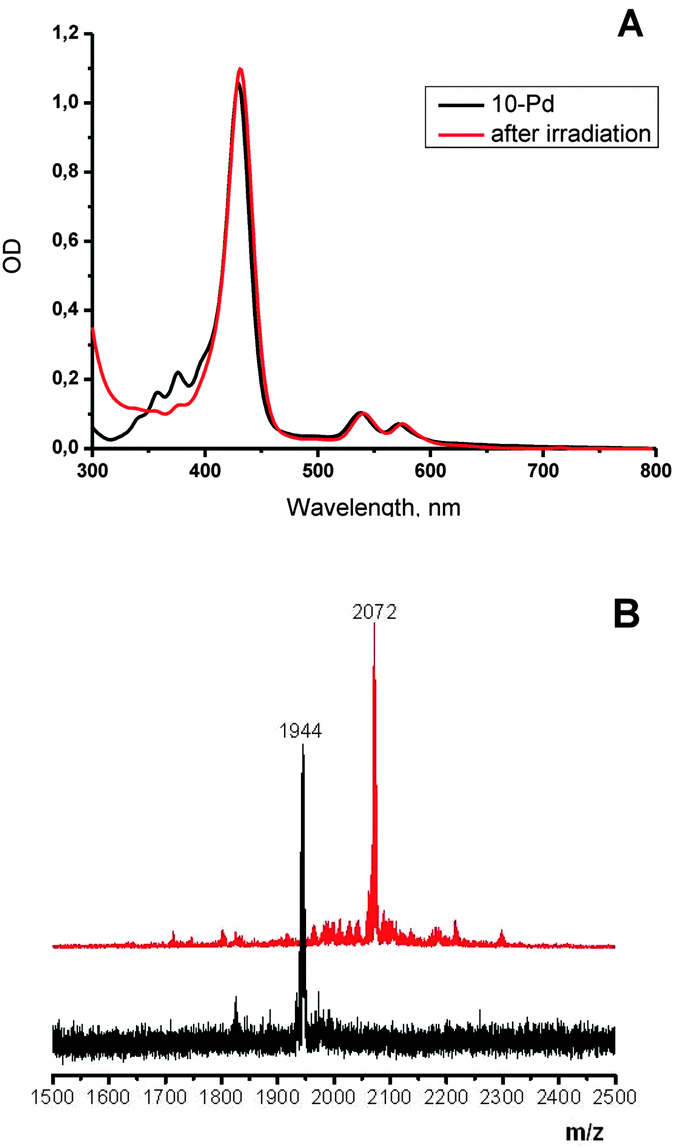 | ||
| Fig. 2 Absorption spectrum of 10-Pd and its change after light irradiation of air-saturated toluene solution (green-line HeNe laser, 3 μW cm−2, 10 h) (A); the corresponding mass spectra of 10-Pd (B, black trace) and the product of irradiation (B, red trace). | ||
The mass spectrum of the irradiated solution showed a single peak with a mass of 2072 Da, corresponding to an adduct of 10-Pd with four oxygen molecules (Scheme 3).
 | ||
| Scheme 3 Photosensitized oxygen addition to porphyrin 10-Pd. | ||
A similar process with compound 11-Pd was monitored by UV-Vis spectroscopy. Its solution (4 × 10−5 M) was irradiated with the red line of HeNe laser (λ = 633 nm, broad beam) at an intensity of only 3 μW cm−2. The laser beam was set to cover the whole cuvette. The complete disappearance of the anthracene absorption in the region of 300–400 nm was observed within 9 h (Fig. 3). Mass spectra of the obtained samples showed a series of (M+ + n × 32) peaks indicating the formation of intermediate products bearing 1, 2, 3 or 4 oxygen molecules attached. At 3 μW cm−2 the addition reaction was complete in 9 h as is evidenced by the absence of spectral change and a sole peak of 2056 Da in the mass spectrum corresponding to the adduct with four oxygen molecules (Scheme 4). Exposure of the solution for longer times of irradiation (up to 40 h) did not result in the change in absorption spectra or origin of other peaks in the mass spectrum.
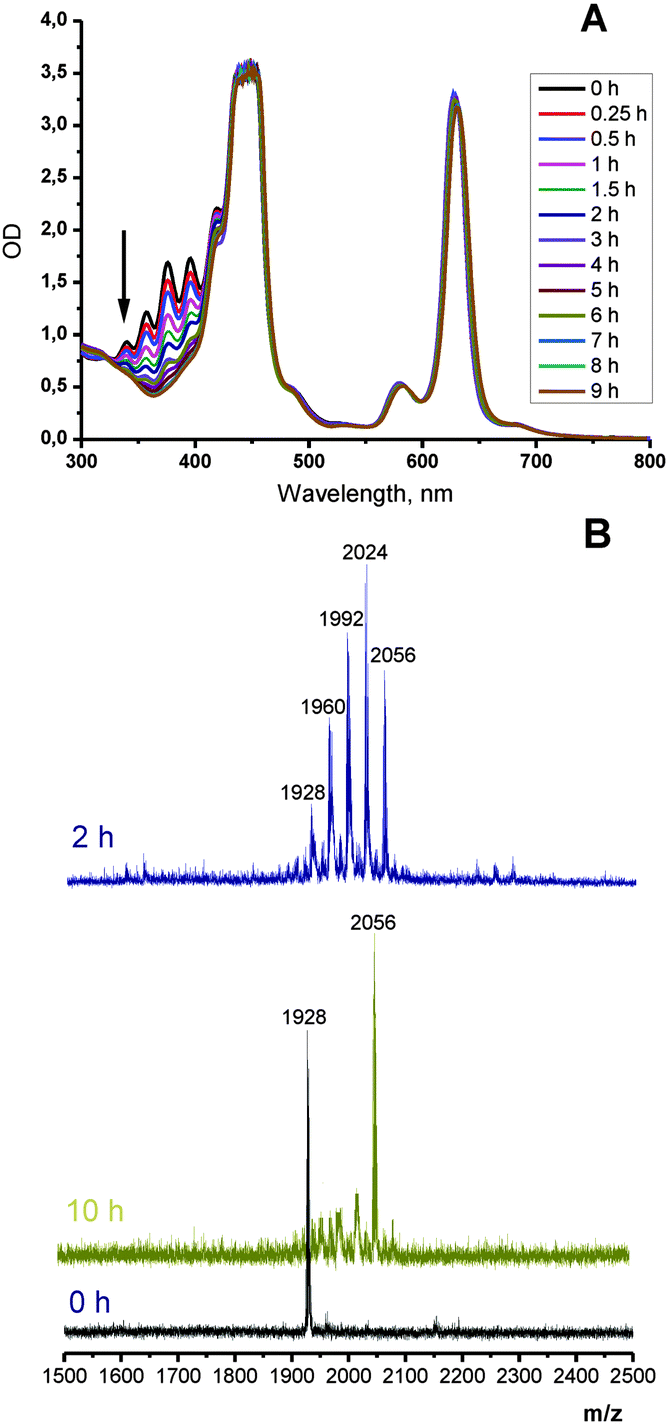 | ||
| Fig. 3 Change in absorption upon irradiation of 4 × 10−5 M toluene solution of 11-Pd using 633 nm laser (3 μW cm−2) (A); the corresponding change in mass spectra (B): 11-Pd (black trace), after 5 h of irradiation (blue trace), and after 10 h of irradiation (yellow trace). Note: saturation of porphyrin absorption is due to high concentration of the solution taken for the experiment, essential for monitoring the relatively weak anthracene absorption. | ||
 | ||
| Scheme 4 Photosensitized oxygen addition to porphyrin 11-Pd. | ||
To further support the idea of photosensitized oxygen addition on anthracene moieties in 11-Pd we performed a similar experiment using a mixture of 9,10-diphenylanthracene and palladium tetraphenyltetrabenzoporphyrin in a molar ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Under the same conditions (concentration, laser intensity) a similar spectral transformation was observed by UV-Vis spectroscopy on a timescale of 6 h (see ESI,† Fig. S14).
:1. Under the same conditions (concentration, laser intensity) a similar spectral transformation was observed by UV-Vis spectroscopy on a timescale of 6 h (see ESI,† Fig. S14).
The release of bound oxygen molecules was achieved when solution of the adduct 13-Pd was concentrated to a solid and then heated at 100–110 °C in vacuum (1 mbar) for 3–5 h. A complete recovery of anthracene absorption (with respect to 11-Pd) as well as a perfect overlap with the original spectrum of 11-Pd were observed. The mass spectrum showed a 1928 Da parent peak. Thus a completely reversible oxygenation–deoxygenation process takes place for 11-Pd.
It should be mentioned that the decomposition temperature (and time) of the adduct depends on the substitution pattern of the aromatic subunits. In our case the parameters of oxygen release match those previously described for 9,10-diphenylanthracene decomposition.20 However, modification of the starting materials would allow varying decomposition parameters. Particularly, it can be made irreversible (by means of introducing one or two alkoxy-groups into the parent anthracene derivative). Alternatively, the temperature of endoperoxide decomposition can be adjusted to 30–40 °C (by using naphthalene derivatives as oxygen traps, instead of anthracenes).
Optical properties: protection of phosphorescence against oxygen quenching
Absorption and emission spectra of 11-Pd are shown in Fig. 4. Its optical properties are very similar to those of tetraphenyltetrabenzoporphyrin-Pd (see ESI†). The sample of 11-Pd prepared in a glove-box (containing <1 ppm oxygen) exhibits strong phosphorescence centered at 796 nm with a quantum yield of 0.18 (with respect to Ph4TBPPd in toluene (0.21)30). No phosphorescence was observed in the sample open to air. The triplet excited state lifetime was measured to be 217 μs that is close to parent Ph4TBPPd (286 μs).30 Thus no significant loss of emissivity was observed despite conjugation of the porphyrin π-system to four anthracene residues.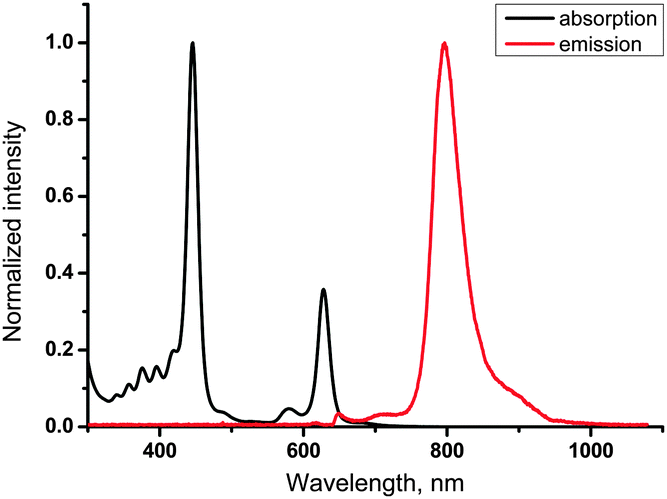 | ||
| Fig. 4 Absorption and phosphorescence spectra of 11-Pd in toluene (1 × 10−5 M). Phosphorescence was excited at 633 nm. | ||
Due to very similar chemical properties, products of photosensitized oxygen addition to porphyrin 11-Pd cannot be isolated. In order to investigate their emission properties a series of 11-Pd solutions were illuminated (633 nm, 3 μW cm−2) at different time intervals (1–9 h). The obtained samples were concentrated to solids, transferred into the glove-box (<1 ppm of oxygen) and dissolved in toluene to prepare a set of solutions of equal concentration for phosphorescence measurement. No significant difference in the phosphorescence intensity between starting 11-Pd and irradiated samples was observed (Fig. 5). Only a moderate decrease of the emission intensity (approximately 25%) takes place upon addition of oxygen molecules to all four anthracenes. This is probably due to a decrease in the phosphorescence quantum yield upon increasing molecule's complexity usually resulting in an increase of non-radiative decay rates.
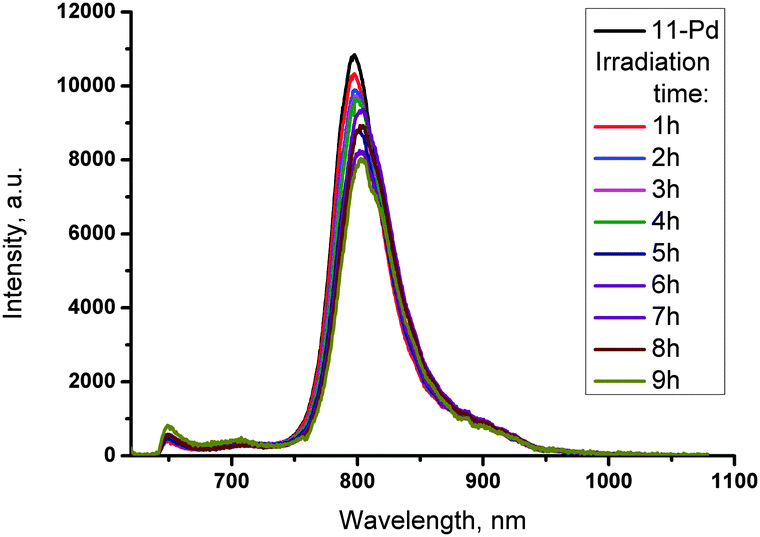 | ||
| Fig. 5 Phosphorescence spectra of 11-Pd and mixtures of oxygen addition products obtained after different periods of irradiation. All phosphorescence spectra are registered for samples of the same concentration keeping excitation parameters (toluene, 633 nm, 1 mW cm−2, integration time of 50 ms) constant. | ||
The fact that adducts with a different number of bound oxygen molecules possess similar emission properties enables us to use the photo-activated oxygen addition to enhance the phosphorescence signal intensity of oxygen contaminated (e.g. as a result of leaking) samples. Under laser irradiation during a short time (minutes or less, depending on excitation intensity), oxygen molecules are being bound to the sensitizer, performing local and real-time “deoxygenation” of the sample. Due to the decrease of the quencher concentration, the phosphorescence efficiency increases.
To demonstrate such an effect, a solution of 11-Pd was prepared and sealed in an atmosphere containing 100 ppm of oxygen. The phosphorescence spectrum registered has shown a peak intensity of 2160 cps (Fig. 6, black line). Then the cuvette was placed under a laser beam with 250 μW cm−2 intensity for a period of 10 min. The phosphorescence spectrum registered after irradiation has shown 100% increase of the peak intensity (Fig. 6, red line). Obviously, the increase of the signal intensity is due to the binding of residual amounts of oxygen present in the sample.
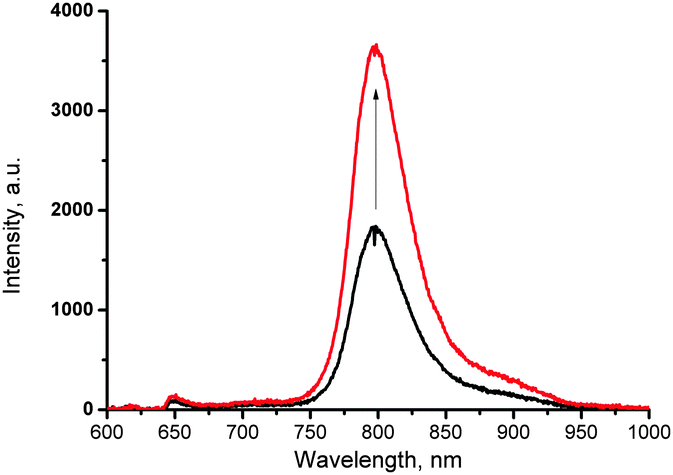 | ||
| Fig. 6 Increase of phosphorescence of a sample of 11-Pd prepared in the atmosphere containing 100 ppm of oxygen (black line) after 10 minutes of continuous irradiation with red laser (toluene, 4 × 10−5 M, 633 nm, 250 μW cm−2) (red line). | ||
Moreover, an enhancement of the phosphorescence originating from porphyrin 11-Pd can be achieved in a local area (spot diameter d = 400 μm) of excitation without preliminary irradiation of the whole sample. In the course of the measurement, the same laser beam being used for the excitation of phosphorescence can simultaneously cause local deoxygenation. The results obtained from the corresponding time-resolved experiment are shown in Fig. 7; an increase of the phosphorescence signal intensity from sample 11-Pd, which increases more than 60% at continuous irradiation by laser beam with different intensities. As is seen from Fig. 7 (green line) at a moderate intensity of 1 mW cm−2 and local excitation, the process of local oxygen scavenging needs only a few seconds, if the excitation intensity is lower, the necessary time is substantially longer (Fig. 7, the black line). The experimental atmosphere was contaminated by 100 ppm of oxygen.
 | ||
| Fig. 7 Change in phosphorescence intensity at 798 nm for the sample of 11-Pd registered in an oxygen contaminated atmosphere (100 ppm), taken at noted periods of time during continuous irradiation by red laser (toluene, 4 × 10−5 M, 633 nm, different excitation intensities). | ||
Owing to the deoxygenation ability, molecules such as 11-Pd can become promising “self-healing” sensitizers for the process of TTA-UC, which is known to crucially depend on the oxygen content. In order to confirm the suitability of 11-Pd as a sensitizer for TTA-UC, it was mixed with perylene as an emitter in an inert atmosphere (<1 ppm of oxygen) and then used for the generation of the upconversion fluorescence. Upon the red excitation at 638 nm, along with some residual phosphorescence of the sensitizer, a blue upconverted emission of perylene in the region 430–550 nm with the reasonable quantum yield (∼3%) was observed (Fig. 8). A corresponding study devoted to the effect of oxygen on the upconversion efficiency of the system will be reported in due course.
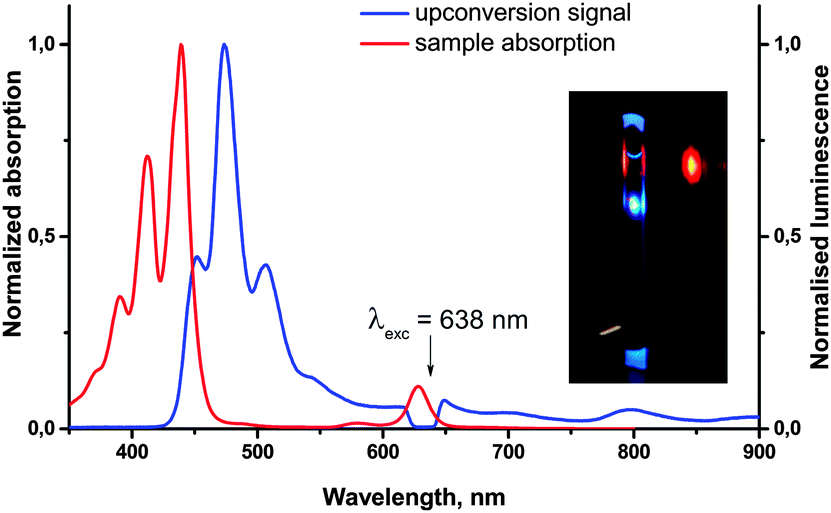 | ||
| Fig. 8 Absorption and emission spectra of the upconversion sample containing 3 × 10−5 M of 11-Pd and using 6 × 10−4 M (toluene) perylene as an emitter. Quantum yield of UC at an excitation intensity of ∼1 W cm−2 is 3%. Inset: a photo of the upconversion sample being irradiated with 638 nm laser. | ||
Conclusions
While in many studies devoted to phosphorescence and triplet excited state dynamics the problem of oxygen quenching is being solved by physical or chemical protection of the sample from oxygen contamination, the development of “self-protective” triplet sensitizer molecules represents an almost unexplored approach.In this work, palladium(II) porphyrin bearing four anthracene subunits in meso-positions was synthesized. Optimization of structural features delivered a material with high solubility and strong near-IR absorption. It was found to possess oxygen-binding properties upon irradiation with either daylight or lasers. There was no effect on the absorption features of porphyrin, and only a weak decrease of phosphorescence intensity was observed during addition of oxygen. Such properties enable us to perform partial or complete deoxygenation of oxygen-contaminated samples, thus enhancing their phosphorescence intensity. Two regimes of deoxygenation were explored: a conventional one – oxygen scavenging of the whole sample, and second – local, and real-time oxygen scavenging, suitable for sensing applications. The starting sensitizer material can be regenerated by means of heating in vacuum. A palette of observed properties makes the material promising for application as phosphorescent sensing probes and a self-protecting sensitizer for the process of TTA-UC.
Although such a protection strategy is limited by the capacity of the sensitizer to bind no more than four oxygen molecules, it may have a potential in applications which demand very low oxygen concentration levels. For example, most of the materials comprising the OLEDs suffer from degradation effects due to the presence of environmental oxygen and water, as both compounds can penetrate into the device. Much research and effort have been put into fabrication methods and appropriate device encapsulation to help mitigate these environmental effects. Thus the demonstrated oxygen protection strategy on a molecular level might become complementary to those already developed.
Experimental
4,5,6,7-Tetrahydroisoindole31 and pyrrole-2-carboxaldehyde32 were prepared according to published synthetic protocols. 9-Phenylanthracene, 4-formylphenylboronic acid, bis(benzonitrile)palladium(II) chloride, and DDQ, were purchased from Sigma-Aldrich. The handling of all air/water sensitive materials was carried out using standard high vacuum techniques. All solvents and reagents were obtained from commercial sources and used as received. Extra dry DMF, toluene and THF were purchased from Sigma-Aldrich. Triethylamine and DCM were distilled from CaH2. Where mixtures of solvents were used, ratios are reported by volume. Column chromatography was carried out on silica gel 60 at normal pressure. NMR spectra were recorded on Bruker DPX 250, Bruker AC300 NMR and Bruker Avance 500 spectrometers, with the solvent proton or carbon signal as an internal standard. Elemental analysis was carried out using a Foss Heraeus Vario EL. Electronic absorption spectra were recorded on a PerkinElmer Lambda 25 instrument. MALDI-TOF spectra were obtained on a Bruker Reflex spectrometer III instrument using dithranol as a matrix. HR ESI spectrometry was performed on a QTof Ultima 3 Fa. Melting points were determined on a Büchi hot stage apparatus and are uncorrected. The emission spectra and phosphorescence lifetime of substances were measured using our home-built spectrometer.33 For the excitation and irradiation of studied materials a temperature stabilized diode laser with λ = 638 nm (QL63H5S, average power 20 mW) was used. In order to irradiate the samples in the quartz cuvettes (2 × 1 × 1 cm), the laser beam size was increased using a negative lens, resulting in the average intensity of 5 μW cm−2 on the entrance surface of the cuvette for 638 nm laser. The suppression of the excitation radiation during phosphorescence spectrum measurements was done using a notch filter NF 03-633U-25 or NF03-532E-25 (Semrock Inc.).10-Phenyl-9-bromoanthracene 3
The title compound was prepared following a modified literature procedure.34 9-Phenylanthracene (3.5 g, 13.8 mmol) was dissolved in acetic acid (150 mL). The solution was heated to 65 °C under nitrogen, and bromine (2.2 g, 13.8 mmol) was added dropwise over a period of 10 min. The reaction mixture was allowed to cool to room temperature and evaporated in vacuum. The residual solid was recrystallized from ethanol to give 10-phenyl-9-bromoanthracene (4.59 g, 95%) as yellow crystals with m.p. 154–155 °C (lit. 153–155 °C). 1H NMR (250 MHz, CD2Cl2) δ 8.64–8.57 (m, 2H), 7.69–7.55 (m, 7H), 7.44–7.35 (m, 4H).Porphyrin 6
Butyllithium (1.6 M solution in hexane, 3.1 mL, 5 mmol) was added dropwise to a mixture of 10-phenyl-9-bromoanthracene (1.5 g, 4.5 mmol) in diethyl ether (30 mL) and the mixture was stirred at room temperature for 30 min. Then a solution of pyrrole-2-carboxaldehyde (0.285 g, 3 mmol) in diethyl ether (5 mL) was added, the resulting solution was stirred for 1 h and poured into water (100 mL). The organic phase was separated, washed with water (3 × 10 mL) and evaporated in vacuum. The residue was then dissolved in a mixture of toluene (15 mL) and propionic acid (5 mL). The reaction mixture was stirred for 3 h at 100 °C then allowed to cool to room temperature. Solvents were evaporated in vacuum, the residue was then redissolved in CHCl3 and passed through a filter with silica. The first red-brown fraction was collected and the solvents were removed. The crude product was recrystallized from DCM–methanol to give porphyrin 6 (0.148 g, 15% with respect to pyrrole-2-carboxaldehyde) as a red-brown solid. 1H NMR (300 MHz, CD2Cl2) δ 8.40–8.34 (m, 8H), 7.96 (d, J = 8.9 Hz, 8H), 7.78–7.65 (m, 20H), 7.40 (ddd, J = 8.9, 6.0, 1.6 Hz, 8H), 7.29–7.17 (m, 16H). 13C NMR (75 MHz, CD2Cl2) δ 147.63, 141.88, 139.16, 135.74, 132.58, 131.87, 130.50, 130.22, 129.22, 128.57, 127.97, 127.77, 127.64, 125.99, 125.87, 119.50. UV/vis (toluene) λmax (logε): 430 (5.54), 519 (4.58), 551 (4.05), 592 (4.09), 649 (3.73). MALDI-TOF: m/z found 1319.4076, calcd for [M+] C100H62N4 1319.5008.
4-(10-Phenyl-anthracen-9-yl)-benzaldehyde 8
The title compound was prepared following a modified literature procedure.35 A mixture of 10-phenyl-9-bromoanthracene (2.5 g, 7.5 mmol), 4-formylphenylboronic acid (1.35 g, 9 mmol), tetrakis(triphenylphosphine)palladium (0.433 g, 0.375 mmol), K2CO3 (2.48 g, 18 mmol), benzene (100 mL), ethanol (20 mL) and water (40 mL) was refluxed under nitrogen for 24 h. The organic phase was separated, washed with brine, and dried over Na2SO4. The solvent was evaporated in vacuum and the remaining solid was purified on a silica gel column using the EtOAc–pentane (1:5) mixture as an eluent. Fractions containing the product (determined by TLC) were concentrated to give the aldehyde (2.36 g, 88%) as yellow crystals with m.p. 234–236 °C. 1H NMR (250 MHz, C2D2Cl4) δ 10.20 (s, 1H), 8.15 (d, J = 8.1 Hz, 2H), 7.76–7.68 (m, 4H), 7.66–7.57 (m, 5H), 7.50 (dd, J = 7.6, 1.7 Hz, 2H), 7.42–7.33 (m, 4H).
Porphyrin 10
4,5,6,7-Tetrahydroisoindole (0.3 g, 2.48 mmol) was dissolved in CH2Cl2 (250 mL) freshly distilled from CaH2, and 4-(10-phenyl-anthracen-9-yl)-benzaldehyde (0.888 g, 2.48 mmol) was added. The mixture was stirred under nitrogen for 10 min in the dark at room temperature. BF3·Et2O (0.035 g, 0.248 mmol) was added in one portion, and the mixture was stirred for an additional 2 h. DDQ (0.422 g, 1.86 mmol) was added followed by additional stirring for 2 h in aqueous Na2SO3, dried over Na2SO4 and concentrated in vacuum. The residue was purified on a silica gel column (eluent CH2Cl2, then CH2Cl2–AcOH, green band collected). Additional purification by repetitive precipitation from CH2Cl2–AcOH (10:1) with hexane delivered the product (0.279 g, 23%) as a dark-green powder. 1H NMR (250 MHz, CD2Cl2) δ 8.80 (d, J = 7.9 Hz, 2H), 8.10–8.01 (m, 4H), 7.84–7.77 (m, 2H), 7.73–7.40 (m, 9H), 3.09 (d, J = 17.5 Hz, 8H), 2.44 (d, J = 17.6 Hz, 8H), 2.03 (d, J = 5.4 Hz, 8H), 1.63–1.35 (m, 14H). 13C NMR (75 MHz, CD2Cl2) δ 144.65, 141.27, 139.55, 138.53, 138.39, 137.31, 136.83, 135.96, 132.47, 131.91, 130.62, 130.46, 129.10, 128.23, 127.76, 127.30, 126.13, 125.80, 118.72, 25.51, 23.26. UV/vis (toluene) λmax (logε): 357 (4.64), 375 (4.79), 396 (4.81), 470 (5.45), 617 (4.06), 675 (4.45). MALDI-TOF: m/z found 1840.9465, calcd for [MH+] C140H103N4 1840.8216.
Porphyrin 10-Pd
Bis(benzonitrile)palladium(II) chloride (0.078 g, 0.204 mmol) and triethylamine (0.101 g, 1 mmol) were added to a solution of porphyrin 10 (0.2 g, 0.102 mmol) in benzonitrile (10 mL), and the mixture was heated at 180 °C for 1–2 h. The conversion was monitored by UV-vis spectroscopy (solvent CH2Cl2–TFA) and was considered complete after the absorption band of the dication at 470 nm disappeared. The mixture was allowed to cool then evaporated in vacuum to dryness. The residue was diluted with CH2Cl2, filtered through a thin layer of Celite to remove Pd black, and the solvent was evaporated. The product was purified by chromatography on silica gel using CH2Cl2 as an eluent (dark red band collected). The solvent was evaporated and the residue was either recrystallized from CH2Cl2–ether to give porphyrin 10-Pd (0.151 g, 76%) as a dark-brown powder. 1H NMR (300 MHz, C2D2Cl4) δ 8.48 (d, J = 7.4 Hz, 8H), 8.18 (d, J = 8.6 Hz, 8H), 7.88 (dd, J = 17.7, 8.1 Hz, 16H), 7.74–7.43 (m, 36H), 2.95 (s, 16H), 1.94 (s, 16H). 13C NMR (126 MHz, C2D2Cl4) δ 141.95, 141.16, 140.53, 139.08, 138.87, 137.66, 137.31, 136.92, 134.32, 131.36, 130.59, 130.08, 130.00, 128.87, 128.26, 128.05, 127.38, 127.09, 126.82, 125.16, 124.94, 120.24, 118.96, 29.42, 23.87. UV/vis (toluene) λmax (logε): 358 (4.37), 376 (4.48), 430 (5.11), 538 (4.21), 572 (4.09). HRMS (ESI): m/z found 1943.7002, calcd for [M+] C140H100N4Pd 1943.7016.
Porphyrin 11-Pd
Porphyrin 10-Pd (0.1 g, 0.051 mmol) was dissolved in THF (10 mL), DDQ (0.187 g, 0.82 mmol) was added, and the mixture was refluxed for 20–40 min. During refluxing the color changed from red-brown to deep green. The mixture was allowed to cool, diluted with CH2Cl2, washed with 10% aqueous solution of Na2SO3, with brine, and dried over Na2SO4. The solvent was removed in a vacuum, and the remaining solid was purified on a silica gel column using CH2Cl2 as an eluent. The first dark-green fraction was collected. The solvent was evaporated and the residue was recrystallized from CH2Cl2–Et2O to give the product (0.06 g, 60%) as blue-green crystals. 1H NMR (300 MHz, C2D2Cl4) δ 8.70 (d, J = 8.0 Hz, 8H), 8.46 (d, J = 8.8 Hz, 8H), 8.15 (d, J = 8.0 Hz, 8H), 7.90 (d, J = 8.8 Hz, 8H), 7.86–7.51 (m, 52H). 13C NMR (126 MHz, C2D2Cl4) δ 141.03, 139.63, 138.75, 138.35, 138.24, 137.65, 136.37, 133.99, 133.17, 132.52, 131.30, 129.97, 129.70, 128.46, 127.57, 127.35, 126.70, 125.60, 125.23, 124.42, 120.19, 117.93. UV/vis λmax(toluene)/nm (logε): 340 (4.29), 357 (4.4), 375 (4.53), 395 (4.54), 446 (5.33), 628 (4.89). HRMS (ESI): m/z found 1927.5749, calcd for [M+] C140H84N4Pd 1927.5764.
Acknowledgements
M. A. Filatov acknowledges the POLINNOVA project (FP7-REGPOT-2012-2013-1) for the financial support. I. Z. Ilieva acknowledges the DFNI E 02/11 – SunStore-project “Molecular solar thermal systems, enhanced by annihilation upconversion” of the Bulgarian National Science Fund for the financial support. S. Baluschev gratefully acknowledges the FCFP FRIAS COFUND Fellowship Programme (FP7-MCA-609305) for the financial support.Notes and references
- (a) Y. Sun, C. Borek, K. Hanson, P. I. Djurovich, M. E. Thompson, J. Brooks, J. J. Brown and S. R. Forrest, Appl. Phys. Lett., 2007, 90, 213503 CrossRef; (b) J. R. Sommer, R. T. Farley, K. R. Graham, Y. Yang, J. R. Reynolds, J. Xue and K. S. Schanze, ACS Appl. Mater. Interfaces, 2009, 1, 274 CrossRef CAS PubMed; (c) K. R. Graham, Y. Yang, J. R. Sommer, A. H. Shelton, K. S. Schanze, J. Xue and J. R. Reynolds, Chem. Mater., 2011, 23, 5305 CrossRef CAS; (d) F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707 CrossRef CAS PubMed; (e) F. Dumur, M. Lepeltier, B. Graff, E. Contal, G. Wantz, J. Lalevee, C. R. Mayer, D. Bertin and D. Gigmes, Synth. Met., 2013, 182, 13 CrossRef CAS; (f) C. S. Oh and J. Y. Lee, Dyes Pigm., 2013, 99, 374 CrossRef CAS; (g) A. M. Bunzli, H. J. Bolink, E. C. Constable, C. E. Housecroft, J. M. Junquera-Hernandez, M. Neuburger, E. Orti, A. Pertegas, J. J. Serrano-Perez, D. Tordera and J. A. Zampese, Dalton Trans., 2014, 43, 728 RSC.
- (a) T. V. Esipova, A. Karagodov, J. Miller, D. F. Wilson, T. M. Busch and S. A. Vinogradov, Anal. Chem., 2011, 83, 8756 CrossRef CAS PubMed; (b) A. Y. Lebedev, A. V. Cheprakov, S. Sakadzic, D. A. Boas, D. F. Wilson and S. A. Vinogradov, ACS Appl. Mater. Interfaces, 2009, 1, 1292 CrossRef CAS PubMed; (c) O. S. Finikova, A. Galkin, V. Rozhkov, M. Cordero, C. Hagerhall and S. A. Vinogradov, J. Am. Chem. Soc., 2003, 125, 4882 CrossRef CAS PubMed; (d) D. F. Wilson, W. M. F. Lee, S. Makonnen, O. S. Finikova, S. Apreleva and S. A. Vinogradov, J. Appl. Physiol., 2006, 101, 1648 CrossRef CAS PubMed; (e) Q. Zhao, M. X. Yu, L. X. Shi, S. J. Liu, C. Y. Li, M. Shi, Z. G. Zhou, C. H. Huang and F. Y. Li, Organometallics, 2010, 29, 1085 CrossRef CAS; (f) Q. Liu, B. R. Yin, T. S. Yang, Y. C. Yang, Z. Shen, P. Yao and F. Y. Li, J. Am. Chem. Soc., 2013, 135, 5029 CrossRef CAS PubMed; (g) M. Chen, Z. Lei, W. Feng, C. Y. Li, Q. M. Wang and F. Y. Li, Biomaterials, 2013, 34, 4284 CrossRef CAS PubMed; (h) K. Koren, R. Dmitriev, S. Borisov, D. Papkovsky and I. Klimant, ChemBioChem, 2012, 13, 1184 CrossRef CAS PubMed; (i) A. V. Kondrashina, R. I. Dmitriev, S. Borisov, I. Klimant, I. O'Brien, Y. M. Nolan, A. Zdanov and D. B. Papkovsky, Adv. Funct. Mater., 2012, 22, 4931 CrossRef CAS; (j) A. Fercher, S. Borisov, A. Zdanov, I. Klimant and D. B. Papkovsky, ACS Nano, 2011, 5, 5499 CrossRef CAS PubMed; (k) S. Hess, A. Becker, S. Baluschev, V. Yakutkin and G. Wegner, Macromol. Chem. Phys., 2007, 208, 2173 CrossRef CAS.
- (a) J. F. Sun, F. F. Zhong and J. Z. Zhao, Dalton Trans., 2013, 42, 9595 RSC; (b) S. Guo, L. H. Ma, J. Z. Zhao, B. Kucukoz, A. Karatay, M. Hayvali, H. G. Yaglioglu and A. Elmali, Chem. Sci., 2014, 5, 489 RSC; (c) J. F. Sun, F. F. Zhong, X. Y. Yi and J. Z. Zhao, Inorg. Chem., 2013, 52, 6299 CrossRef CAS PubMed; (d) J. Kyriakopoulos, A. T. Papastavrou, G. D. Panagiotou, M. D. Tzirakis, D. Manolis, K. S. Triantafyllidis, M. N. Alberti, K. Bourikas, C. Kordulis, M. Orfanopoulos and A. Lycourghiotis, J. Mol. Catal. A: Chem., 2014, 381, 9 CrossRef CAS; (e) K. Mori, Y. Kubota and H. Yamashita, Chem. – Asian J., 2013, 8, 3207 CrossRef CAS PubMed; (f) S. Tombe, E. Antunes and T. Nyokong, J. Mol. Catal. A: Chem., 2013, 371, 125 CrossRef CAS.
- (a) A. Turshatov, D. Busko, S. Baluschev, T. Miteva and K. Landfester, New J. Phys., 2011, 10, 083035 CrossRef; (b) C. Wohnhaas, A. Turshatov, V. Mailaender, S. Lorenz, S. Baluschev, T. Miteva and K. Landfester, Macromol. Biosci., 2011, 11, 772 CrossRef CAS PubMed; (c) C. Wohnhaas, V. Mailander, M. Droge, M. A. Filatov, D. Busko, Y. Avlasevich, S. Baluschev, T. Miteva, K. Landfester and A. Turshatov, Macromol. Biosci., 2013, 13, 1422 CrossRef CAS PubMed; (d) S. K. Sugunan, C. Greenwald, M. F. Paige and R. P. Steer, J. Phys. Chem. A, 2013, 117, 5419 CrossRef CAS PubMed; (e) X. Cao, B. Hu and P. Zhang, J. Phys. Chem. Lett., 2013, 4, 2334 CrossRef CAS; (f) J. S. Lissau, D. Nauroozi, M. P. Santoni, S. Ott, J. M. Gardner and A. Morandeira, J. Phys. Chem. C, 2013, 117, 14493 CrossRef CAS; (g) P. C. Boutin, K. P. Ghiggino, T. L. Kelly and R. P. Steer, J. Phys. Chem. Lett., 2013, 4, 4113 CrossRef CAS; (h) S. Borisov, R. Saf, R. Fischer and I. Klimant, Inorg. Chem., 2013, 52, 1206 CrossRef CAS PubMed; (i) Y. Y. Cheng, B. Fuckel, T. Khoury, R. G. C. R. Clady, M. J. Y. Tayebjee, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, J. Phys. Chem. Lett., 2010, 1, 1795 CrossRef CAS.
- Y. Y. Cheng, B. Fuckel, R. W. MacQueen, T. Khoury, R. G. C. R. Clady, T. F. Schulze, N. J. Ekins-Daukes, M. J. Crossley, B. Stannowski, K. Lips and T. W. Schmidt, Energy Environ. Sci., 2012, 5, 6953 CAS.
- M. Filatov, S. Ritz, I. Ilieva, V. Mailander, K. Landfester and S. Baluschev, SPIE Newsroom. DOI: http://10.1117/2.1201403.005378, published online: April 7, 2014, http://spie.org/x106642.xml.
- C. Schweitzer and R. Schmidt, Chem. Rev., 2003, 103, 1685 CrossRef CAS PubMed.
- (a) R. R. Islangurov, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2007, 129, 12652 CrossRef PubMed; (b) S. Hess, M. Demir, V. Yakutkin, S. Baluschev and G. Wegner, Macromol. Rapid Commun., 2009, 30, 394–401 CrossRef CAS PubMed; (c) A. J. Tilley, M. J. Kim, M. Chen and K. P. Ghiggino, Polymer, 2013, 54, 2865 CrossRef CAS; (d) E. Stanislovaityte, J. Simokaitiene, S. Raisys, H. Al-Attar, J. V. Grazulevicius, A. P. Monkman and V. Jankus, J. Mater. Chem. C, 2013, 1, 8209 RSC.
- (a) H.-C. Chen, C.-Y. Hung, K.-H. Wang, H.-L. Chen, W. S. Fann, F.-C. Chien, P. Chen, T. J. Chow, C.-P. Hsu and S.-S. Sun, Chem. Commun., 2009, 4064 RSC; (b) C. Wohnhaas, K. Friedemann, D. Busko, K. Landfester, S. Baluschev, D. Crespy and A. Turshatov, ACS Macro Lett., 2013, 2, 446 CrossRef CAS; (c) Y. C. Simon and C. Weder, Chimia, 2012, 66, 878 CrossRef.
- Q. Liu, T. Yang, W. Feng and F. Li, J. Am. Chem. Soc., 2012, 22, 4360 Search PubMed.
- (a) N. J. Turro, G. Sidney and X. Li, Photochem. Photobiol., 1983, 37, 149 CrossRef CAS; (b) Z. W. Gao, X. Feng, L. Mu, X. L. Ni, L. L. Liang, S. F. Xue, Z. Tao, X. Zeng, B. E. Chapman, P. W. Kuchel, L. F. Lindoy and G. Wei, Dalton Trans., 2013, 42, 2608 RSC; (c) P. F. Duan, N. Yanai and N. Kimizuka, J. Am. Chem. Soc., 2013, 135, 19056 CrossRef CAS PubMed; (d) K. Tanaka, H. Okada, W. Ohashi, J. H. Jeon, K. Inafuku and Y. Chujo, Bioorg. Med. Chem., 2013, 21, 2678 CrossRef CAS PubMed; (e) F. Marsico, A. Turshatov, R. Peköz, Yu. Avlasevich, M. Wagner, K. Weber, D. Donadio, K. Landfester, S. Baluschev and F. R Wurm, J. Am. Chem. Soc., 2014, 136, 11057 CrossRef CAS PubMed.
- (a) S. A. Vinogradov, L.-W. Lo and D. F. Wilson, Chem. – Eur. J., 1999, 5, 1338 CrossRef CAS; (b) A. Y. Lebedev, T. Troxler and S. A. Vinogradov, J. Porphyrins Phthalocyanines, 2008, 12, 1261 CrossRef CAS PubMed; (c) I. B. Rietveld, E. Kim and S. A. Vinogradov, Tetrahedron, 2003, 59, 3821 CrossRef CAS; (d) B. W. Pedersen, L. E. Sinks, T. Breitenbach, N. B. Schack, S. A. Vinogradov and P. R. Ogilby, Photochem. Photobiol., 2011, 87, 1077 CrossRef CAS PubMed.
- (a) A. Segura Carretero, C. Cruces Blanco and A. Fernandez Gutierrez, Anal. Sci., 1996, 12, 653 CrossRef; (b) A. Segura Carretero, C. Cruces Blanco, B. Canabate Diaz and A. Fernandez Gutierrez, Anal. Chim. Acta, 1998, 361, 217 CrossRef.
- R. R. Islangulov, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2007, 129, 12652 CrossRef CAS PubMed.
- F. Marsico, A. Turshatov, R. Peköz, Yu. Avlasevich, M. Wagner, K. Weber, D. Donadio, K. Landfester, S. Baluschev and F. R. Wurm, J. Am. Chem. Soc., 2014, 136, 11057 CrossRef CAS PubMed.
- A. J. Svagan, D. Busko, Yu. Avlasevich, G. Glasser, S. Baluschev and K. Landfester, ACS Nano, 2014, 8, 8198 CrossRef CAS PubMed.
- M. A. Filatov, S. Baluschev, I. Z. Ilieva, V. Enkelmann, T. Miteva, K. Landfester, S. E. Aleshchnkov and A. V. Cheprakov, J. Org. Chem., 2012, 77, 11119 CrossRef CAS PubMed.
- D. Eastwood and M. Gouterman, J. Mol. Spectrosc., 1970, 35, 359 CrossRef CAS.
- J. Zhao, S. Jia and H. Guoa, RSC Adv., 2011, 1, 937 RSC.
- (a) J.-M. Aubry, C. Pierlot, J. Rigaudy and R. Schmidt, Acc. Chem. Res., 2003, 36, 668 CrossRef CAS PubMed; (b) L. Slavetinska, J. Mosinger and P. Kubat, J. Photochem. Photobiol., A, 2008, 195, 1 CrossRef CAS.
- (a) D. Zehm, W. Fudickar and T. Linker, Angew. Chem., Int. Ed., 2007, 46, 7689 CrossRef CAS PubMed; (b) W. Jiang, M. Han, H.-Y. Zhang, Z.-J. Zhang and Y. Liu, Chem. – Eur. J., 2009, 15, 9938 CrossRef CAS PubMed; (c) W. Fudickar and T. Linker, Chem. – Eur. J., 2006, 12, 9276 CrossRef CAS PubMed; (d) D. Zehm, W. Fudickar, M. Hans, U. Schilde, A. Kelling and T. Linker, Chem. – Eur. J., 2008, 14, 11429 CrossRef CAS PubMed; (e) W. Fudickar and T. Linker, Chem. – Eur. J., 2011, 17, 13661 CrossRef CAS PubMed; (f) C. G. Collins, J. M. Baumes and B. D. Smith, Chem. Commun., 2011, 47, 12352 RSC; (g) W. Fudickar and T. Linker, J. Am. Chem. Soc., 2012, 134, 15071 CrossRef CAS PubMed.
- (a) G. R. Martinez, J.-L. Ravanat, M. H. G. Medeiros, J. Cadet and P. Di Mascio, J. Am. Chem. Soc., 2000, 122, 10212 CrossRef; (b) D. Costa, E. Fernandes, J. L. M. Santos, D. C. G. A. Pinto, A. M. S. Silva and J. L. F. C. Lima, Anal. Bioanal. Chem., 2007, 387, 2071 CrossRef CAS PubMed.
- (a) M. Matsumoto, M. Yamada and N. Watanabe, Chem. Commun., 2005, 483 RSC; (b) C. Changtonga, D. W. Carneya, L. Luoa, C. A. Zotoa, J. L. Lombardib and R. E. Connors, J. Photochem. Photobiol., A, 2013, 260, 9 CrossRef; (c) S. Benz, S. Notzli, J. S. Siegel, D. Eberli and H. J. Jessen, J. Med. Chem., 2013, 56, 10171 CrossRef CAS PubMed.
- V. Nardello and J.-M. Aubry, Methods Enzymol., 2000, 319, 50–58 CAS.
- A. Tohara and M. Sato, J. Porphyrins Phthalocyanines, 2007, 11, 513 CrossRef CAS.
- (a) M. Angrick and E. O. Riecken, Chem.-Ztg., 1985, 109, 308 CAS; (b) M. Davis, M. O. Senge and O. B. Locos, Z. Naturforsch., B: J. Chem. Sci., 2010, 65, 1472 CAS; (c) N. K. S. Davis, A. L. Thompson and H. L. Anderson, J. Am. Chem. Soc., 2011, 133, 30 CrossRef CAS PubMed; (d) M. O. Senge, Chem. Commun., 2011, 47, 1943 RSC.
- (a) N. K. S. Davis, M. Pawlicki and H. L. Anderson, Org. Lett., 2008, 10, 3945 CrossRef CAS PubMed; (b) N. K. S. Davis, A. L. Thompson and H. L. Anderson, Org. Lett., 2012, 12, 2124 CrossRef PubMed; (c) O. S. Finikova, A. V. Cheprakov and S. A. Vinogradov, J. Org. Chem., 2005, 70, 9562 CrossRef CAS PubMed; (d) Y. Zagranyarski, L. Chen, Y. Zhao, H. Wonneberger, C. Li and K. Mullen, Org. Lett., 2012, 14, 5444 CrossRef CAS PubMed.
- (a) O. M. Senge, Highly Substituted Porphyrins. The Porphyrin Handbook, Academic Press, Boston, 2011, vol. 1, p. 239 Search PubMed; (b) A. Y. Lebedev, M. A. Filatov, A. V. Cheprakov and S. A. Vinogradov, J. Phys. Chem. A, 2008, 112, 7723 CrossRef CAS PubMed.
- (a) O. S. Finikova, S. Y. Chernov, A. V. Cheprakov, M. A. Filatov, S. A. Vinogradov and I. P. Beletskaya, Dokl. Chem., 2003, 391, 222 CrossRef CAS; (b) M. A. Filatov, A. V. Cheprakov and I. P. Beletskaya, Eur. J. Org. Chem., 2007, 2468 Search PubMed; (c) M. A. Filatov, A. Y. Lebedev, S. A. Vinogradov and A. V. Cheprakov, J. Org. Chem., 2008, 73, 4175 CrossRef CAS PubMed; (d) M. A. Filatov and A. V. Cheprakov, Tetrahedron, 2011, 67, 3559 CrossRef CAS; (e) A. V. Cheprakov and M. A. Filatov, J. Porphyrins Phthalocyanines, 2009, 13, 291 CrossRef CAS.
- S. Borisov, G. Nuss, W. Haas, R. Saf, M. Schmuck and I. Klimant, J. Photochem. Photobiol., A, 2009, 201, 128 CrossRef CAS.
- O. S. Finikova, A. V. Cheprakov, I. P. Beletskaya, P. J. Carroll and S. A. Vinogradov, J. Org. Chem., 2004, 69, 522 CrossRef CAS PubMed.
- R. M. Silverstein, E. E. Ryskiewicz and C. Willard, Org. Synth., 1956, 36, 74 CrossRef CAS.
- A. Turshatov, D. Busko, Y. Avlasevich, T. Miteva, K. Landfester and S. Baluschev, ChemPhysChem, 2012, 13, 3112 CrossRef CAS PubMed.
- X.-M. Zhang, F. G. Bordwell, J. E. Bares, J.-P. Cheng and B. C. Petrie, J. Org. Chem., 1993, 58, 3051 CrossRef CAS.
- W. Fudickar and T. Linker, Chem. – Eur. J., 2006, 12, 9276 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, mass and optical spectroscopy data. See DOI: 10.1039/c4cp05025h |
| This journal is © the Owner Societies 2015 |