
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Orthogonal breaking and forming of dynamic covalent imine and disulfide bonds in aqueous solution†
Michael E.
Bracchi
and
David A.
Fulton
*
A Chemical Nanoscience Laboratory, School of Chemistry, Newcastle University, Bedson Building, Newcastle Upon Tyne, NE1 7RU, UK. E-mail: david.fulton@ncl.ac.uk; Fax: +44 (0)191 208 6929; Tel: +44 (0)191 208 7065
First published on 5th June 2015
Abstract
Orthogonal bond-breaking and forming of dynamic covalent disulfide and imine bonds in aqueous solution is demonstrated. Through judicious choice of reaction partners and conditions, it is possible to cleave and reform selectively these bonds in the presence of each other in the absence of unwanted competing processes.
The design and study of functional systems of molecules is an area of interest within the growing field of systems chemistry.1 An approach to increase system complexity is to exploit multiple orthogonal supramolecular interactions, yet only in recent years have chemists made significant efforts2 to emulate this feature which is ubiquitous in natural systems. Orthogonal interactions in functional systems3 have been exploited in increasingly complex systems e.g. molecular machines,4 interlocked molecules5 and responsive materials,6 and to continue this development there is a clear need for well-understood orthogonal interactions.
Chemical bonds which can reversibly break and reform in response to stimuli are well-known7 in chemistry, and these so-called “dynamic covalent bonds” (DCBs) can be utilized as “modules”8 to introduce stimuli-responsiveness into functional systems. Of particular interest to us is reversible imine and disulfide bonds. Imine bonds are formed from the condensation of amines and carbonyls (Fig. 1a), and the position of the equilibrium is pH dependent, with work by Lehn demonstrating9 that the position of the imine equilibrium can be shifted from almost complete imine to starting materials over about three pH units. Redox-sensitive disulfide bonds can be reduced to their corresponding thiols in the presence of a reducing stimulus, and re-oxidized to form the disulfide (Fig. 1b).10 Since pH and redox can be controlled independent of each other it should be possible to selectively cleave and reform one of these bonds in the presence of the other, and thus these bonds can be considered to be orthogonal.‡ Assuming complete orthogonality, and considering only situations in which DCBs are in either “broken” or “formed” states, there are four distinct scenarios. It is convenient to map these states onto a four-node network (Fig. 2) where each node represents one of four possible scenarios regarding whether the disulfide and imine bonds are “broken” or “formed” and the vertices display the orthogonal stimuli required to drive the bond forming and breaking processes. The condition for orthogonality is that it is possible to successfully navigate between all nodes through the application of orthogonal stimuli with no undesired reactions occurring between the molecules in the mixture i.e. alternative combinations of the reaction partners are not detectable by 1H NMR spectroscopy. In particular, the reaction between thiols and aldehydes to form hemithioacetals (Fig. 1c) was identified as a potential competing process, and our investigation showed that careful choice of reaction partners is necessary to avoid this problem.
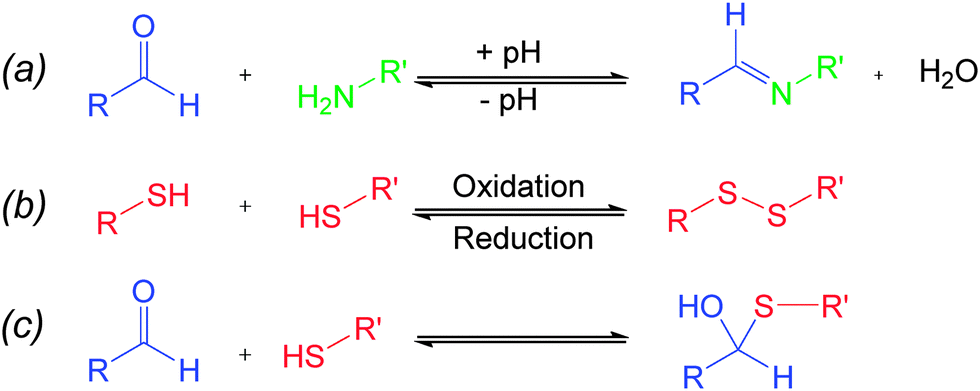 | ||
| Fig. 1 (a) pH-sensitive imine formation and hydrolysis, (b) redox-sensitive disulfide formation and cleavage, (c) hemithioacetal formation. | ||
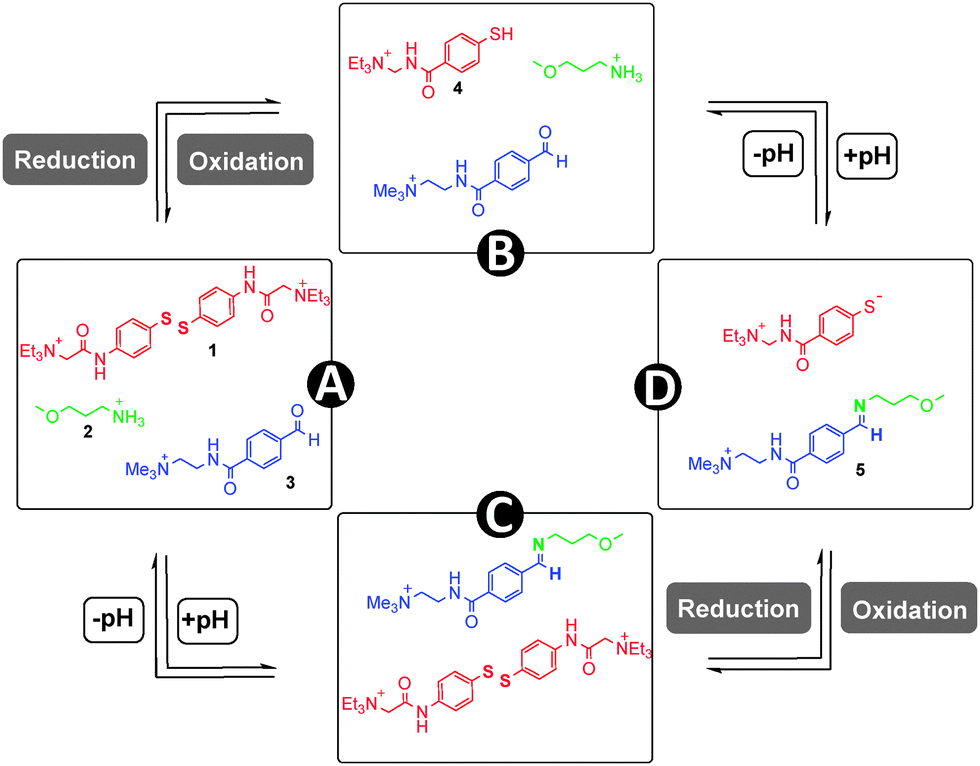 | ||
| Fig. 2 Considering only situations in which imine and disulfide DCBs are in either “broken” or “formed” states, there are four distinct scenarios which can be mapped onto a four-node network. In node A, the disulfide bond is “formed” and the imine “broken”. In node B both the disulfide and imine bonds are “broken”. In node C both the disulfide and imine bonds are “formed”, and in node D the disulfide bond is “broken” the imine “formed”. It is possible to successfully navigate between all nodes through the application of the appropriate orthogonal stimuli. | ||
In this work we investigate the orthogonality of the bond breaking and forming of imine and disulfide DCBs by establishing a set of conditions under which sequential application of stimuli allows interconversion between nodes within a 4-node network in the absence of unwanted, interfering processes.11 We also highlight how careful choice of reaction partners is required in order to avoid formation of undesired products and ‘cross-talk’ between our chosen DCB motifs.
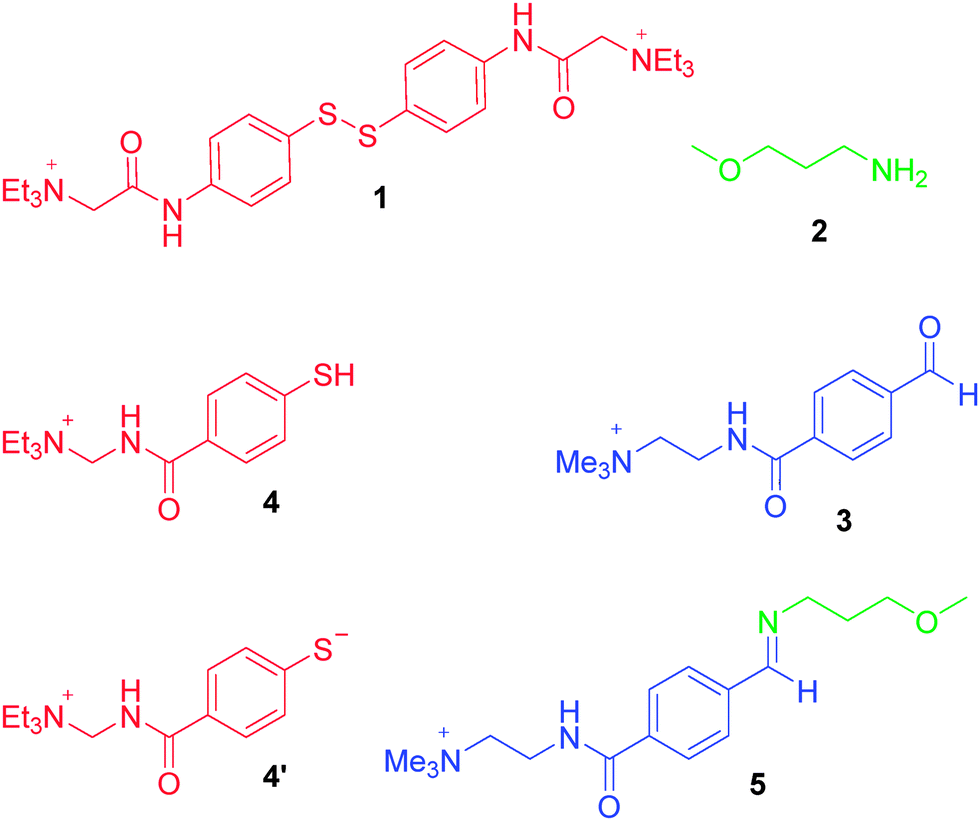
As a redox-sensitive DCB, we chose the disulfide 1 whose quaternary ammonium groups impart water solubility. It is possible to reversibly interconvert this species with thiol 4 upon application of reducing and oxidising agents. As a pH-sensitive DCB we chose imine 5, which can be reversibly interconverted into its water-soluble reaction partners amine 2 and aldehyde 3 by modulation of pH. The imine is formed almost exclusively at pH 12.0, and the reaction partners at pH 6.5.
The four-node network can be analyzed starting at any node, and for the sake of experimental simplicity we started at node A. A solution of disulfide 1, amine 2 and aldehyde 3 in D2O (15 mM of each of these three species) at pH 6.5 was prepared and analyzed by 1H NMR spectroscopy (Fig. 3a). The presence of aldehyde 3 is confirmed by a singlet at δ = 9.9 ppm and a pair of aromatic doublets at δ = 7.7–8.0 ppm. Signals at δ = 7.4 ppm indicates the presence of the aromatic disulfide 1. Importantly, the spectrum indicates disulfide 1 is stable at pH 6.5 and the absence of a signal at δ = 8.4 ppm indicates that there is no unwanted imine formed at this pH. Closer examination of this spectrum does reveal a second pair of doublets of extremely low intensity between δ = 7.5–7.7 ppm and a singlet at ∼δ = 6.0 ppm suggesting the presence of a small amount of hydrate. The hydrate of 3 exists merely as a “spectating” species in low concentration at pH 6.5 and does not influence the orthogonality of the imine and disulfide bonds.
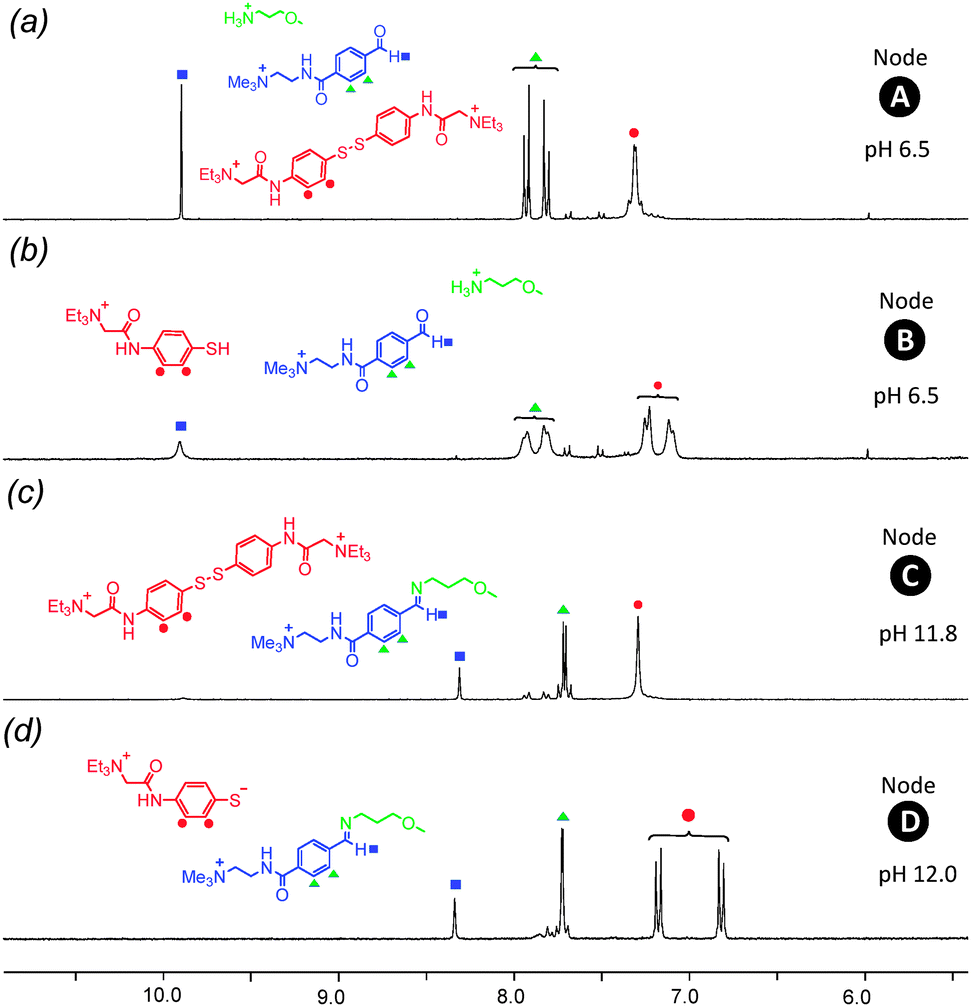 | ||
| Fig. 3 1H NMR spectra (300 MHz, D2O) collected at each ‘node’ in the 4-node network. (a) In node A, the disulfide bond is “formed” and the imine “broken”. (b) In node B both the disulfide and imine bonds are “broken”. (c) In node C both the disulfide and imine bonds are “formed”, (d) in node D the disulfide bond is “broken” the imine “formed”. | ||
To drive the transition from node A to node B, a reductive stimulus was applied through the addition of a slight excess of the organic reductant dithiothreitol (DTT). Analysis by 1H NMR spectroscopy (Fig. 3b) reveals the appearance of a broad multiplet at δ = 7.8 ppm associated with thiol 4 and the disappearance of the pair of doublets at δ = 7.3–7.5 ppm associated with disulfide 1. This observation indicates the successful and complete reduction of the disulfide 1 into thiol 4. The signals corresponding to aldehyde 3 remain unchanged suggesting no unwanted hemithioacetal formation has occurred as through reaction of thiol 4 with aldehyde 3. Furthermore, signals corresponding to amine 2 remain unchanged between nodes A to B (see ESI† for expanded spectra), indicating no unwanted processes occur involving 2. A degree of unexpected spectra broadening was observed which further NMR work indicates is attributable to dynamic processes (see ESI† Fig. S7, S8 for full details), however, this spectrum is still sufficiently informative to fully support the conclusions drawn.
The transition from node B to node D was driven by applying an increase in pH, serving as a stimulus to favor the condensation of 2 with 3 to form imine 5. The pH was raised from 6.5 to 12.0 using 10 μl aliquots of 0.1 M NaOH, and subsequent analysis by 1H NMR spectroscopy revealed (Fig. 3d) the disappearance of the aldehyde proton signal at δ = 9.9 ppm and emergence of a new singlet at δ = 8.4 ppm corresponding to imine proton in 5. This change was accompanied by the appearance of a new pair of aromatic doublets at δ = 6.8–7.2 ppm corresponding to the aromatic protons of imine 5 and the disappearance of the aromatic protons associated with aldehyde 3. These observations suggest near-quantitative formation of imine 5 from aldehyde 3 and amine 2 at pH 12.0 without the formation of any unwanted side-products. In particular, there is no evidence for the formation of unwanted hemithioacetal as observed by the absence of a singlet at ∼δ = 6.1–6.2 ppm. Because the increase in pH causes deprotonation of the aromatic thiol 4 resulting in thiolate 4′, its aromatic signals appear as a pair of doublets at δ = 6.7–7.2 ppm. It is also worthwhile to note that at pH 12 aldehyde 3 is not susceptible to hydrate formation, as evidenced by the lack of a signal at ∼δ = 6.0–6.1 ppm.
The transition from node D to node C was completed by slow addition of 0.25 M H2O2 to drive the oxidation of the thiolate 4′ whilst maintaining pH at 11.8. The resulting 1H NMR spectrum (Fig. 3c) displays a pair of aromatic doublets at δ = 7.3 ppm corresponding to aromatic protons in 1 and the disappearance of the pair of doublets corresponding to the thiolate 4′. A singlet corresponding to the imine proton at δ = 8.4 ppm of 5 accompanied by the characteristic pair of aromatic doublets at δ = 7.6 ppm suggests the imine bond has been successfully retained. The presence of low-intensity doublets at ∼δ = 8.0 ppm suggest the presence of a very small fraction of unreacted aldehyde 3. These observations indicate that successful oxidation of thiol 4 to form stable disulfide 1 at pH 11.8 in the presence of imine 5.
To ensure the complete reversibility of every step within the network, an anti-clockwise cycle was performed. 1H NMR spectroscopic analysis of the system at each node displayed the expected resonances, suggesting that the system is reversible at each transition (see ESI† Fig. S4).
Taken together, these experiments demonstrate the high degree of orthogonality which can be displayed in the bond breaking and forming processes of disulfide and imine DCBs.
To further explore the limitations of orthogonality, additional experiments were performed suggesting that the disulfide and imine bonds are orthogonal only below pH 12.0. At pH values higher than 12.0 unwanted decomposition of disulfide 1 to yield the thiolate 4′ occurs i.e. the stimuli which modulates the formation and hydrolysis of the imine bond is actually interfering with the disulfide bond. Direct attack by hydroxide resulting in cleavage of aromatic disulfide bonds has been reported by Danehy,12 and this undesired process sets an upper operational limit regarding pH upon the system.
Alternative reaction partners investigated by us failed to deliver orthogonality. When electron-rich aldehydes such as 4-hydroxybenzaldehyde were used, it was found that there is very little reaction with amine 2 to form imine at pH 12.0. The chosen electron-deficient aromatic aldehyde 3 appears to be both relatively resistant to hydrate formation and is capable of forming imines at pH 12.0. A series of alkyl thiols were also investigated as substitutes for thiol 4. At pH 6.5 in D2O these thiols engaged in nucleophilic attack at aldehyde 3, with further 1H NMR spectroscopic studies providing evidence for the formation of unwanted hemithioacetals (Fig. 4). The propensity to form hemithioacetals with electron-deficient aldehydes rules alkyl thiols out as potential thiol-disulfide system components. The chosen aromatic thiol 4 does not form hemithioacetal with aldehyde 3, probably on account of aromatic thiols being poorer nucleophiles than alkyl thiols and thus less likely to form hemithioacetals.
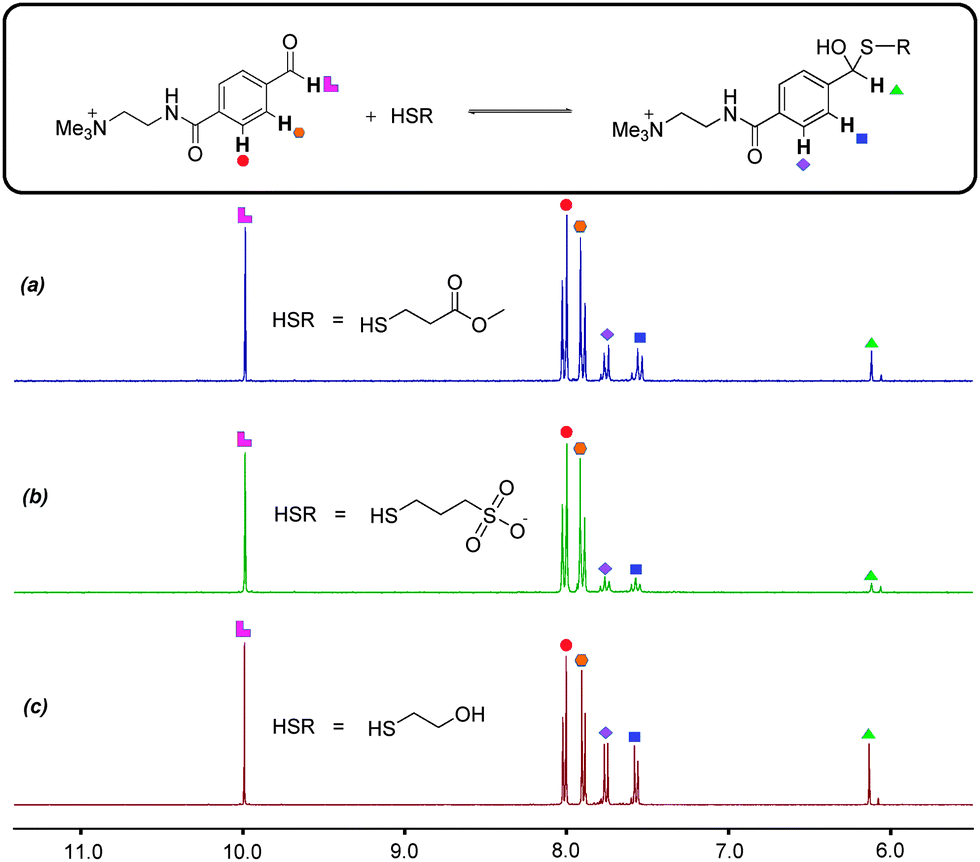 | ||
| Fig. 4 1H NMR spectra (300 MHz, D2O) demonstrating formation of hemithioacetal at pH 6.5. Aldehyde 3 and RSH present in 15 mM concentration (30 mM total). RSH = (a) 2-mercaptoethyl acetate, (b) sodium 3-mercapto-1-propanesulfonate, (c) 2-mercaptoethanol. The low intensity singlet observed just upfield of the diagnostic hemithioacetal signal at δ = 6.2 ppm indicates the presence of expected hydrate formed by reaction the aldehyde with water. | ||
In summary, our model investigation has shown the orthogonal nature of the bond-forming and bond-breaking processes of imine and disulfide DCBs. This study highlights the importance of carefully testing orthogonal systems through the application of well-considered models, and that careful choice of reaction partners is important to ensure the absence of any unwanted competing processes. We are now applying this knowledge to develop multi-stimuli responsive polymer materials.
We thank Dr Corinne Wills and Prof. William McFarlane for invaluable help and guidance with NMR spectroscopy.
Notes and references
- R. F. Ludlow and S. Otto, Chem. Soc. Rev., 2008, 37, 101–108 RSC
.
- M. L. Saha, S. De, S. Pramanik and M. Schmittel, Chem. Soc. Rev., 2013, 42, 6860–6909 RSC
- A. Wilson, G. Gasparini and S. Matile, Chem. Soc. Rev., 2014, 43, 1948–1962 RSC
- M. von Delius, E. M. Geertsema and D. A. Leigh, Nat. Chem., 2010, 2, 96–101 CrossRef CAS PubMed
- M. A. A. Garcıa and N. Bampos, Org. Biomol. Chem., 2013, 11, 27–30 Search PubMed
- A. W. Jackson and D. A. Fulton, Macromolecules, 2012, 45, 2699–2708 CrossRef CAS
-
(a) W. Zhang, R. J. Denman, C. Yu and Y. Jin, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC
- J. M. Spruell and C. J. Hawker, Chem. Sci., 2011, 2, 18–26 RSC
- C. Godoy-Alcántar, A. K. Yatsimirsky and J. M. Lehn, J. Phys. Org. Chem., 2005, 18, 979–985 CrossRef PubMed
- B. T. Tuten, D. Chao, C. K. Lyon and E. B. Berda, Polym. Chem., 2012, 3, 3068–3071 RSC
- Although this work has a focus upon orthogonality in bond forming and bond breaking processes, it is important highlight that orthogonality of component exchange processes, which have been utilized within dynamic combinatorial libraries or other supramolecular systems, has previously been reported. For further information, see:
(a) A. M. Escalante, A. G. Orrillo, I. Cabezudo and R. L. E. Furlan, Org. Lett., 2012, 14, 5816–5819 CrossRef CAS PubMed
- J. P. Danehy and K. N. Parameswaran, J. Org. Chem., 1968, 33, 568–572 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc02716k |
| ‡ We follow the definition of orthogonality as proposed2 by Schmittel et al. as “two (or more) dynamic interactions without crosstalk”, where without cross-talk denotes that “two or more interactions are mutually compatible and that alternative combinations are not detectable by means of the applied spectroscopic techniques”. |
| This journal is © The Royal Society of Chemistry 2015 |