 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The one pot synthesis of heterobimetallic complexes from a homoditopic pyrimidine–hydrazone ligand†
Daniel J. Hutchinson,
Lyall R. Hanton* and
Stephen C. Moratti
Department of Chemistry, University of Otago, PO Box 56, Dunedin, New Zealand. E-mail: lhanton@chemistry.otago.ac.nz
First published on 11th March 2014
Abstract
The symmetrical, homoditopic, pyrimidine-hydrazone (pym-hyz) ligand L1 was used to synthesise three new heterobimetallic complexes, CuPbL1(ClO4)4, CuAgL1(SO3CF3)3, and CuZnL1(SO3CF3)4. Each of the complexes was produced in a one-pot reaction in CH3CN, and was isolated in high yield and purity simply by precipitation through the addition of diethyl ether. Analysis was carried out by IR, UV-Vis and ESMS spectroscopy, as well as microanalysis. Crystals were also grown for the purposes of X-ray diffraction studies, which yielded the structures [CuPbL1(ClO4)(CH3CN)2(H2O)](ClO4)3 (1), [CuAgL1(SO3CF3)(CH3CN)2](SO3CF3)2·CH3CN (2), and CuZnL1(SO3CF3)2(CH3CN)(H2O)](SO3CF3)2·CH3CN (3), all of which were linear complexes containing a Cu(II) ion in one of the pym-hyz-py coordination sites, and either a Pb(II), Ag(I), or Zn(II) ion in the other.
Introduction
The cooperation of two different metal centres imbues heterobimetallic complexes with distinctly different physical and chemical properties from their mono- and homobimetallic analogues.1 As a result, synthetic heterobimetallics displaying novel properties have been extensively studied and exploited in the fields of catalysis,2 mixed spin magnetic systems,3 and molecular sensing and imaging.4 Additionally, they act as model compounds for studying the mechanisms and cooperative effects seen in important metalloenyzmes which utilise mixed metal ion active sites.5Supramolecular self-assembly can be employed to efficiently synthesise heterobimetallic complexes through the use of ditopic ligands with disparate metal binding sites which are selective towards different metal ions.6 The desired heterobimetallic complex can then be achieved by reacting the heteroditopic ligand with the metal ions in a sequential fashion. However, even with carefully designed heteroditopic ligands, these reactions often result in the formation of multiple complexes, necessitating yield limiting purification steps.7 Evidently, the synthesis of heterobimetallic complexes is not trivial and is aided by an adept understanding of molecular recognition.
We have previously reported that the addition of Cu(II) ions to a homoditopic pyrimidine-hydrazone (pym-hyz) ligand (L1) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 metal to ligand ratio resulted in discrete Cu(L1H)(ClO4)3 and CuL1(SO3CF3)2 complexes in which one of the coordination sites was occupied by a Cu(II) ion while the other one remained vacant (Fig. 1).8 Usually, ditopic pym-hyz ligands self-assemble into [2 × 2] grids when reacted with Cu(II) ions in a 1:1 metal to ligand9 ratio as the terpyridine like pym-hyz–py coordination pockets are well suited to binding octahedral metal ions in a coplanar mer fashion. However, the addition of hydroxymethyl arms to the terminal py rings of L1 results in tetradentate coordination to the Cu(II) ion, which prevents the perpendicular arrangement of ligand molecules required to form a grid complex.10
:1 metal to ligand ratio resulted in discrete Cu(L1H)(ClO4)3 and CuL1(SO3CF3)2 complexes in which one of the coordination sites was occupied by a Cu(II) ion while the other one remained vacant (Fig. 1).8 Usually, ditopic pym-hyz ligands self-assemble into [2 × 2] grids when reacted with Cu(II) ions in a 1:1 metal to ligand9 ratio as the terpyridine like pym-hyz–py coordination pockets are well suited to binding octahedral metal ions in a coplanar mer fashion. However, the addition of hydroxymethyl arms to the terminal py rings of L1 results in tetradentate coordination to the Cu(II) ion, which prevents the perpendicular arrangement of ligand molecules required to form a grid complex.10
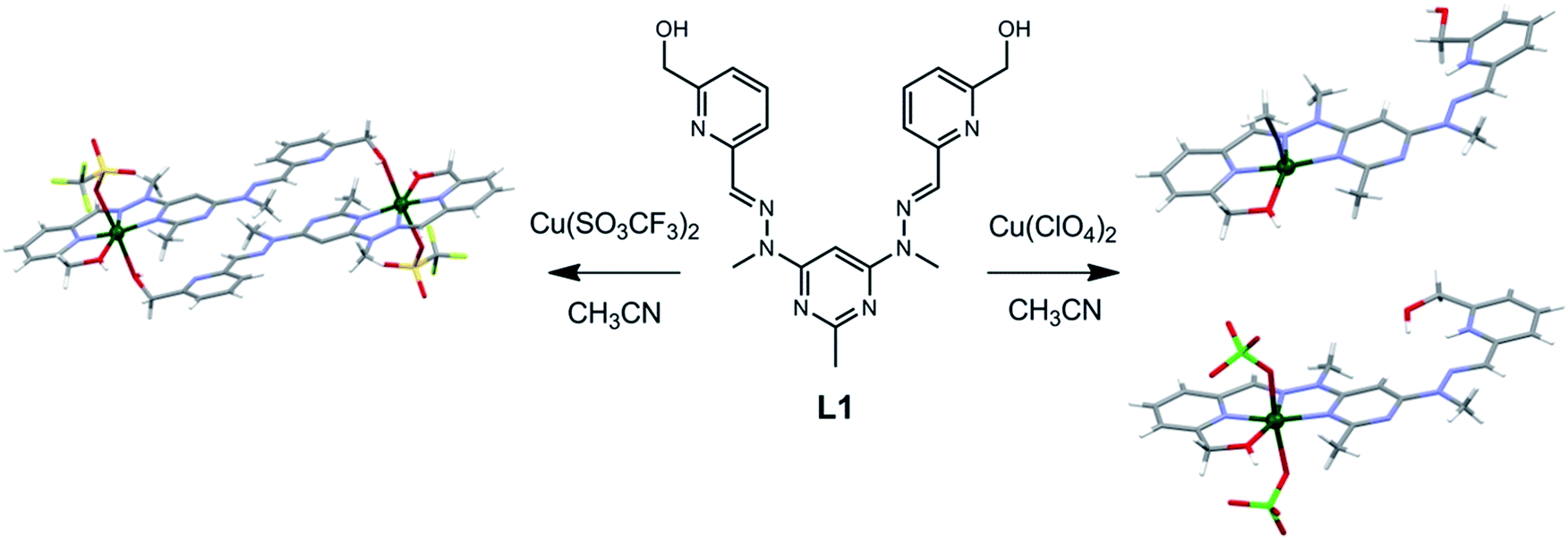 | ||
| Fig. 1 Solid state structures of the (right) Cu(L1H)(ClO4)3 and (left) CuL1(SO3CF3)2 complexes synthesised by reacting L1 with either Cu(ClO4)2 or Cu(SO3CF3)2 in a 1:1 metal to ligand ratio.8 | ||
At the time of publication we envisioned that these monocopper L1 complexes could be useful precursors in the formation of heterobimetallic complexes.8 The facile synthesis of L1 and the high purity and yield at which the monocopper complexes are synthesised makes them readily accessible. Herein we report that reacting the Cu(L1H)(ClO4)3 and CuL1(SO3CF3)2 complexes with either Pb(II), Ag(I) or Zn(II) ions resulted in the formation of heterobimetallic CuMn+L1A(2+n) complexes (where A = ClO4− or SO3CF3−; Fig. 2). Each of the complexes was formed in a one pot reaction in which L1 was first mixed with a solution of Cu(II) ions, before a solution of the other metal ion was added. The heterobimetallic complexes were isolated in high yield without the need for extensive purification steps. They were characterised by microanalysis, mass spectrometry, IR and UV-Vis spectroscopy, and X-ray crystallography.
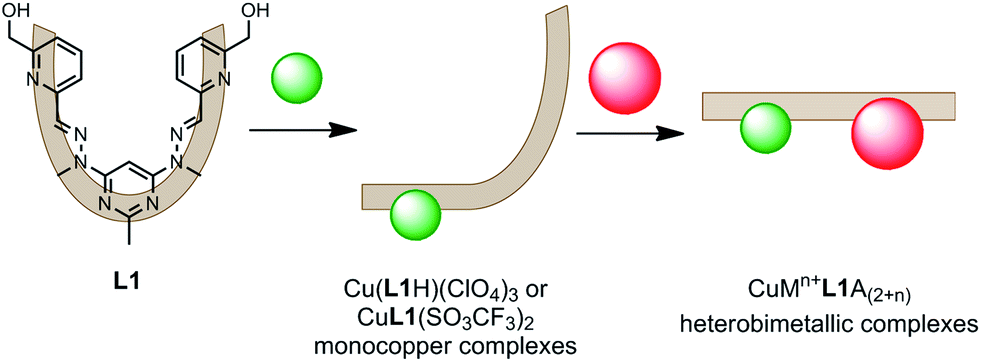 | ||
| Fig. 2 The two step, one pot synthetic strategy employed to form the heterobimetallic CuMn+L1A(2+n) complexes (where Mn+ = Pb(II), Ag(I) or Zn(II), and A = ClO4− or SO3CF3−). | ||
Results
Synthesis and analysis of complexes
The CuMn+L1A(2+n) heterobimetallic complexes were all produced in high yields and purity by first synthesising the monocopper Cu(L1H)(ClO4)3 or CuL1(SO3CF3)2 complexes, then filling the vacant coordination site with either a Pb(II), Ag(I) or Zn(II) ion (Fig. 2). As previously described, adding a CH3CN solution of either Cu(ClO4)2·6H2O or Cu(SO3CF3)2·4H2O to L1 in a 1:1 metal to ligand ratio resulted in dark green solutions of the monocopper complexes.8 Solutions of either Pb(ClO4)2·3H2O, AgSO3CF3, or Zn(SO3CF3)2 in CH3CN in a 1:1 metal to ligand ratio were then added, and the heterobimetallic complexes were isolated as green precipitates simply by adding diethyl ether to the resulting solutions.
The microanalytical results from the CuMn+L1A(2+n) precipitates were consistent with the formulae CuPbL1(ClO4)4, CuAgL1(SO3CF3)3·CH3CN and CuZnL1(SO3CF3)4, while their ESMS spectra showed peaks due to either the [PbL1–H]+, [CuAgL1(SO3CF3)–H]+, or [CuL1–H]+ molecular ions, respectively. The IR spectra of the CuPbL1(ClO4)4, CuAgL1(SO3CF3)3 and CuZnL1(SO3CF3)4 complexes showed an O–H stretching mode at either 3449, 3436 or 3324 cm−1, respectively, which were all typical frequencies of the Cu(II), Pb(II), Ag(I) and Zn(II) complexes of L1.8,11,12 The CuPbL1(ClO4)4 and CuAgL1(SO3CF3)3 complexes each showed two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching modes, one of which was at 1565 cm−1, while the other was at either 1541 or 1539 cm−1. The CuZnL1(SO3CF3)4 complex showed only the one CN stretching mode at 1564 cm−1, however it did have a distinct shoulder at 1554 cm−1.
N stretching modes, one of which was at 1565 cm−1, while the other was at either 1541 or 1539 cm−1. The CuZnL1(SO3CF3)4 complex showed only the one CN stretching mode at 1564 cm−1, however it did have a distinct shoulder at 1554 cm−1.
Samples of the complexes were dissolved in CH3CN and analysed by UV-Vis spectroscopy. Each of the complexes showed a single, broad, featureless d–d transition, in a similar fashion to the mono- and homobicopper L1 complexes.8 The d–d transitions of the CuPbL1(ClO4)4, CuAgL1(SO3CF3)3 and CuZnL1(SO3CF3)4 complexes had λmax values of either 697, 670, or 699 nm, and extinction coefficients of either 147, 144, or 135 L mol−1 cm−1, respectively. By comparison the transitions of the mono- and homobicopper complexes of L1 had λmax values of 656 nm and 699 nm, respectively, with extinction coefficients of 170 and 250 L mol−1 cm−1, respectively.8
X-ray crystallography
Crystals of the CuMn+L1A(2+n) complexes were also produced through the diffusion of diethyl ether vapour into the solutions, resulting in the solid state structures of [CuPbL1(ClO4)(CH3CN)2(H2O)](ClO4)3 (1), [CuAgL1-(SO3CF3)(CH3CN)2](SO3CF3)2·CH3CN (2), and [CuZnL1-(SO3CF3)2(CH3CN)–(H2O)](SO3CF3)2·CH3CN (3; Fig. 3–5). The overall structures of these complexes were similar to the previously reported Cu2L1A4, Pb2L1A4, Ag2L1A2 and Zn2L1A4 structures, as well as other pym–hyz ditopic complexes, in that they were linear with both pym–hyz–py bonds in the cisoid–cisoid conformation.8,11–13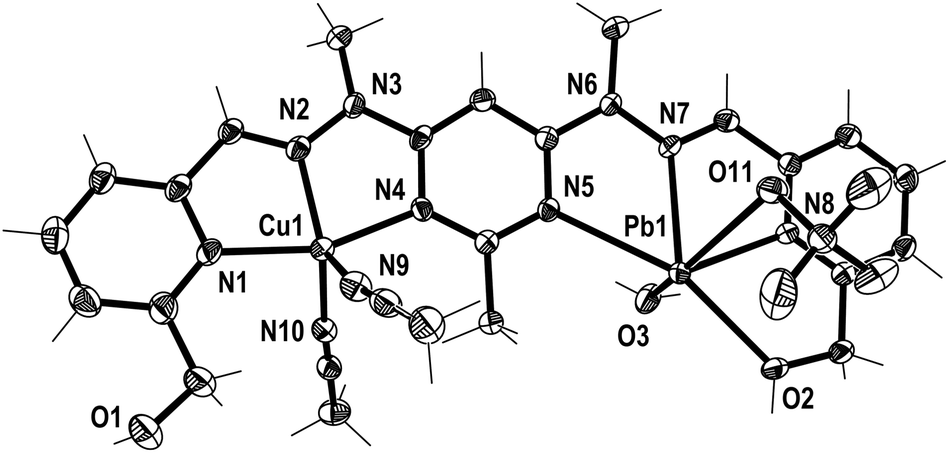 | ||
| Fig. 3 View of the [CuPbL1(ClO4)(CH3CN)2(H2O)]3+ cation of complex 1 (crystallographic numbering). Thermal ellipsoids are drawn at the 50% probability level. | ||
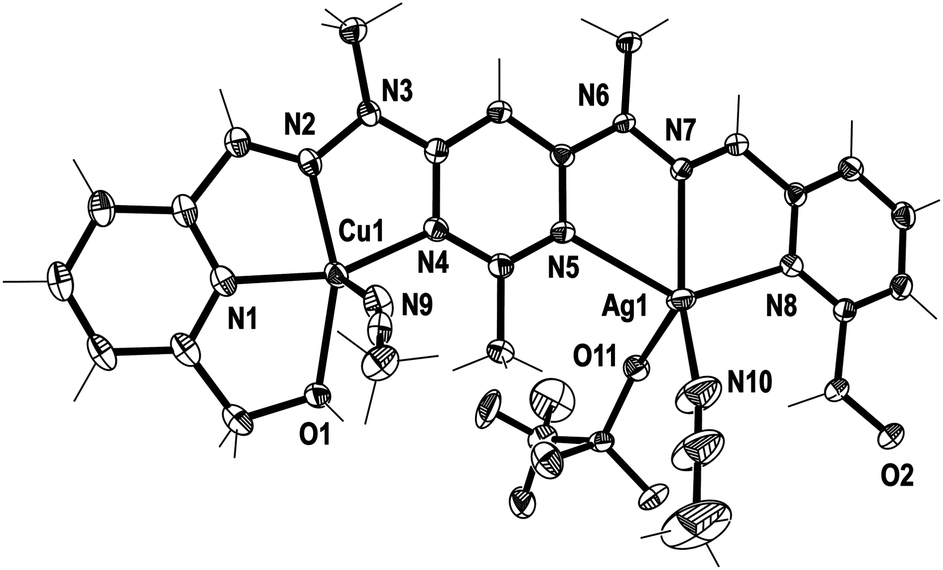 | ||
| Fig. 4 View of the [CuAgL1(SO3CF3)(CH3CN)2]2+ cation of complex 2 (crystallographic numbering). Thermal ellipsoids are drawn at the 50% probability level. | ||
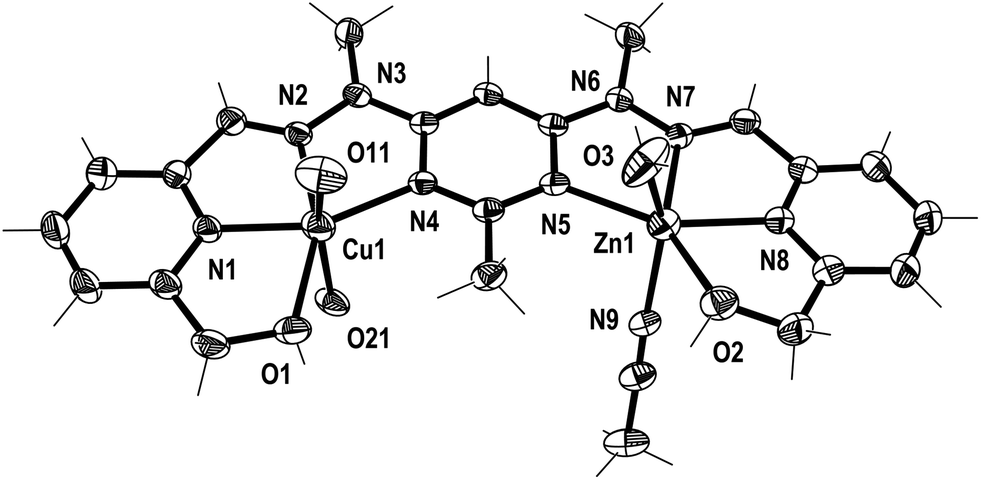 | ||
| Fig. 5 View of the [CuZnL1(SO3CF3)2(CH3CN)(H2O)]2+ cation of complex 3 (crystallographic numbering). Thermal ellipsoids are drawn at the 50% probability level (the SO3CF3− anions coordinated to Cu1 have been simplified to O11 and O21 for clarity). | ||
Additionally, in complexes 1 and 2, the halves of L1 which were bound to the Cu(II) ions were slightly curved in the mean plane of the central pym ring, while the halves bound to either Pb(II) or Ag(I) were straighter, as seen previously in the homobimetallic L1 complexes.8,11,12 As a result the lengths of complexes 1 and 2, 13.45 and 13.19 Å, respectively, were longer than the curved Cu2L1A48 complexes but shorter than the straight Pb2L1A411 and Ag2L1A212 complexes. Both halves of complex 3 were slightly curved in the plane of the central pym ring, resulting in a length of 12.82 Å, which was slightly longer than the Cu2L1A4 complexes and similar in length to the curved Zn2L1(SO3CF3)4 complex. The intermetallic distances of complexes 1, 2 and 3 were 6.55, 6.36 and 6.11 Å, respectively, which were longer than the Cu–Cu distances previously reported for L1 homobimetallic complexes, but shorter than the Pb–Pb, Ag–Ag and Zn–Zn distances.8,11,12
The crystal structures of the monocopper complexes of L1 all contained a Cu(II) ion bound to the hydroxymethyl arm and N donors of L1 in a tetradentate fashion.8 However, when comparing complexes 1–3 it appeared that the tendency of the hydroxymethyl arm to bind to the Cu(II) ion was controlled by whether the other hydroxymethyl arm was bound to the other metal ion. In complex 1 the Cu(II) ion was not bound to the hydroxymethyl arm, while the Pb(II) ion was, as has been consistently observed in other Pb(II) L1 complexes.11 Conversely, in complex 2 the hydroxymethyl arm was coordinated to the Cu(II) ion, but not to the Ag(I) ion, which commonly does not bind to the hydroxymethyl arms of L1.12 The asymmetric binding behaviour of the hydroxymethyl arms of L1 was previously observed in the Cu2L1A4 complexes in which only one of the Cu(II) ions in each complex was bound to the hydroxymethyl arms.8 In contrast, complex 3 had the hydroxymethyl arms bound to both the Cu(II) and Zn(II) ions.
In addition to the hydroxymethyl arms, each of the metals in 1–3 was bound to the three N donors of the pym–hyz–py sites, CH3CN solvent molecules, H2O molecules and either ClO4− or SO3CF3− anions. The Cu(II) ions in 1 and 2 adopted either a distorted trigonal bipyrimidal (τ5 = 0.59)14 or perfect square pyramidal (τ5 = 0.01)14 geometry, respectively. The Cu(II) ion in complex 3 had an octahedral geometry which was tetragonally distorted, with elongated bonds to the axially positioned SO3CF3− anions, as is typical of Jahn–Teller distorted octahedral Cu(II) ions.15 The geometries of the Pb(II) and Zn(II) ions in 1–3 were typical of other Pb(II), and Zn(II) complexes of L1. The Pb(II) ion in 1 adopted a six coordinate geometry, which resembled a distorted pentagonal bipyrimid due to the presence of a stereochemically active lone pair of electrons,16 while the Zn(II) ion in 3 was present in a distorted octahedral geometry. The Ag(I) ion in 2 occupied a distorted square pyramidal environment (τ5 = 0.25),14 which is a coordination geometry rarely displayed by Ag(I) ions.17
Both complexes 1 and 3 displayed H-bonding networks which arranged them into one dimensional chains (Fig. 6 and 7) while complex 2 was H-bonded into dimers (Fig. 8). The [CuPbL1(ClO4)(CH3CN)2(H2O)]3+ cations of complex 1 were joined by H-bonds between one of the ClO4− counterions, and the hydroxymethyl arm, and the ClO4− and the H2O molecule, both of which were bound to the Pb(II) ion. Additionally, there was an intermolecular anion–π interaction18 in complex 1 between the ClO4− anion bound to Pb1 and the central pym ring of L1. The distance from O14 to the centroid of the pym ring was 3.159(7) Å (Fig. 6). The H-bonding network of complex 3 involved bonds between the H2O molecules coordinated to Zn(II), the SO3CF3− anion bound to Cu(II) in a neighbouring [CuZnL1(SO3CF3)2(CH3CN)(H2O)]2+ cation, and a free SO3CF3− anion which was in turn H-bonded to one of the hydroxymethyl arms of another [CuZnL1(SO3CF3)2(CH3CN)(H2O)]2+ cation. The other hydroxymethyl arms were H-bonded to a CH3CN molecule. The [CuAgL1(SO3CF3)(CH3CN)2]2+ cations of complex 2 were paired into dimers by H-bonding between the hydroxymethyl arm coordinated to Cu(II) and the SO3CF3− anion coordinated to Ag(I) (Fig. 8).
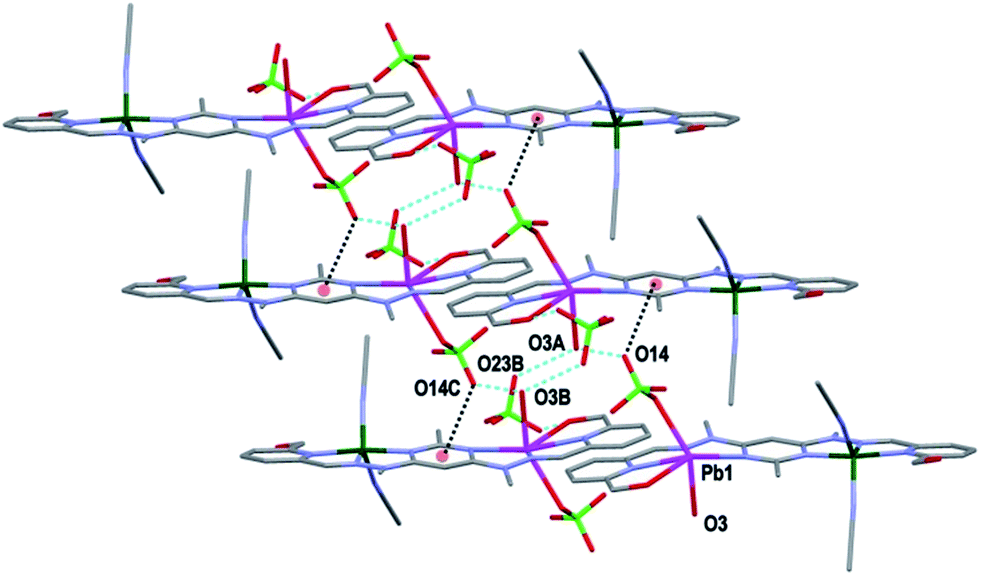 | ||
| Fig. 6 View of the arrangement of complex 1 into a one dimensional chain in the [1 0 0] direction through H-bonding. The anion-π interaction between O14 and the central pym rings is also shown (hydrogens have been omitted for clarity; symmetry codes (A) −1 + x, y, z; (B) −x, 1 − y, −z). | ||
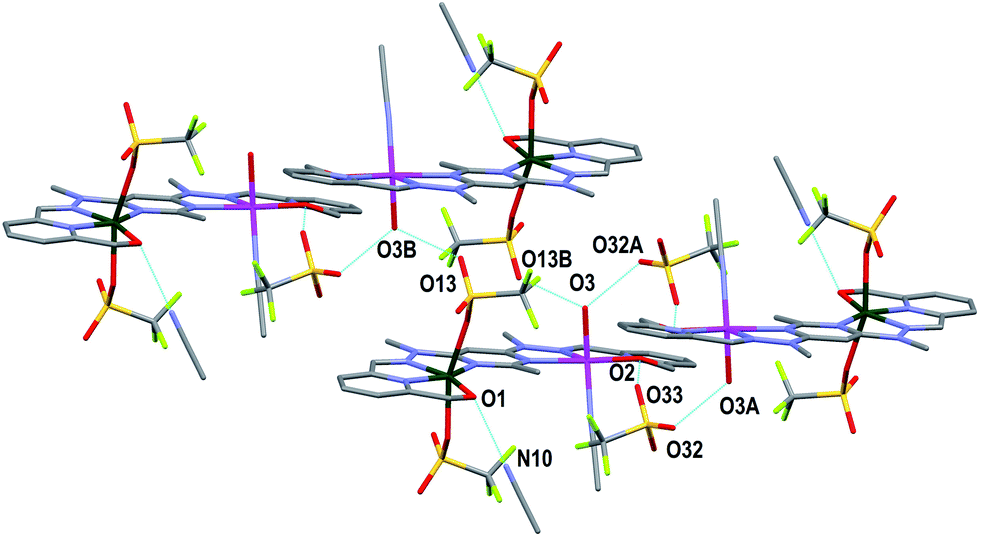 | ||
| Fig. 7 View of the arrangement of complex 3 into a one dimensional chain in the [1 0 0] direction through H-bonding. The H-bonding interaction between O1 and a CH3CN molecule is also shown (hydrogens have been omitted for clarity; symmetry codes A: −x, −y, 1 − z; B: 1 − x, −y, 1 − z). | ||
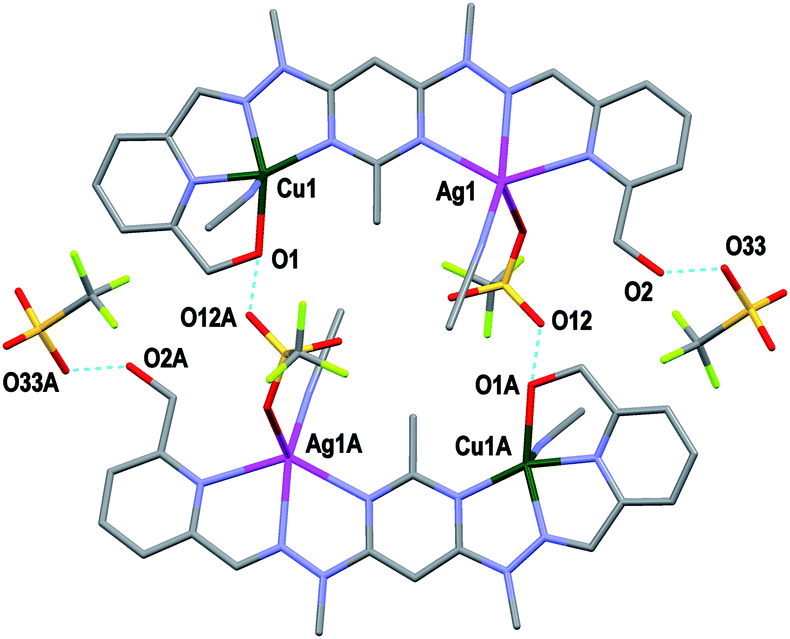 | ||
| Fig. 8 View of two [CuAgL1(SO3CF3)(CH3CN)2]2+ cations of complex 2 joined as a dimer through H-bonding. Also shown is the H-bonding between O2 and the third SO3CF3− anion (symmetry codes A: 2 − x, 1 − y, 1 − z). | ||
Experimental
General
All metal salts were purchased from commercial sources and were used as received without further purification with the exception of Cu(SO3CF3)2·4H2O, which was produced by the treatment of CuCO3·Cu(OH)2·H2O with aqueous triflic acid. Ligand L1 was synthesised according to its literature method.8 All solvents were used as received, and were of LR grade or better.Microanalyses were carried out in the Campbell Microanalytical Laboratory, University of Otago. All measured microanalysis results had an uncertainty of ±0.4. Electrospray mass spectrometry (ESMS) was carried out on a Bruker microTOFQ instrument (Bruker Daltronics, Bremen, Germany) by employing direct infusion into an ESI source in positive mode. Infrared (IR) spectra were recorded on a Bruker Alpha-P ATR-IR spectrometer. UV-Vis spectra were recorded on an Agilent 8453 spectrophotometer against a CH3CN background using quartz cells with a 1 cm path length.
CuPbL1(ClO4)4
Cu(ClO4)2·6H2O (28.4 mg, 0.0768 mmol) was dissolved in CH3CN (2.00 mL) and added to a suspension of L1 (32.0 mg, 0.0762 mmol) in CH3CN (2.00 mL) stirring at 75 °C. This resulted in complete dissolution of the ligand material and the formation of a clear, green solution. Pb(ClO4)2·3H2O (37.1 mg, 0.0807 mmol) in CH3CN (2.00 mL) was then added to the stirring solution at 75 °C. The solution was cooled to rt and diethyl ether (20.0 mL) was added, resulting in the precipitation of a green solid. The solid was washed with diethyl ether and dried in vacuo (63.9 mg, 78%): Anal. found: C 23.42; H 2.76; N 10.41. Calc. for C21H24N8O18Cl4CuPb: C 23.16; H 2.22; N 10.29. ESMS m/z found: 627.1653. Calc. for C21H23N8O2Pb+: 627.1707. Selected IR ν cm−1: 3449br (OH), 3067w (CH), 1600m, 1565m, 1541s (CN), 1497m, 1431m, 1386w, 1289m, 1269m, 1158m, 1032s (ClO4−). UV-Vis (CH3CN) λmax (ε)/nm (L mol−1 cm−1): 697 (147). Green crystals suitable for X-ray determination were grown by the slow diffusion of diethyl ether into a CH3CN solution of Cu(ClO4)2·6H2O, Pb(ClO4)2·3H2O and L1 in a 1:1:1 ratio. These crystals gave the structure [CuPbL1(ClO4)(CH3CN)2(H2O)](ClO4)3 (1).
CuAgL1(SO3CF3)3
As described for CuPbL1(ClO4)4 but with Cu(SO3CF3)2·4H2O (32. 3 mg, 0.0745 mmol), L1 (29.5 mg, 0.0701 mmol), and AgSO3CF3 (20.5 mg, 0.0801 mmol); green solid (58.8 mg, 83%): Anal. found: C 29.12; H 2.69; N 11.50. Calc. for C24H24N8O11F9S3CuAg·CH3CN: C 28.91; H 2.52; N 11.67. ESMS m/z found: 737.9842, 482.1249. Calc. for C21H23N8O2CuAg(SO3CF3)+: 737.9811. Calc. for C21H23N8O2Cu+: 482.1240. Selected IR ν cm−1: 3436br (OH), 3068w (CH), 1619m, 1599m, 1565m (CN), 1539m (CN), 1485m, 1456m, 1430m, 1384m, 1272s, 1235s (SO3CF3−), 1220s, 1152s, 1045m, 1023s. UV-Vis (CH3CN) λmax (ε)/nm (L mol−1 cm−1): 670 (144). Green crystals suitable for X-ray determination were grown by the slow diffusion of diethyl ether into a CH3CN solution of Cu(SO3CF3)2·4H2O, AgSO3CF3, and L1 in a 1:1:1 ratio. These crystals gave the structure [CuAgL1(SO3CF3)(CH3CN)2](SO3CF3)2·CH3CN (2).
CuZnL1(SO3CF3)4
As described for CuPbL1(ClO4)4 but with Cu(SO3CF3)2·4H2O (32.8 mg, 0.0758 mmol), L1 (31.2 mg, 0.0743 mmol), and Zn(SO3CF3)2 (27.3 mg, 0.0752 mmol); green solid (58.3 mg, 69%): Anal. found: C 26.32; H 2.49; N 9.84. Calc. for C25H24N8O14F12S4CuZn: C 26.21; H 2.11; N 9.78. ESMS m/z found: 482.1114. Calc. for C21H23N8O2Cu+: 482.1240. Selected IR ν cm−1: 3324br (OH), 3087w (CH), 1602m, 1564m (CN), 1489w, 1433w, 1387w, 1365w, 1272s, 1220s (SO3CF3−), 1155s, 1056m, 1023s. UV-Vis (CH3CN) λmax (ε)/nm (L mol−1 cm−1): 699 (135). Green crystals suitable for X-ray determination were grown by the slow diffusion of diethyl ether into a CH3CN solution of Cu(SO3CF3)2·4H2O, Zn(SO3CF3)2, and L1 in a 1:1:1 ratio. These crystals gave the structure CuZnL1(SO3CF3)2(CH3CN)(H2O)](SO3CF3)2·CH3CN (3).
X-ray crystallography information
Crystallographic data are summarised in Table 1, while selected bond lengths and angles for complexes 1–3 are available in the supporting information. X-ray diffraction data for complexes 1 and 2 were collected on a Bruker APEX II CCD diffractometer, with graphite monochromated Mo-Kα (λ = 0.71073 Å) radiation. Intensities for complexes 1 and 2 were corrected for Lorentz polarisation effects19 and a multiscan absorption correction20 was applied. The X-ray diffraction data for complex 3 were collected on an Agilent Supernova dual radiation source XRD with an Atlas detector and mirror monochromated Cu (λ = 1.5418 Å) radiation. The data for complex 3 was processed using Agilent CrysAlisPro software (version 1.171.36),21 and an analytical absorption correction22 was applied.| 1 | 2 | 3 | |
|---|---|---|---|
| Formula | C25H32Cl4CuN10O19Pb | C30H33AgCuF9N11O11S3 | C29H32CuF12N10O25S4Zn |
| M | 1189.16 | 1162.26 | 1245.80 |
| Crystal system | Triclinic | Triclinic | Triclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
| a/Å | 8.8553(8) | 10.0293(2) | 13.2894(2) |
| b/Å | 12.1810(11) | 14.0756(2) | 13.8254(3) |
| c/Å | 18.5657(16) | 16.8259(3) | 15.4186(2) |
| α/° | 88.849(4) | 70.9120(6) | 91.4555(15) |
| β/° | 84.339(4) | 87.5816(6) | 111.9600(15) |
| γ/° | 76.798(5) | 72.0006(6) | 115.552(2) |
| V/Å3 | 1940.2(3) | 2130.25(7) | 2310.61(9) |
| Z | 2 | 2 | 2 |
| T/K | 90(2) | 90(2) | 100(2) |
| μ mm−1 | 5.248 | 1.215 | 4.028 |
| Reflections collected | 25898 |
51464 |
52974 |
| Unique reflections (Rint) | 7205 (0.0221) | 7879 (0.0295) | 9368 (0.0295) |
| R1 indices [I > 2σ(I)] | 0.0361 | 0.0299 | 0.0601 |
| wR2 (all data) | 0.1069 | 0.0749 | 0.1659 |
| Crystal size/mm | 0.50 × 0.12 × 0.11 | 0.51 × 0.46 × 0.35 | 0.15 × 0.13 × 0.06 |
| Max theta | 25.50 | 25.50 | 66.49 |
| Goodness of fit | 1.095 | 1.057 | 1.077 |
The structures of complexes 1–3 were solved by direct methods (SHELXS23 or SIR-9724) and refined on F2 using all data by full-matrix least-squares procedures (SHELXL 9725). All calculations were performed using the WinGX interface.26 Detailed analyses of the extended structure were carried out using PLATON27 and MERCURY28 (Version 2.4).
Conclusions
The hetereobimetallic complexes CuPbL1(ClO4)4, CuAgL1(SO3CF3)3, and CuZnL1(SO3CF3)4 were synthesised by reacting the homoditopic ligand, L1, with either Cu(ClO4)2·6H2O or Cu(SO3CF3)2·4H2O, followed by either Pb(ClO4)2·3H2O, AgSO3CF3, or Zn(SO3CF3)2. The reactions were carried out in CH3CN, in a one pot reaction, and the heterobimetallic complexes were isolated in high yield and purity simply by precipitating them out of solution through the addition of diethyl ether. Crystals of the complexes were also grown through vapour diffusion of diethyl ether. The X-ray diffraction studies of these showed the solid state structure of the complexes were all linear, with a Cu(II) ion in one coordination pocket and either a Pb(II), Ag(I) or Zn(II) ion in the other. The ability of L1 to produce these complexes in high yield and purity from one pot reactions, despite its symmetrical, homoditopic nature, makes it an interesting addition to the array of ligands currently employed to synthesise heterobimetallic complexes.Acknowledgements
We thank the Department of Chemistry, University of Otago and the New Economic Research Fund of the Foundation for Research, Science and Technology (NERF Grant No UOO-X0808) for financial support.Notes and references
- (a) B. G. Cooper, J. W. Napoline and C. M. Thomas, Catal. Rev.: Sci. Eng., 2012, 54, 1–40 CrossRef CAS; (b) D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959 CrossRef CAS PubMed; (c) V. Ritleng and M. J. Chetcuti, Chem. Rev., 2007, 107, 797–858 CrossRef CAS PubMed; (d) N. Wheatley and P. Kalck, Chem. Rev., 1999, 99, 3379–3420 CrossRef CAS PubMed; (e) D. W. Stephan, Coord. Chem. Rev., 1989, 95, 41–107 CrossRef CAS.
- (a) W. Guan, S. Yamabe and S. Sakaki, Dalton Trans., 2013, 42, 8717–8728 RSC; (b) S. Gu, D. Xu and W. Chen, Dalton Trans., 2011, 40, 1576–1583 RSC; (c) A. Zanardi, J. A. Mata and E. Peris, J. Am. Chem. Soc., 2009, 131, 14531–14537 CrossRef CAS PubMed; (d) S. Maggini, Coord. Chem. Rev., 2009, 253, 1793–1832 CrossRef CAS PubMed; (e) A. Fihri, V. Artero, M. Razavet, C. Baffert, W. Leibl and M. Fontecave, Angew. Chem., Int. Ed., 2008, 47, 564–567 CrossRef CAS PubMed.
- (a) S. Öz, J. Titiš, H. Nazir, O. Atakol, R. Boča, I. Svoboda and H. Fuess, Polyhedron, 2013, 59, 1–7 CrossRef PubMed; (b) J. Černák, I. Kočanová and M. Orendá, Comments Inorg. Chem., 2012, 33, 2–54 CrossRef; (c) T. D. Pasatoiu, C. Tiseanu, A. M. Madalan, B. Jurca, C. Buhayon, J. P. Sutter and M. Andruh, Inorg. Chem., 2011, 50, 5879–5889 CrossRef CAS PubMed.
- (a) C.-F. Chow, M. H. W. Lam and W.-Y. Wong, Anal. Chem., 2013, 85, 8246–8253 CrossRef CAS PubMed; (b) A. Boulay, S. Laine, N. Leygue, E. Benosit, S. Laurent, L. V. Elst, R. N. Muller, B. Mestre-Voegtle and C. Picard, Tetrahedron Lett., 2013, 54, 5395–5398 CrossRef CAS PubMed.
- (a) L. M. K. Dassama, A. K. Boal, C. Krebs, A. C. Rosenzweig and J. M. Bollinger, J. Am. Chem. Soc., 2012, 134, 2520–2523 CrossRef CAS PubMed; (b) P. A. Lindahl, Inorg. Biochem., 2012, 172–178 CrossRef CAS PubMed; (c) V. R. I. Kaila, M. I. Verkhovsky and M. Wikström, Chem. Rev., 2010, 110, 7062–7081 CrossRef CAS PubMed; (d) W. Jiang, D. Yun, L. Saleh, E. W. Barr, G. Xing, L. M. Hoffart, M. A. Maslak, C. Krebs and J. M. Bollinger, Science, 2007, 316, 1188–1191 CrossRef CAS PubMed; (e) J. A. Tainer, E. D. Getzoff, J. S. Richardson and D. C. Richardson, Nature, 1983, 306, 284–287 CrossRef CAS.
- (a) S. K. Goforth, R. C. Walroth and L. McElwee-White, Inorg. Chem., 2013, 52, 5692–5701 CrossRef CAS PubMed; (b) M. R. Halvagar, B. Neisen and W. B. Tolman, Inorg. Chem., 2013, 52, 793–799 CrossRef CAS PubMed; (c) R. Maity, H. Koppetz, A. Hepp and F. E. Hahn, Inorg. Chem., 2013, 135, 4966–4969 CAS; (d) N. Wang, J.-S. Lu, T. M. McCormick and S. Wang, Dalton Trans., 2012, 41, 5553–5561 RSC; (e) Z.-L. You, Y. Lu, N. Zhang, B.-W. Ding, H. Sun, P. Hou and C. Wang, Polyhedron, 2011, 30, 2186–2194 CrossRef CAS PubMed; (f) D. Huang and R. H. Holm, J. Am. Chem. Soc., 2010, 132, 4693–4701 CrossRef CAS PubMed; (g) D. L. M. Suess and J. C. Peters, Chem. Commun., 2010, 46, 6554–6556 RSC.
- (a) Y. Sano, A. C. Weitz, J. W. Ziller, M. P. Hendrich and A. S. Borovik, Inorg. Chem., 2013, 52, 10229–10231 CrossRef CAS PubMed; (b) C. Uyeda and J. C. Peters, Chem. Sci., 2013, 4, 157–163 RSC.
- D. J. Hutchinson, L. R. Hanton and S. C. Moratti, Inorg. Chem., 2010, 49, 5923–5934 CrossRef CAS PubMed.
- (a) N. Parizel, J. Ramírez, C. Burg, S. Choua, M. Bernard, S. Gambarelli, V. Maurel, L. Brelot, J.-M. Lehn, P. Turek and A.-M. Stadler, Chem. Commun., 2011, 47, 10951–10953 RSC; (b) A. R. Stefankiewicz, J. Harrowfield, A. Madalan, K. Rissanen, A. N. Sobolev and J.-M. Lehn, Dalton Trans., 2011, 40, 12320–12332 RSC.
- (a) L. N. Dawe, K. V. Shuvaev and L. K. Thompson, Chem. Soc. Rev., 2009, 38, 2334–2359 RSC; (b) M. Ruben, J. Rojo, F. J. Romero-Salguero, L. H. Uppadine and J.-M. Lehn, Angew. Chem., Int. Ed., 2004, 43, 3644–3662 CrossRef CAS PubMed.
- D. J. Hutchinson, L. R. Hanton and S. C. Moratti, Inorg. Chem., 2011, 50, 7637–7649 CrossRef CAS PubMed.
- D. J. Hutchinson, S. A. Cameron, L. R. Hanton and S. C. Moratti, Inorg. Chem., 2012, 51, 5070–5081 CrossRef CAS PubMed.
- A.-M. Stadler, N. Kyritsakas, R. Graff and J.-M. Lehn, Chem. –Eur. J., 2006, 12, 4503–4522 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, Dalton Trans., 1984, 1349–1356 RSC.
- (a) B. Murphy and B. J. Hathaway, Coord. Chem. Rev., 2009, 243, 237–262 CrossRef; (b) B. J. Hathaway, in Comprehensive Coordination Chemistry: The synthesis, reactions, properties & applications of coordination compounds, ed. G. Wilkinson, R. Gillard and J. A. McCleverty, Pergamon Press, Oxford, 1987, vol. 5, ch. 53, pp. 594–600 Search PubMed; (c) H. A. Jahn and E. Teller, Proc. R. Soc. A, 1937, 161, 220–223 CrossRef CAS.
- (a) J. Harrowfield, Helv. Chim. Acta, 2005, 88, 2430–2432 CrossRef CAS PubMed; (b) L. Shimoni-Livny, J. P. Glusker and C. W. Bock, Inorg. Chem., 1998, 37, 1853–1867 CrossRef CAS.
- (a) D. J. Hutchinson, M. P. James, L. R. Hanton and S. C. Moratti, Inorg. Chem., 2014, 53, 2122–2132 CrossRef CAS PubMed; (b) K. Chainok, S. M. Neville, C. M. Forsyth, W. J. Gee, K. S. Murray and S. R. Batten, CrystEngComm, 2012, 14, 3717–3726 RSC; (c) A. G. Young and L. R. Hanton, Coord. Chem. Rev., 2008, 252, 1346–1386 CrossRef CAS PubMed.
- (a) T. J. Mooibroek, C. A. Black, P. Gamez and J. Reedijk, Cryst. Growth Des., 2008, 8, 1082–1093 CrossRef CAS; (b) C. A. Black, L. R. Hanton and M. D. Spicer, Inorg. Chem., 2007, 46, 3669–3679 CrossRef CAS PubMed.
- (a) Z. Otwinowski and W. Minor, in Processing of X-Ray Diffraction Data Collected in Oscillation Mode, Methods in Enzymology, Molecular Crystallography, Part A, ed. C. W. Carter and R. M. Sweet, Academic Press, New York, 1997, vol. 276, pp. 307–326 Search PubMed; (b) SAINT V4, Area Dectector Control and Integration Software, Siemens Analytical X-ray Systems Inc., Madison, 1996 Search PubMed.
- G. M. Sheldrick, SADABS, Program for Absorption Correction, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- Agilent Technologies, Xcalibur/Supernova CCD system, CrysAlisPro Software System, version 1.171.36.XX, 2012, Agilent Technologies UK Ltd, Oxford, UK Search PubMed.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 887–897 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. M. Sheldrick, SHELXL-97, Program for the Solution of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 153–165 Search PubMed.
- (a) C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS; (b) I. J. Bruno, J. C. Cole, P. R. Edgington, M. K. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data for complexes 1–3 including a discussion of how disorder was handled, pictorial views with thermal ellipsoids at the 50% probability level and selected bond lengths and angles in tabulated form. CCDC 976905–976907. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra47735e |
| This journal is © The Royal Society of Chemistry 2014 |