Structure-related differences in the temperature-regulated fluorescence response of LCST type polymers†
Sahika Inala,
Jonas D. Kölschb,
Leonardo Chiappisic,
Dietmar Janietzd,
Michael Gradzielskic,
André Laschewsky*b and
Dieter Neher*a
aInstitute of Physics and Astronomy, University of Potsdam, Karl-Liebknecht-Str. 24-25, Potsdam, 14476, Germany. E-mail: neher@uni-potsdam.de
bInstitute of Chemistry, University of Potsdam, Karl-Liebknecht-Str. 24-25, Potsdam, 14476, Germany. E-mail: laschews@uni-potsdam.de
cStranski-Laboratory for Physical and Theoretical Chemistry, Institute of Chemistry, Technische Universität Berlin, Sekr. TC7, Str. des 17. Juni 124, Berlin, 10623, Germany
dFraunhofer Institute for Applied Polymer Research (IAP), Geiselberg-Str. 69, Potsdam, 14476, Germany
First published on 8th August 2013
Abstract
We demonstrate new fluorophore-labelled materials based on acrylamide and on oligo(ethylene glycol) (OEG) bearing thermoresponsive polymers for sensing purposes and investigate their thermally induced solubility transitions. It is found that the emission properties of the polarity-sensitive (solvatochromic) naphthalimide derivative attached to three different thermoresponsive polymers are highly specific to the exact chemical structure of the macromolecule. While the dye emits very weakly below the LCST when incorporated into poly(N-isopropylacrylamide) (pNIPAm) or into a polyacrylate backbone bearing only short OEG side chains, it is strongly emissive in polymethacrylates with longer OEG side chains. Heating of the aqueous solutions above their cloud point provokes an abrupt increase of the fluorescence intensity of the labelled pNIPAm, whereas the emission properties of the dye are rather unaffected as OEG-based polyacrylates and methacrylates undergo phase transition. Correlated with laser light scattering studies, these findings are ascribed to the different degrees of pre-aggregation of the chains at low temperatures and to the extent of dehydration that the phase transition evokes. It is concluded that although the temperature-triggered changes in the macroscopic absorption characteristics, related to large-scale alterations of the polymer chain conformation and aggregation, are well detectable and similar for these LCST-type polymers, the micro-environment provided to the dye within each polymer network differs substantially. Considering sensing applications, this finding is of great importance since the temperature-regulated fluorescence response of the polymer depends more on the macromolecular architecture than the type of reporter fluorophore.
Introduction
Water soluble thermoresponsive polymers undergo a morphological transition when heating their solution above their lower critical solution temperature (LCST).1,2 This phase transition is associated with the transformation of the polymer chains from a hydrophilic, well hydrated and thus an expanded state to a relatively more hydrophobic and collapsed one. The result is a two-phase system with aggregates comprising multiple chains de-mixed from the aqueous solution. Still in many cases, full macroscopic phase separation is not observed due to the formation of stable colloidal aggregates in the 100 nm up to lower micrometer range, the so-called “mesoglobules”.3,4 The interest in utilizing such polymers for the design and development of new functional materials has stimulated intense investigations of the phase transition phenomenon. In this context, a widespread approach has been the incorporation of fluorophores into thermoresponsive polymer chains, enabling sensitive, easy and fast detection of molecular events via fluorescence spectroscopy.5–7 Such fundamental studies have incited the development of molecular sensors, e.g., of fluorescent thermometers,8–10 the majority of which included polarity-sensitive (solvatochromic) fluorophores responding to changes in the polarity of the environment with alterations in their emission properties.5,11–19The sensing strategy based on a solvatochromic dye and a thermoresponsive polymer relies on the fact that the solubility transition of the polymer results in marked changes in the polarity and viscosity of the dye's micro-environment. At lower temperatures, the fluorophore is rather mobile and exposed to water. When heated above the phase transition temperature, the release of water from the solvent shell and the subsequent formation of less polar, polymer-rich, and dense micro-domains change the steady-state and transient fluorescence properties (Scheme 1). For a positive solvatochromic fluorophore, a decrease in environmental polarity, as is encountered upon heating the polymer above its LCST, is manifested via a hypsochromic (blue) shift in the emission (and absorption) spectrum.20 Governed by specific dynamic interactions between the fluorophore and the solvent molecules upon photoexcitation, and depending on the chemical structure of the fluorophore,21,22 this spectral shift can as well be accompanied by an enhancement of the emission intensity. Consequently, events triggering the phase transition can be detected via the emission output. This sensing concept has been exemplified by various polarity-sensitive fluorophores covalently attached to thermoresponsive polymers, as in the form of hydro/micro/nanogels,11–13 of linear chains in aqueous solution,5,14–17 or of micelles.18,19
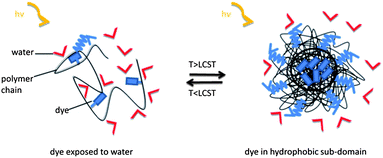 | ||
| Scheme 1 Idealized mechanism of fluorescence sensing in aqueous media through the coil-to-globule transition of a thermoresponsive polymer. As long as the (solvatochromic) fluorophore is exposed to water, the fluorescence intensity is weak. When heated above the LCST, the fluorophores get embedded in less polar and less fluidic sub-domains formed of collapsed chains, and emit more strongly at a blue-shifted maximum. | ||
1,8-Naphthalimide, in particular 4-amino-1,8-naphthalimide, derivatives are a well-studied class of environmental-sensitive dyes.23–29 This a priori non-ionic chromophore shows intense absorbance and emission in a convenient range of the visible spectrum, while it is rather insensitive to photooxidation and to quenching by oxygen. Also, due to the relatively small size and the rather polar, yet non-ionic character, it is less prone to hydrophobic self-aggregation in water and to unspecific electrostatic interactions than most of the standard fluorescent labels, such as pyrene, fluorescein or rhodamine. The strong polarity-sensitivity of 4-amino-1,8-naphthalimide derivatives is attributed to the internal charge transfer (ICT) from the electron donating amino substituent at the 4-position to the electron withdrawing imide ring.22,26,30 Increasing solvent polarity stabilizes the ICT state, which is observed as a red-shift of the emission (and absorption) maximum. It was further proposed that intermolecular hydrogen-bonding of the carbonyl oxygen and/or the imide nitrogen with solvent molecules in protic solvents results in a drop of the quantum yield26,29 accompanied by a pronounced red-shift of emission maxima.21,23,31 Due to this strong environmental sensitivity, besides several reports regarding their use in chemosensors for detection of metal ions,30,32,33 these fluorophores were also exploited as a tool for monitoring protein–protein interactions,27,34 or as a potential sensor for DNA.35–37 Also, they have recently been used to signal the pH-induced conformational changes of a protein.38
In a recent study, a 4-amino-1,8-naphthalimide functionalized monomer has been integrated within the well-known thermoresponsive poly(N-isopropylacrylamide) (pNIPAm)-based nanogels with the aim to develop an aqueous fluorescent sensor for temperature and metal ions (Hg2+).13 Utilizing the change that the metal ions induce in the ICT strength of the fluorophore upon binding, the authors reported that the nanogel acts as a ratiometric sensor at the nanomolar level at room temperature and that the detection sensitivity can be enhanced above the phase transition temperature of the nanogel.13 As for such platforms, pNIPAm has been generally the accepted prototype for the majority of fundamental research and the choice of “smart” polymer for the development of functional systems.3,39 Yet, its role has been increasingly challenged by vinyl polymers bearing oligo(ethylene glycol) (OEG) side chains, which seem to present superior anti-fouling behaviour as well as particularly good biocompatibility.40–43
Having general interest in thermoresponsive polymer systems in aqueous media, we envisaged to incorporate a naphthalimide label into thermoresponsive polymers based on acrylamide and vinyl polymers. This was done primarily to develop an aqueous functional system reflecting the thermally induced conformational transition of chains through the emission profile, which can also easily be improved for sensing stimuli other than temperature. In addition, we wanted to address questions regarding the influence of polymer structure on the solubility–insolubility transition, knowing that the LCST behaviour of various non-ionic polymers in aqueous solution may differ substantially.1,2 Considering the application of these materials for optical sensing purposes, a thorough understanding of the macromolecular assembly behaviour is crucial; however, for the particular case of detection through a fluorescence response, such aspects have not been stressed in detail. To cover these key points, we designed a series of three analogous thermoresponsive polymers which exhibit an LCST in the physiologically interesting range of ±10 °C around body temperature, namely pNIPAm (P-1), pMEO2A (P-2), and p(MEO2MA-co-OEGMA) (P-3). P-1, P-2, and P-3 are based on the monomers N-isopropylacrylamide (NIPAm), 2-(methoxyethoxy)ethylacrylate (MEO2A), and on an appropriate mixture of 2-(methoxyethoxy)ethylmethacrylate (MEO2MA) and oligo(ethylene glycol)methacrylate (OEGMA), respectively. All three thermoresponsive polymers were labelled via copolymerization with the new 4-dimethylamino-1,8-naphthalimide (4-DMN) bearing monomer 2-(6-(dimethylamino)-1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)ethyl methacrylate (3) (Fig. 1).
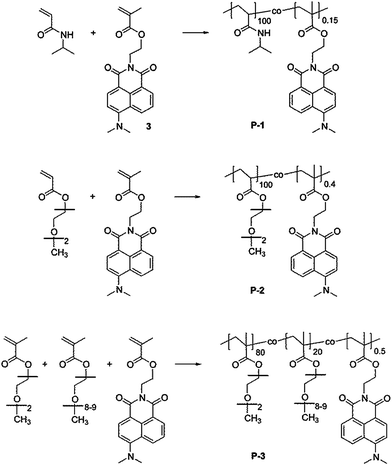 | ||
| Fig. 1 Chemical structure of the thermoresponsive copolymers comprising the 4-dimethylamino-1,8-naphthalimide (4-DMN) functionalized monomer (3). From top to bottom: pNIPAm (P-1), pMEO2A (P-2), and p(MEO2MA-co-OEGMA) (P-3). | ||
Amidst the various possibilities for thermoresponsive polymers,2 we chose pNIPAm (P-1) because it is currently the most studied representative of this group of polymers. Polyacrylate pMEO2A (P-2) was chosen for our comparative investigation as it is structurally very close to pNIPAm with respect to backbone, size and LCST.44 The third polymer (P-3), p(MEO2MA-co-OEGMA), was designed with a composition that provides a similar LCST to the other two polymers. While such copolymers have inherently a somewhat more complex composition than pMEO2A, they possess a poly(methacrylate) backbone which is more resistant to hydrolysis (a priori an advantage for any intended application) and have, therefore, been studied on several occasions in recent years.40–42 Belonging to the same polymer family, a comparative study of P-2 and P-3 can reveal the impact of small variations in the chemical structure, e.g. the presence of longer OEG side chains, or of α-methyl groups at the backbone on the aqueous solubility behaviour.
Materials and methods
Synthetic procedures
2-(2-Methoxyethoxy)ethyl methacrylate (MEO2MA, Aldrich, 95%) and (oligo(ethylene glycol)monomethylether)methacrylate (OEGMA, Aldrich, Mn = 475 g mol−1) were passed before use through a basic alumina column to remove inhibitors. 2-(2-Methoxyethoxy)ethyl methacrylate (“methoxydiethylene glycol acrylate”, MEO2A) was a gift from A. Miasnikova.44 2,2′-Azobisisobutyronitrile (AIBN, Acros, 98%) was crystallized from methanol before use. Other chemicals were purchased and used as received without further purification: N-isopropylacrylamide (NIPAm) (Acros, 99%, stabilized with 500 ppm 4-methoxyphenol (MEHQ)), ethanolamine (Acros, 99%), 6-bromobenzo[de]isochromene-1,3-dione (“4-bromo-1,8-naphthalic anhydride”, Acros), 3-dimethylaminopropionitrile (Aldrich, 98%), iso-amylalcohol (Acros, 98%), triethylamine (Acros, 99%), methacryloyl chloride (Fluka, >97%), stabilized by about 0.02% 2,6-di-tert-butyl-4-methylphenol (BHT), and phosphate buffered saline (PBS, Fluka).Synthesis of 6-(dimethylamino)benzo[de]isochromene-1,3-dione (1)
6-Bromobenzo[de]isochromene-1,3-dione (1.108 g, 4 mmol) and 3-dimethylaminopropionitrile (1.82 mL, 16 mmol) were stirred in 28 mL iso-amylalcohol at 140 °C overnight. After cooling the solution to room temperature, the resulting precipitate was filtered off, washed with water, dried and recrystallized from iso-amylalcohol to give yellow needle-like crystals. Yield: 0.69 g (2.9 mmol, 71%); mp 130 °C. Mass spectrum (ESI): signal at 242.0832 [M + H]+ (calcd: 242.0817). Elemental analysis (C14H11NO3): calcd: C = 69.70%, H = 4.60%, N = 5.81%; found: C = 69.68%, H = 4.51%, N = 6.76%; FT-IR (cm−1), selected bands: 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 450 ν(C
450 ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1718 ν(CO), 1583 ν(CC)aryl; 1H NMR (300 MHz, CDCl3) δ = 8.57–8.43 (m, 3H, CHaryl), 7.70–7.65 (m, 1H, CHaryl), 7.10 (d, J = 8.3 Hz, 1H, CHaryl), 3.18 (s, 6H, N–CH3) 13C NMR (75 MHz, CDCl3) δ = 161.7, 160.8, 158.0, 135.0, 133.2, 132.9, 125.1, 125.0, 119.4, 113.3, 44.7.
O), 1718 ν(CO), 1583 ν(CC)aryl; 1H NMR (300 MHz, CDCl3) δ = 8.57–8.43 (m, 3H, CHaryl), 7.70–7.65 (m, 1H, CHaryl), 7.10 (d, J = 8.3 Hz, 1H, CHaryl), 3.18 (s, 6H, N–CH3) 13C NMR (75 MHz, CDCl3) δ = 161.7, 160.8, 158.0, 135.0, 133.2, 132.9, 125.1, 125.0, 119.4, 113.3, 44.7.
6-(Dimethylamino)-2-(2-hydroxyethyl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (2)
6-(Dimethylamino)benzo[de]isochromene-1,3-dione (1.5 g, 6.21 mmol) and aminoethanol (0.5 mL, 8.2 mmol) were stirred in ethanol at 100 °C overnight. The solvent was evaporated and the remaining solid was purified by flash-column chromatography (eluent CH2Cl2). Yield: 1.5 g (5.27 mmol, 84%), mp >300 °C. Mass spectrum (ESI) signal at 285.1211 [M + H]+ (calcd: 285.1239). Elemental analysis (C16H16N2O3): calcd: C = 67.59%, H = 5.67%, N = 9.85%; found: C = 67.17%, H = 5.52%, N = 9.56% FT-IR (cm−1), selected bands: 3459 ν(OH), 2953 ν(CH2), 2877 ν(CH2), 1688 ν(CO), 1645 ν(CO), 1580 ν(CC)aryl. 1H NMR (300 MHz, CDCl3) δ = 8.57 (dd, J1 = 7.3 Hz, J2 = 1.1 Hz, 1H, CHaryl), 8.49–8.43 (m, 2H, CHaryl), 7.66 (dd, J1 = 8.5 Hz, J2 = 7.3 Hz, 1H, CHaromatic), 7.11 (d, J = 8.3 Hz, 1H, CHaryl), 4.47–4.42 (m, 2H, N–CH2–), 3.99–3.97 (m, 2H, –CH2–O–), 3.12 (s, 6H, N–CH3) 13C NMR (75 MHz, CDCl3) δ = 165.6, 165.1, 157.4, 133.1, 131.7, 131.5, 130.5, 125.3, 125.0, 122.9, 114.5, 113.3, 62.4, 44.9, 42.9.
2-(6-(Dimethylamino)-1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)ethyl methacrylate (3)
6-(Dimethylamino)-2-(2-hydroxyethyl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (500 mg, 1.76 mmol) and triethylamine (1 mL, 7.04 mmol) were dissolved in CH2Cl2. Methacryloyl chloride (0.34 mL, 3.52 mmol) was dropped slowly to the solution at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred overnight. After evaporating the solvent, the residue was dissolved in a small amount of acetone. This solution was poured into water and the yellow precipitate formed was collected by filtration. Yield: 620 mg (1.76 mmol, 99%), mp 108 °C. Mass spectrum (ESI) signal at 353.1471 [M + H]+ (calcd: 353.1501). Elemental analysis (C20H20N2O4): calcd: C = 68.17%, H = 5.72%, N = 7.95%, found: C = 67.04%, H = 5.73%, N = 8.08% FT-IR (cm−1), selected bands: 2956 ν(CH2), 2796 ν(CH2), 1719 ν(CO)imide, 1693 ν(CO)imide, 1654 ν(CO)methacrylate, 1582 ν(CC)aromatic. 1H NMR (300 MHz, CDCl3) δ = 8.57 (dd, J1 = 7.3 Hz, J2 = 1.1 Hz, 1H, CHaryl), 8.50–8.43 (m, 2H, CHaryl), 7.69–7.64 (m, 1H, CHaryl), 7.12 (d, J = 8.2 Hz, 1H, CHaryl), 6.06 (s, 1H, CCH2,methacrylate), 5.50 (s, 1H, CCH2,methacrylate), 4.57–4.46 (m, 4H, N–CH2–CH2–O), 3.11 (s, 6H, –N–CH3), 1.87 (s, 3H, CC–CH3) 13C NMR (75 MHz, CDCl3) δ = 167.4, 164.7, 164.1, 157.2, 136.3, 132.8, 131.4, 131.2, 130.5, 125.7, 125.5, 125.0, 123.1, 115.1, 113.5, 62.2, 44.9, 38.8, 18.3.
Synthesis of NIPAm-based copolymer
4-DMN bearing monomer (3) and NIPAm were dissolved in ethanol. The flask was sealed with a rubber septum, and the mixture was purged with dry nitrogen for 30 min and heated to 65 °C for 17 h in an oil bath. The solution was then cooled down, the solvent evaporated, and the residue was dissolved in a small amount of acetone. The solution was precipitated into diethyl ether. The solid precipitate was redissolved in acetone and precipitated at least thrice, until the supernatant solvent was colorless. The polymer was dried to give a yellowish powder. (Labelled pNIPAm: 1.5 g NIPAm, 6.5 mg AIBN, 11 mg naphthalimide, 10 mL ethanol, yield: 451 mg (30%) of P-1.)Synthesis of MEO2A and MEO2MA (co)polymers
The fluorescent monomer (3) and MEO2A (or a mixture of OEGMA (475 g mol−1) and MEO2MA) were dissolved in DMF (10 wt%). The flask was sealed with a rubber septum, and the mixture was purged with dry nitrogen for 30 min and heated to 65 °C for 17 h in an oil bath. The mixture was then diluted with de-ionized water and dialyzed against de-ionized water (Roth, ZelluTrans membrane, molecular weight cut-off 4000–6000 g mol−1). The purified polymer was isolated by freeze drying, to yield a yellow gluey mass. (Labelled pMEO2A: 1 g MEO2A, 20 mg naphthalimide, 9.4 mg AIBN, 8 mL ethanol, yield: 820 mg (82%) of P-2; labelled p(MEO2MA-co-OEGMA): 875 mg MEO2MA, 555 mg OEGMA, 6.5 mg AIBN, 22.9 mg naphthalimide, 8 mL ethanol, yield: 1.26 g (86%) of P-3.)Photophysical characterization
Absorption spectra of solutions were measured with a Varian Cary 5000 spectrometer. Turbidity measurements were performed on a Varian Cary 50 UV-VIS photometer. Transmittance of polymer solutions at 700 nm was monitored as a function of temperature (with heating–cooling cycle at rates of 0.1 °C min−1, cell path length of 12 mm). Steady-state fluorescence spectra were recorded with a HORIBA Jobin Yvon Fluorolog-3. The samples in PMMA cells of 1 cm path length were excited at the wavelength of their maximum absorption and the emission was detected at front face. PL quantum efficiencies were determined with a Hamamatsu C9920 set-up, including an integrating sphere combined with a photonic multi-channel analyzer. The time-resolved fluorescence measurements were performed by using a single photon counting setup (TCSPC) with a Becker & Hickl PML-spectrometer (modified Oriel MS-125) with a laser repetition rate of 20 MHz. The detector comprises a Becker & Hickl PML-16-C-1 (modified Hamamatsu) multi-alkaline photomultiplier. Considering that
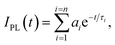 | (1) |
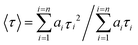 | (2) |
A cuvette holder equipped with a Peltier element was used to perform temperature-dependent measurements. Each spectrum is acquired after reaching equilibrium, i.e., ensuring that both the intensity and the shape of the spectrum are stable 15 minutes after reaching the desired temperature. Note that we did not observe any change in the shape of the emission spectra at 50 °C after keeping the solutions at this temperature overnight.
Laser light scattering studies
Static light scattering (SLS) experiments were performed on an ALV/CGS-3 instrument, equipped with a He–Ne laser with a wavelength of 632.8 nm at scattering angles in the range of 30° to 135° (15° steps) with an ALV-SP 125 goniometer. All measurements were performed in a thermostatted toluene bath (±0.1 °C) for aqueous polymer solutions (0.1 g L−1) at 15 °C, 25 °C, 30 °C, and 50 °C. The scattering intensity was normalized using the scattering of toluene at 25 °C as a reference, having a Rayleigh ratio of 1.34 × 10−5 cm−1 at 632.8 nm.45 The radius of gyration Rg and the scattering intensity (I) for q → 0 were obtained by means of the Guinier approximation:|
I(q) ≈ I(0)exp(−Rg2q2/3),
| (3) |
sin(θ/2)/λ, n the refractive index of the solution, θ the scattering angle, and λ the incident wavelength of the light). The apparent weight-averaged molecular weight (Mw,app) was determined as
 | (4) |
The apparent chain density (ρ) defined as the polymer concentration within the spherical volume occupied by its chains was calculated according to3
| ρ = 3Mw,app/4πNAR3 | (5) |
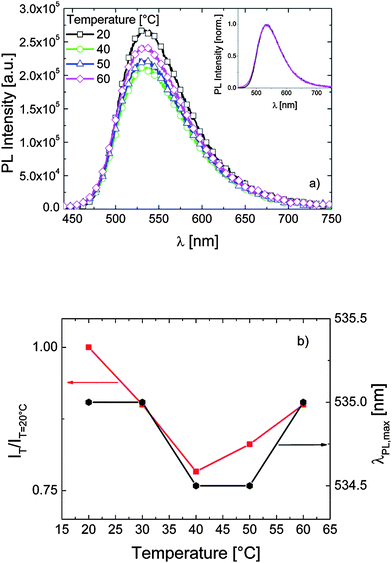
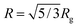 .
.
Results
In analogy with a recently reported procedure,47 the new naphthalimide-functionalized monomer (3) was synthesized starting from 4-bromo-naphthalic anhydride, by subsequent reactions with dimethylaminopropionitrile, aminoethanol, and finally with methacryloyl chloride. The fluorophore-labelled polymers were obtained by free radical copolymerization of the 4-DMN labelled methacrylate with appropriate amounts of the monomers, namely NIPAm, MEO2A, or the appropriate mixture of MEO2MA and OEGMA, respectively. Conditions were chosen such that the molar masses obtained were comparably high, and that the polymers contain on average roughly 1 chromophore per polymer chain. The low dye content was preferred to ensure that the optical properties of the isolated fluorophore are conserved upon incorporation into the polymer, and that possible effects on the solubility and phase transition properties were minimized.The incorporation of the dye into the copolymers was quantified via UV-VIS spectroscopy due to the low content (<1 mol%), and could be qualitatively corroborated by 1H NMR spectroscopy (Fig. S1†). The phase transition temperatures increase in the order P-1 < P-2 < P-3. While P-1 undergoes the coil-to-globule transition and subsequent aggregation at a temperature of about 30 °C, the cloud point of P-2 is 38 °C, and that of P-3 is 44 °C (see Table 1 and Fig. S2† for the temperature–transmission profiles). Although the acrylate backbone is more hydrophilic than the methacrylate one and additional α-methyl groups might prevent water from forming hydrogen bonds due to steric effects,48 the cloud point is obviously more strongly affected by the length of the pendant OEG chains.41 Table 1 summarizes the analytical data of these fluorescent copolymers. The macromolecules differ with regard to the molar mass, PDI and DP. While these differences certainly affect some thermodynamic properties such as the LCST, according to our preliminary results on polymers analogous to P-1 and P-3, they do not influence the temperature dependent fluorescence properties.
| Polymer | Mappna [kg mol−1] | Mappw/Mappna | DPb | Dye contentc [% mol] | NCd | Cloud pointe [°C] | T (50%)f [°C] |
|---|---|---|---|---|---|---|---|
| a Determined by size exclusion chromatography in dimethylformamide (DMF), using polystyrene standards.b Nominal number average of monomer repeat units per polymer chain.c Calculated from the UV-VIS spectra of the monomer and the copolymer solutions in CHCl3 according to Beer–Lambert's law (molar extinction coefficient (ε): 13000 L mol−1 cm−1).d Average number of chromophores per polymer chain.e Onset of the decrease of transmission in PBS solution (0.1 g L−1).f Temperature at which the transmission is reduced by 50% in PBS solution (0.1 g L−1). |
|||||||
| P-1 | 30 | 2.4 | 265 | 0.15 | 0.4 | 30 | 34 |
| P-2 | 21 | 1.9 | 120 | 0.4 | 0.5 | 38 | 52 |
| P-3 | 59 | 4.3 | 240 | 0.5 | 1.2 | 44 | 51 |
The optical properties of the 4-DMN bearing monomer were studied in solvents of varying polarity (cf. Table S1 and Fig. S3a†). The absorption spectrum of the monomer is dominated by a broad and intense band with its maximum appearing between 391 and 440 nm. A broad emission band is analogously seen in the fluorescence spectra of the monomer in all solvents other than hexane. A change of the solvent from hexane to dimethylsulfoxide results in bathochromic shifts of the absorption and of the emission maximum (Δλabsmax = 46 nm and ΔλPLmax = 86 nm, Table S1†), accompanied by a significant reduction of the fluorescence quantum efficiency (from 61 to 1.9%, Table S1†). The magnitude of these changes is characteristic of a pronounced ICT from the electron-rich amino group (methyl-substitution increasing the inductive effect) and the electron-poor imide moiety upon excitation.26 An additional red-shift in λPLmax and a further drop in quantum yield are measured for the solutions of the monomer in protic solvents, e.g. in ethanol or water. This is indicative of the sensitivity of optical spectra to specific solvent effects such as hydrogen bonding, which are responsible for an increased non-radiative decay rate (Fig. S3b†).29
The basic photophysical characteristics of the copolymers are given in Table 2. Note that at this concentration (0.5 g L−1) the spectral properties of the otherwise more diluted polymers (0.1 g L−1) are fully preserved. For all polymers in PBS, we observe a blue-shift in λPLmax compared to the value recorded for the aqueous monomer solution (Table S1†). Considering the ICT character of the dye, this might indicate that the dye attached to the polymers is inherently located in a less polar environment than water.49 We, therefore, compared the optical properties of the monomer solution in diethylene glycol dimethyl ether and N-methylformamide, which mimic the side chains of the respective polymers, MEO2A and MEO2MA (co)polymers and, pNIPAm, respectively, with those of the aqueous polymer solutions. The P-1 solution in PBS has a very low quantum yield (φ = 1.6%), comparable to that of the monomer dissolved in N-methylformamide, indicating strong exposure of the dye to water. Interestingly, aqueous P-3 exhibits a quantum efficiency of 24%, which is even higher than that of the monomer in diethylene glycol dimethyl ether (φ = 12%). This implies that 4-DMN attached to P-3 interacts only weakly with water molecules and, also, that the dipoles of the organic micro-environment consisting of the P-3 backbone and covalently attached OEG side chains are not able to relax or reorient around the 4-DMN dipole moment upon excitation. In accordance with this interpretation, the quantum efficiency of P-3, and also of P-2 and P-1, in chloroform, which is known to be a good solvent for these polymers, is rather high (up to 63%). It is only slightly lower than the efficiency of the fluorescent monomer in the same solvent. We reason that in the well-dissolved/expanded state of polymer chains, even polar side chains are not able to massively affect the emission properties of the solvatochromic dye. In this context, the fact that the aqueous solution of P-2 has a quantum yield (φ = 1.5%) significantly lower than that of the monomer in diethylene glycol dimethyl ether manifests the impact of the length of OEG side chains and/or the backbone flexibility on the extent of exposure of the fluorophores to water. It is, therefore, the conformation/co-aggregation of the chains with the ability to take up or to expel water which determines the local environment surrounding the dye.
The steady-state and transient fluorescence properties of all aqueous polymer solutions were studied as a function of temperature, below and above the LCST. The results are summarized in Fig. 2–4. It should be noted that the solution becomes cloudy above the phase transition temperature, but due to the dilute solution conditions, no macroscopic separation is observed during the course of the measurements. To ensure that all measurements are performed on an equilibrated sample, steady-state and transient PL measurements started not earlier than 15 minutes after a new temperature was reached.
Fluorescence spectra of aqueous P-1 solution at various temperatures are shown in Fig. 2a. Upon raising the solution temperature of pNIPAm chains beyond their phase transition temperature, we observe a drastic increase in the fluorescence intensity. The enhancement of emission intensity is about 25 fold, and thus much higher than that reported for a naphthalimide derivative embedded in pNIPAm nanogels.13 The increase in quantum efficiency along with a blue-shift of the emission maximum from 544 nm to 530 nm indicates important changes in the dye micro-environment. These changes are most prominent between 25 and 35 °C, as seen in Fig. 2b. We measure a longer average fluorescence lifetime for 4-DMN after the pNIPAm phase transition (Fig. S4†). It should be noted that no temperature-induced emission enhancement is observed for the monomer solution in PBS (cf. Fig. S5†). This indicates that the changes in fluorescence properties of P-1 are attributed to the phase transition of the polymer. Additionally, the onset of changes in the PL spectrum coincides well with that in the absorbance spectrum of the solution (Fig. S6†). Upon further heating (T > 40 °C), the decrease of the PL intensity is related to the temperature-activated non-radiative channels. Overall, it is the abrupt change of the chain conformation that leads to the drastic changes observed in the photophysical properties. Since the coil–globule transition is reversible, the working principle of this pNIPAm-based temperature sensor can be repeated. The reversible fluorescence on/off response renders the polymer useful for sensing applications.
 | ||
| Fig. 2 (a) The evolution of the fluorescence spectrum of P-1 in PBS (0.1 g L−1) with temperature, recorded at an excitation wavelength of 450 nm. (b) The correlation between λPLmax, the relative emission intensity (IT/IT=20 °C), and the temperature of the solution. Data are taken from the spectra in (a). | ||
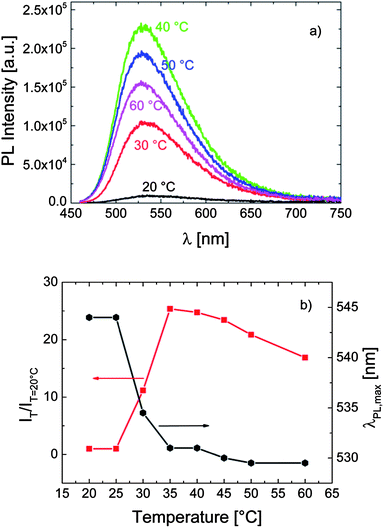
The temperature dependent fluorescence properties of P-2 bearing only short OEG side chains are shown in Fig. 3. As for P-1, heating the solution of this polymer in PBS above its cloud point (ca. 38 °C) leads to a sudden increase in its fluorescence intensity accompanied by a blue-shift in λPLmax. However, the effect is rather weak: the quantum efficiency increased only by a factor of 2, while λPLmax shifted bathochromically only by ca. 5 nm.
 | ||
| Fig. 3 (a) The effect of temperature on the fluorescence characteristics of P-2 in PBS (0.1 g L−1). The excitation wavelength was 435 nm. (b) The correlation between λPLmax, the relative emission intensity (IT/IT=20 °C), and the temperature of the solution. Data are taken from the spectra in (a). | ||
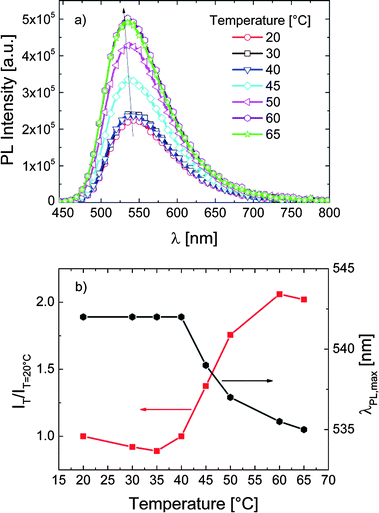
For the same dye now attached to a methacrylate backbone bearing both short and long OEG side chains, we do not observe marked changes in the intensity of emission associated with the phase transition of the P-3 chains (Fig. 4a). Also, as displayed in Fig. 4b, λPLmax is not affected by the temperature rise. Accordingly, transient PL measurements (cf. Fig. S7†) display a slight decrease of the 4-DMN lifetime as the temperature increases. This decrease does not follow a specific pattern and is associated with the fact that the non-radiative processes related to thermal agitation are more efficient at higher temperatures.20,50 In contrast to the case in P-1 and P-2, the dye of P-3 does not sense a change in its micro-environment with increasing temperature, i.e., the probe environment is not perturbed by the chain collapse and subsequent aggregation.
 | ||
| Fig. 4 (a) The fluorescence spectra of P-3 in PBS (0.1 g L−1) at various temperatures. Excitation was at 430 nm. The inset depicts the emission spectra at 20 and 60 °C normalized to their maximum emission intensity. (b) The correlation between λPLmax, the relative emission intensity (IT/IT=20 °C), and the temperature of the solution. Data are taken from the spectra in (a). | ||
The above findings show that the response of the dye to the changes in solution structure is specific to the type of the host polymer. This points to substantial differences between the aqueous solubility and phase transition phenomena of these polymers. Although the fundamental mechanisms and grounds of the “responsive” behaviour are comparable, properties such as water affinity, degree of chain contraction and dehydration, and the structure of the mesoglobules may differ significantly depending on the polymer architecture.40,51,52 We, therefore, anticipated that static light scattering (SLS) studies could provide additional insight into the association/aggregation behaviour of these labelled chains below and above their LCST, which could assist in explaining the observed fluorescence characteristics.
Table 3 summarizes the values of Rg, Mw,app, ρ, and the aggregation number (Nagg = Mw,app/Mw) for all polymers at three different temperatures deduced from SLS studies. Note that 15 °C is well below the cloud point of all polymers, while 30 °C corresponds to the onset of the decrease of P-1 transmission, and 50 °C corresponds to the temperature at which the transmission from P-2 and P-3 solutions is reduced by roughly 50%.
| Polymer | T [°C] | Rg [nm] | Mw,app [106 g mol−1] | ρ [10−3 g cm−3] | Nagg |
|---|---|---|---|---|---|
| a The calculated value of ρ for Rg = 25 nm. | |||||
| P-1 | 15 | <25 | 0.18 | 2.1a | 2 |
| P-1 | 30 | 120 | 1040 | 110 | 14500 |
| P-1 | 50 | 127 | 2920 | 262 | 40600 |
| P-2 | 15 | <25 | 0.04 | 0.5a | 1 |
| P-2 | 30 | <25 | 0.06 | 0.7a | 2 |
| P-2 | 50 | 130 | 1040 | 86 | 26000 |
| P-3 | 15 | 54 | 2.9 | 3.4 | 11 |
| P-3 | 30 | 54 | 3.2 | 3.7 | 13 |
| P-3 | 50 | 134 | 670 | 51 | 2600 |
The difference in the aggregation behaviour of these thermoresponsive polymer chains before their solubility transition is reflected in intensity-autocorrelation curves recorded at 15 °C (Fig. S8†). As reported in Table 3, P-1 and P-2 solutions at 15 °C contain well-hydrated unimers or assemblies of only a few chains. For these solutions, the scattered light was independent of the scattering angle in the examined q range, which agrees with a model of short flexible chains (Rg < 25 nm) that do not form larger assemblies. Similar solubility behaviour was observed for other fluorescent NIPAm-based polymers.3 In contrast to P-1 and P-2, P-3 solution at 15 °C already contains loose aggregates consisting of ca. 11 chains with an Rg of 54 nm. The apparent chain density of these water-draining structures is similar to that reported for an analogue OEGMA-based copolymer (ρ = 10−3 g cm−3).47
The phase transition of P-1 solution is manifested via a huge increase of all structural parameters upon raising the temperature from 15 to 30 °C. On heating the solution further to 50 °C, the globules become bigger, denser and heavier. When the solution of OEG-based copolymers is heated to 30 °C, which is below their cloud point, we observe a slight increase in the number of associating chains, indicating that the polymer chains do not undergo a temperature-induced precontraction process below the cloud point, in contrast to what is usually reported for NIPAm-based polymers (cf. Table 3).53 This evidences that the phase transition is dominated by intermolecular interactions.40 Upon raising the temperature of these solutions up to 50 °C, the intensity of the scattered light increases to a great extent, and so do Rg, Mw,app, and Nagg.
However, comparing the values of Mw,app, ρ, and Nagg of P-1 at 30 °C with those of P-2 and particularly of P-3 at 50 °C, we see a clear trend emerging: at a very early stage of the transition (30 °C), the labelled pNIPAm chains form much denser aggregates compared to the OEG-based copolymers at 50 °C, i.e., it corresponds to a later phase of their solubility transition. It appears that pNIPAm chains dehydrate to a much larger extent considering that the experimental conditions are the same for all polymers. Compared to P-3, the change in the chain packing density triggered by the phase transition is much more drastic for P-1 solution. It is worth noting that the number of P-2 chains in one aggregate at 50 °C is 10 fold higher than that of P-3 at the same temperature.
Discussion
As for P-1, the quantum efficiency in PBS at room temperature is very low (1.5%) and λPLmax is considerably red-shifted, we conclude that 4-DMN fluorophores are in intimate contact with water. Also, SLS gives no evidence for interchain association of P-1 chains at 15 °C. These findings imply that the aqueous solution consists of well-hydrated single chains (cf. Table 3). Upon heating this solution above its LCST, the emission becomes much more intense. In fact, it is even more intense than that of the monomer dissolved in its structurally analogous solvent, N-methylformamide (cf. Table 2). This suggests that the densification of the micro-environment upon phase transition inhibits the dye from forming hydrogen bonds with water. Also, densification may increase the quantum yield by restricting certain vibrational modes.27 Apparently in the P-1 network, 4-DMN fluorophores are in a rigid environment surrounded by mostly dehydrated pNIPAm chains.Similar to P-1, the aqueous solution of P-2 consists of well-swollen chains at low temperatures (cf. Table 3). Since the emission is red-shifted and displays efficiency much smaller than that of the monomer in diethylene glycol dimethyl ether, or that of the polymer in chloroform, we deduce that the dye in aqueous P-2 solution must be well exposed to water. In contrast to P-1, these photophysical properties do not change strongly upon the LCST-type transition, meaning that the dye environment becomes only slightly less polar than that provided by swollen chains.
One of the several parameters leading to the observed differences in the photophysical response of NIPAm and OEG based polymers is that the aggregates of pNIPAm are stabilized by strong intramolecular and intermolecular N–H⋯OC hydrogen bonds, whereas no hydrogen donator site exists in OEG-based polymers.40,54 The role of intrachain hydrogen bonding in the compactness of folded chains is well established for proteins.55 Concerning a comparative investigation on thermoresponsive polymers, Zhou et al. concluded that the individual single chain globules of a poly(N,N-diethylacrylamide) are less compact compared to those formed by pNIPAm due to the lack of interchain hydrogen bonding.56 Moreover, from a recent study of an analogous, yet unlabelled, p(MEO2MA-co-OEGMA) by temperature-variable 1H NMR and IR analysis, Wu et al. demonstrated the presence of soluble and mobile side chains after phase transition, pointing to only a weak dehydration of the OEG chains.40 This finding is in accordance with earlier reports devoted to OEGMA-based copolymers showing that longer OEG units preserve their mobility upon heating the solution above its LCST.57,58 From the aspect of sensing capability, Hoogenboom et al. demonstrated the influence of the hydrophilicity of the polymer on the efficiency of fluorescent thermometers based on a MEO2MA-type copolymer bearing a polarity-sensitive dye.5 When the fluorescent copolymer included longer OEG chains, the sensing capability was lost. The authors proposed that the hydrophilic copolymer remains well-hydrated even in the collapsed state. Consequently, we reason that in addition to the lack of protein-like intra-hydrogen-bonding, hydrophilic OEG side chains of MEO2A and MEO2MA copolymers hinder an effective dehydration process. Thus, the micro-environment of the probe is not dramatically affected by phase transition.
A rather different scenario is observed in the case of P-3. This polymer exhibits a “pre-association” at room temperature. The 4-DMN fluorophores within these associates are rather immobilized and in some way protected from the high polarity and H-bonding interactions of water and other possible quenching mechanisms. Such a unique environment of the probe is presumably related to the more rigid methacrylate backbone and, despite their low amount in P-3 composition, to the pre-aggregation attributed to OEGMA side chains. In addition to the argument above based on the incomplete dehydration of OEG side chains, the complete loss of the sensing capability of P-3 can as well be reasoned by the limited flexibility of the polymer backbone. As reported in Table 3, aggregates of P-3 contain 10 times fewer chains in number compared to those of P-2. The α-methyl groups can cause restrictions on the free rotation of chains resulting in less flexible conformations and this may prevent dense aggregation.
Conclusions
In summary, we demonstrated new fluorophore-labelled functional materials from acrylamide- and oligo(ethylene glycol) bearing thermoresponsive polymers for sensing purposes and investigated their temperature triggered solubility transitions. We based our arguments on the fact that the fluorescence properties of the ICT-type probe are extremely sensitive to the aggregation properties of polymer chains, which govern the contribution from the polarities near the main chains, the degree of water exposure, and the mobility of the dye. Through the optical spectra, we reveal that each of the studied polymers has peculiar solvation behaviour providing a different local micro-environment to the dye, and that the LCST-type phase transition of each polymer affects the dye emission in a different way. Compared to OEG-bearing polymers, the labelled pNIPAm experiences a much more drastic change in chain packing density at an early stage of its transition and signals this by a large increase in its fluorescence intensity. On the other hand, OEG-bearing copolymers exhibit either weak (P-2) or negligible (P-3) spectral changes upon phase transition, the effects vanishing with increasing length of the side chains. We propose that in addition to the lack of intra-hydrogen-bonding, the hydrophilic OEG side chains hinder complete dehydration of the dye micro-environment and dye's mobility to reorient. As a special case, associated with the backbone rigidity and the presence of OEGMA chains, P-3 provides a different environment to the fluorophore: the quantum yield is already high below the LCST and hardly changes with phase transition. The results of this comparative work show that the pNIPAm copolymer features ideal hydration properties for applications relying on drastic conformational changes, while the copolymer bearing OEGMA side chains provides a rather stable micro-environment. This manifests that such a photophysical investigation combined with light scattering studies is able to yield valuable insight into the structural changes that occur at the LCST for differently built thermoresponsive polymers. Such findings then are not only of fundamental importance for an enhanced understanding of this interesting process but also for the design of molecular sensors.Acknowledgements
The work was funded by the BMBF-Initiative “Spitzenforschung und Innovation in den neuen Ländern” within the cooperative project “Taschentuchlabor – Impulszentrum für Integrierte Bioanalyse” (FKZ 03IS2201B). SI thanks Prof. Dr Michael Kumke (University of Potsdam) for fruitful discussions and Sina Reiter (University of Potsdam) for her lab assistance. The help by C. Wieland and E. Wischerhoff (Fraunhofer IAP) for molar mass determination is gratefully acknowledged.Notes and references
- V. Aseyev, H. Tenhu and F. M. Winnik, Adv. Polym. Sci., 2006, 196, 1–85 CrossRef CAS.
- V. Aseyev, H. Tenhu and F. M. Winnik, Adv. Polym. Sci., 2011, 242, 29–89 CrossRef CAS.
- P. Kujawa, V. Aseyev, H. Tenhu and F. M. Winnik, Macromolecules, 2006, 39, 7686–7693 CrossRef CAS.
- V. Aseyev, S. Hietala, A. Laukkanen, M. Nuopponen, O. Confortini, F. E. Du Prez and H. Tenhu, Polymer, 2005, 46, 7118–7131 CrossRef CAS PubMed.
- C. Pietsch, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 5653–5656 CrossRef CAS PubMed.
- H. Ringsdorf, J. Venzmer and F. M. Winnik, Macromolecules, 1991, 24, 1678–1686 CrossRef CAS.
- H. G. Schild and D. A. Tirrell, Langmuir, 1991, 7, 1319–1324 CrossRef CAS.
- C. Pietsch, U. S. Schubert and R. Hoogenboom, Chem. Commun., 2011, 47, 8750–8765 RSC.
- J. Hu and S. Liu, Macromolecules, 2010, 43, 8315–8330 CrossRef CAS.
- C. Li and S. Liu, Chem. Commun., 2012, 48, 3262–3278 RSC.
- K. Iwai, Y. Matsumura, S. Uchiyama and A. P. de Silva, J. Mater. Chem., 2005, 15, 2796–2800 RSC.
- C. Gota, K. Okabe, T. Funatsu, Y. Harada and S. Uchiyama, J. Am. Chem. Soc., 2009, 131, 2766–2767 CrossRef CAS PubMed.
- C. Li and S. Liu, J. Mater. Chem., 2010, 20, 10716–10723 RSC.
- S. Uchiyama, Y. Matsumura, A. P. de Silva and K. Iwai, Anal. Chem., 2003, 75, 5926–5935 CrossRef CAS PubMed.
- L. Yin, C. He, C. Huang, W. Zhu, X. Wang, Y. Xu and X. Qian, Chem. Commun., 2012, 48, 4486–4488 RSC.
- Y. Shiraishi, R. Miyamoto, X. Zhang and T. Hirai, Org. Lett., 2007, 9, 3921–3924 CrossRef CAS PubMed.
- M. Onoda, S. Uchiyama and T. Ohwada, Macromolecules, 2007, 40, 9651–9657 CrossRef CAS.
- Q. Yan, J. Yuan, Y. Kang, Z. Cai, L. Zhou and Y. Yin, Chem. Commun., 2010, 46, 2781–2783 RSC.
- F. Li, A. H. Westphal, A. T. M. Marcelis, E. J. R. Sudholter, M. A. Cohen Stuart and F. A. M. Leermakers, Soft Matter, 2011, 7, 11211–11215 RSC.
- B. Valeur, in Molecular Fluorescence, Wiley-VCH Verlag GmbH, 2001, pp. 34–71 Search PubMed.
- A. Marini, A. Muñoz-Losa, A. Biancardi and B. Mennucci, J. Phys. Chem. B, 2010, 114, 17128–17135 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, 2009 Search PubMed.
- S. Dhar, S. Singha Roy, D. K. Rana, S. Bhattacharya, S. Bhattacharya and S. C. Bhattacharya, J. Phys. Chem. A, 2011, 115, 2216–2224 CrossRef CAS PubMed.
- S. L. Dmitruk, S. I. Druzhinin, R. A. Minakova, A. I. Bedrik and B. M. Uzhinov, Russ. Chem. Bull., 1997, 46, 2027–2031 CrossRef CAS.
- A. Pardo, E. Martin, J. M. L. Poyato, J. J. Camacho, M. F. Braña and J. M. Castellano, J. Photochem. Photobiol., A, 1987, 41, 69–78 CrossRef CAS.
- S. Saha and A. Samanta, J. Phys. Chem. A, 2002, 106, 4763–4771 CrossRef CAS.
- G. Loving and B. Imperiali, J. Am. Chem. Soc., 2008, 130, 13630–13638 CrossRef CAS PubMed.
- B. Ramachandram, G. Saroja, B. Sankaran and A. Samanta, J. Phys. Chem. B, 2000, 104, 11824–11832 CrossRef CAS.
- D. Yuan and R. G. Brown, J. Phys. Chem. A, 1997, 101, 3461–3466 CrossRef CAS.
- H. Wang, H. Wu, L. Xue, Y. Shi and X. Li, Org. Biomol. Chem., 2011, 9, 5436–5444 CAS.
- F. A. S. Chipem, A. Mishra and G. Krishnamoorthy, Phys. Chem. Chem. Phys., 2012, 14, 8775–8790 RSC.
- R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936–3953 RSC.
- I. Grabchev, J. M. Chovelon and X. Qian, J. Photochem. Photobiol., A, 2003, 158, 37–43 CrossRef CAS.
- G. S. Loving, M. Sainlos and B. Imperiali, Trends Biotechnol., 2010, 28, 73–83 CrossRef CAS PubMed.
- K. A. Stevenson, S. F. Yen, N. C. Yang, D. W. Boykin and W. D. Wilson, J. Med. Chem., 1984, 27, 1677–1682 CrossRef CAS.
- E. B. Veale and T. Gunnlaugsson, J. Org. Chem., 2010, 75, 5513–5525 CrossRef CAS PubMed.
- Z. Chen, X. Liang, H. Zhang, H. Xie, J. Liu, Y. Xu, W. Zhu, Y. Wang, X. Wang, S. Tan, D. Kuang and X. Qian, J. Med. Chem., 2010, 53, 2589–2600 CrossRef CAS PubMed.
- M. Yang, Y. Song, M. Zhang, S. Lin, Z. Hao, Y. Liang, D. Zhang and P. R. Chen, Angew. Chem., Int. Ed., 2012, 51, 7674–7679 CrossRef CAS PubMed.
- I. Dimitrov, B. Trzebicka, A. H. E. Müller, A. Dworak and C. B. Tsvetanov, Prog. Polym. Sci., 2007, 32, 1275–1343 CrossRef CAS PubMed.
- S. Sun and P. Wu, Macromolecules, 2012, 46, 236–246 CrossRef.
- C. Weber, R. Hoogenboom and U. S. Schubert, Prog. Polym. Sci., 2012, 37, 686–714 CrossRef CAS PubMed.
- J.-F. Lutz, Adv. Mater., 2011, 23, 2237–2243 CrossRef CAS.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- A. Miasnikova and A. Laschewsky, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3313–3323 CrossRef CAS.
- M. Itakura, K. Shimada, S. Matsuyama, T. Saito and S. Kinugasa, J. Appl. Polym. Sci., 2006, 99, 1953–1959 CrossRef CAS.
- X. Yang, Y.-Y. Tong, Z.-C. Li and D. Liang, Soft Matter, 2011, 7, 978–985 RSC.
- S. Inal, J. D. Kölsch, L. Chiappisi, M. Kraft, A. Gutacker, D. Janietz, U. Scherf, M. Gradzielski, A. Laschewsky and D. Neher, Macromol. Chem. Phys., 2013, 214, 435–445 CrossRef CAS.
- Y. Maeda, H. Yamauchi and T. Kubota, Langmuir, 2008, 25, 479–482 CrossRef PubMed.
- K. Iwai, N. Matsumoto, M. Niki and M. Yamamoto, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1998, 315, 53–58 CrossRef.
- C. K. Chee, S. Rimmer, I. Soutar and L. Swanson, React. Funct. Polym., 2006, 66, 1–11 CrossRef CAS PubMed.
- B. Sun, Y. Lin, P. Wu and H. W. Siesler, Macromolecules, 2008, 41, 1512–1520 CrossRef CAS.
- S. Sun and P. Wu, J. Phys. Chem. B, 2011, 115, 11609–11618 CrossRef CAS PubMed.
- C. Wu and X. Wang, Phys. Rev. Lett., 1998, 80, 4092–4094 CrossRef CAS.
- J.-F. Lutz, K. Weichenhan, O. Akdemir and A. Hoth, Macromolecules, 2007, 40, 2503–2508 CrossRef CAS.
- K. A. Dill, Biochemistry, 1990, 29, 7133–7155 CrossRef CAS.
- K. Zhou, Y. Lu, J. Li, L. Shen, G. Zhang, Z. Xie and C. Wu, Macromolecules, 2008, 41, 8927–8931 CrossRef CAS.
- S. Han, M. Hagiwara and T. Ishizone, Macromolecules, 2003, 36, 8312–8319 CrossRef CAS.
- T. Ishizone, A. Seki, M. Hagiwara, S. Han, H. Yokoyama, A. Oyane, A. Deffieux and S. Carlotti, Macromolecules, 2008, 41, 2963–2967 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of the polymers, the photophysical characterization of the monomer in various solvents, turbidity profiles of the polymers, temperature-dependent fluorescence decay curves of the polymers, temperature dependent absorbance spectra of P-1, temperature-dependent emission of the monomer, and the intensity–time autocorrelation functions. See DOI: 10.1039/c3tc31304b |
| This journal is © The Royal Society of Chemistry 2013 |