 Open Access Article
Open Access ArticleFacile synthesis of complex multi-component organic and organic–magnetic inorganic nanocomposite particles†
Marco
Giardiello
a,
Tom O.
McDonald
a,
Phillip
Martin
b,
Andrew
Owen
b and
Steve P.
Rannard
*a
aDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: srannard@liverpool.ac.uk; Fax: +44(0)1517943588; Tel: +44 (0)151 794 3501
bDepartment of Molecular and Clinical Pharmacology, University of Liverpool, Block H (first floor), 70 Pembroke Place, Liverpool L69 3GF, UK. E-mail: aowen@liverpool.ac.uk; Fax: +44 (0)1517945656; Tel: +44 (0)1517948211
First published on 1st October 2012
Abstract
A generic in situ method for producing triple component hydrophobic inorganic–organic nanocomposite particles, using a combination of modified emulsion templating and freeze-drying, is presented. Model nanocomposite particles have been developed bearing up to three hydrophobic ingredients chosen from polystyrene, oil red and 15–20 nm oleic acid-coated super-paramagnetic iron oxide (Fe3O4) nanoparticles. The technique avoids harsh conditions, in situ polymer synthesis and lengthy workup procedures, and results in high incorporation of magnetic particles (approximately 80% of triple-component nanocomposite particles contain magnetite) with retention of super-paramagnetism (>90% preservation). The nanocomposites have been characterised using dynamic light scattering, and studied under static and flow conditions in the presence of magnetic fields. Drug release was demonstrated using model nanocomposite particles bearing ibuprofen with differing hydrophobic polymer; polycaprolactone and poly(n-butyl methacrylate). Drug release varied with temperature, suggesting the synthetic technique could thus be adopted to develop drug carrier particles with tailored drug release properties.
Introduction
Inorganic–organic nanocomposite particles have recently attracted much attention due to their potential for a wide range of applications particularly in the field of biomedical research;1–8 including hyperthermia for cancer therapy,9,10 targeted drug delivery,11,12 triggered drug release,13–18 magnetic resonance imaging contrast agents19–22 and the emerging technique of magnetic particle imaging.23 Super-paramagnetic iron oxide nanoparticle (SPION, e.g. maghemite (γ-Fe2O3) and magnetite (Fe3O4)) loaded nanosystems are of particular interest due to their relatively low toxicity and insensitivity to oxidation, while surface modification techniques can be utilised to prevent agglomeration and enhance bioavailability.24,25 Direct SPION surface modification has been achieved via ligand exchange reactions giving rise to surface bound initiators from which polymer chains can be grown to yield biocompatible polymer functionalised nanoparticles.26–28 Other strategies use miniemulsion polymerisation to encapsulate SPIONs within polymeric matrices.29 Lan et al. recently described a miniemulsion polymerisation technique for the preparation of Fe3O4–poly(methyl methacrylate) composite nanospheres.30 Through the combination of ultrasonication and polymerisation under oxidising conditions, the final materials suffered a loss of super-paramagnetism and achieved only a 65% inclusion of Fe3O4 within the two-component nanocomposites. A further example was described by Ban et al. in which Fe3O4–polystyrene composite nanospheres were prepared but the details of inclusion and resulting super-paramagnetism were not reported.31 In both cases, emulsions containing a mixture of monomers, oleic acid coated magnetite and initiators were prepared prior to thermal polymerisation, separation and washing to isolate magnetic composite nanospheres. Although such strategies are well known and reasonably facile, the necessity for several synthetic and purification steps leads to lengthy and labour intensive preparations. Specifically, the removal of unreacted, toxic monomers is essential for medical use. Herein, a generic method for producing hydrophobic inorganic–organic nanocomposite particles, using a combination of modified emulsion templating and freeze-drying is presented.A series of nanocomposite particles are produced bearing two or three hydrophobic ingredients; namely polystyrene (PS), oil red (OR) and 15–20 nm oleic acid coated magnetite (Fe3O4). The facile, in situ method therefore produces composite particles bearing a choice of polymeric, hydrophobic small molecule and inorganic materials and results in high retention of super-paramagnetism of the embedded SPION. The resulting coloured magnetic nanocomposite dispersions are visibly responsive to magnetic fields under both static and flow conditions. Magnetic nanocomposite particles containing ibuprofen (IB) and either polycaprolactone (PC) or poly(n-butyl methacrylate) (PBM) were also prepared, showing varied drug release properties at 20 °C and 42 °C.
Experimental section
Materials
15–20 nm average size oleic acid coated magnetite (Fe3O4) magnetic nanoparticles, 5 mg ml−1 in toluene, oil red, polystyrene (MW 350![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), polycaprolactone (MW 14000) and poly(n-butyl methacrylate) (MW 370000) were purchased from Sigma-Aldrich® and used without further purification. Ibuprofen was purchased from TCI chemicals. Toluene was purchased from Fisher Scientific LTD. Kollicoat Protect® and Solutol HS® 15 were purchased from BASF®. 0.25 inch, cubic neodymium–iron–boron rare earth magnets were purchased from Magcraft®.
000), polycaprolactone (MW 14000) and poly(n-butyl methacrylate) (MW 370000) were purchased from Sigma-Aldrich® and used without further purification. Ibuprofen was purchased from TCI chemicals. Toluene was purchased from Fisher Scientific LTD. Kollicoat Protect® and Solutol HS® 15 were purchased from BASF®. 0.25 inch, cubic neodymium–iron–boron rare earth magnets were purchased from Magcraft®.
Nanocomposite particle preparation
Stock solutions of polystyrene and oil red were prepared at 40 mg ml−1 and 10 mg ml−1 respectively in toluene. 15–20 nm Fe3O4 was received as 5 mg ml−1 in toluene. These were then used to prepare solutions of a mixture of hydrophobic components containing a total mass of 10 mg ml−1 (total volume 1 ml); PO = 99% polystyrene, 1% oil red, i.e., 247.5 μl (9.9 mg) polystyrene stock, 10 μl (0.1 mg) oil red stock and 742.5 μl toluene; magnetic POFe = 89% polystyrene, 1% oil red and 10% Fe3O4, i.e., 222.5 μl (8.9 mg) polystyrene stock, 10 μl (0.1 mg) oil red stock, 200 μl (1 mg) Fe3O4 stock, and 567.5 μl toluene; PFe = 90% polystyrene and 10% Fe3O4, i.e., 225 μl (9 mg) polystyrene stock, 200 μl (1 mg) Fe3O4 stock, and 575 μl toluene; magnetic OFe = 75% oil red and 25% Fe3O4, i.e., 1500 μl (7.5 mg) 15 mg ml−1 oil red stock, 1500 μl (2.5 mg) Fe3O4 stock, and 575 μl toluene. These solutions were then mixed for 2 hours to ensure homogeneity in the solution. An aqueous solution of the polymer Kollicoat Protect® and the surfactant Solutol HS® 15 were prepared in a 3:1 mass ratio respectively to a total concentration of 22.5 mg ml−1. Final combination mixtures were then prepared by addition of 100 μl of one of the prepared toluene solutions to 400 μl of the aqueous solution. The mixtures were then emulsified via sonication for 12 seconds (Covaris S2x with a duty cycle of 20, an intensity of 10 and 500 cycles per burst in frequency sweeping mode) followed by immediate cryogenic freezing. Both the organic and water solvents were removed via freeze drying to produce a stable, porous composite material bearing the water insoluble hydrophobic compounds as well as the water-soluble polymers and surfactants, resulting in10 mg solid mass composed of 10% combined hydrophobic, 60% polymer and 30% surfactant. Upon addition of water to the porous composites the hydrophobic compounds are released as combination particles containing all added hydrophobic materials as particles.
Ibuprofen loaded nanocomposite particles were prepared following identical techniques but using polycaprolactone (PC) and poly(n-butyl methacrylate) (PBM). Stock solutions of PC or PBM and ibuprofen (IB) were prepared at 40 mg ml−1 in toluene. As above, 15–20 nm Fe3O4 was purchased as 5 mg ml−1 in toluene and hydrophobic mixtures containing a total mass of 10 mg ml−1 (total volume 1 ml) were prepared comprising 200 μl Fe3O4, 6.75 μl ibuprofen, 15.75 μl polymer stock and 777.5 μl toluene. Thus, PCIFe = polycaprolactone based nanocomposites; PBMIFe = poly(n-butyl methacrylate) based nanocomposites. 100 μl of the prepared toluene solutions was added to 400 μl of the aqueous solution as described for POFe and likewise sonication and freeze drying followed the same procedure as described earlier.
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM)
10 mg of POFe and PO were dispersed in 1 ml water. TEM grids were prepared as follows; holey carbon films on 400 mesh TEM grids (Agar Scientific) were cleaned via rinsing with acetone and ethanol. One drop (∼0.08 ml) of solutions of graphene oxide (0.10–0.15 mg ml−1) was deposited onto each grid and left to air-dry for ∼30 min. 10 μl of the particle dispersions were pipetted directly onto a GO grid and immediately blotted away using a piece of filter paper.32 SEM samples of the monoliths were prepared by addition of approximately 1 mg directly onto adhesive carbon disk covered aluminum SEM stubs. The samples were gold coated for 1.5 minutes at 25 μA using a sputter-coater (EMITECH K550X) prior to imaging. Both SEM and TEM characterisation was carried out using a Hitachi S-4800 FE-SEM.Dynamic light scattering (DLS)
Nanoparticles were dispersed in 1 ml of water to give 1 mg ml−1 dispersion, with respect to hydrophobic mass. The hydrodynamic diameter (z-average) was recorded using 1 cm path length disposable cuvettes. Further experiments in the presence and absence of a magnetic field were recorded where a neodymium–iron–boron rare earth magnet was coated with parafilm and suspended inside the cuvette, just touching the top of the liquid. Dynamic light scattering measurements were carried out at 25 °C on a Malvern Instruments Ltd. Zetasizer Nano Series Nano-ZS spectrometer using the automatic attenuator and measurement position settings of the Zetasizer Software.Ultraviolet-visible (UV-vis) spectroscopy
To measure the effects of sonication on OR in the presence of SPIONS, a sample was prepared in toluene containing all ingredients used to prepare emulsions (i.e. Kollicoat Protect® and the surfactant Solutol HS® 15 suspended in toluene). UV-vis using a Thermo Scientific NanoDrop 2000c spectrophotometer monitoring the absorption at λmax = 518 nm was recorded prior to and following sonication for 12 seconds (see Fig. S1, ESI†). For aqueous nanodispersion studies, nanoparticles were dispersed in 1 ml of water to give a1 mg ml−1 dispersion, with respect to hydrophobic mass. UV-vis spectra were recorded using a Perkin Elmer Lambda 25 UV-vis Spectrometer with a 1 cm disposable cuvette. T700nm was recorded between the range 350 to 800 nm both in the presence and absence of a neodymium–iron–boron rare earth magnet, suspended inside the cuvette, coated with parafilm and just touching the top of the liquid. Scans were recorded every 30 minutes until a plateau in T700nm was reached (see Fig. S3, ESI†).Superconducting quantum interference (SQUID)
SQUID analysis was carried out using a MPMS-XL-7 from Quantum Design. The POFe sample was placed into a gel capsule (provided by the same company). 100 μl of the commercially obtained 15–20 nm average size oleic acid coated Fe3O4 nanoparticles in toluene was dripped onto a small strip of filter paper, dried and placed in the gel capsule.33 The accurate mass of the Fe3O4 content in toluene solution was determined by thermal gravimetric analysis (TGA) using a TGA-500 supplied by TA Instruments. The TGA pan was tarred to zero and 50 μl was dropped on; mass = 0.512 mg as pan entered the instrument. 39.9% by mass corresponded to pure Fe3O4, hence 0.20 mg in 50 μl (see Fig. S2, ESI†). Therefore, the overall solution mass of pure Fe3O4 was determined as 4 mg ml−1. Masses were subsequently inferred for all SQUID analysis from this value.Magnetic particles: flow-through experiments
POFe nanocomposite particle aggregation following flow past a magnet; 1 mg ml−1 dispersions (with respect to hydrophobic mass) of 3 ml volume in water were prepared. An initial T700nm was recorded. Two experiments were performed at differing flow rates (1 ml h−1 and 5 ml−1). Samples were passed through a 1 mm diameter, 100 mm length glass capillary tube, alongside which magnets were arranged. The experiment was carried out underneath a Meiji™ MX9300 optical microscope fitted with an Infinity 1 camera. Experiments were run with magnet arrangements as follows; one single magnet with poles horizontal to capillary, one single magnet with poles perpendicular to capillary, two magnets on opposing sides of the capillary (perpendicular poles), two magnets in a row (perpendicular poles). Experiments were continued until 1 ml dispersant was collected in a disposable cuvette. T700nm was then recorded for the resulting dispersion.Oil red calibrations
A 0.1 mg ml−1 (0.2448 mM) solution of oil red was prepared in a 75:25 mixture of THF and water. UV-vis spectra were recorded using a Thermo Scientific NanoDrop 2000c spectrometer monitoring the absorption at λmax = 518 nm. Spectra were recorded following 50% serial dilution. A range of spectra were measured yielding eqn (1) (see Fig. S4, ESI†). | (1) |
An equivalent calibration was carried out using blank, non-magnetic nanoparticles; 1 mg active is 99% polystyrene, 1% oil red, therefore 0.01 mg oil red. First the 10 mg nanoparticles composite was dispersed in 1 ml water; the oil red content is thus 0.01 mg ml−1 (0.0244 8 mM). 0.5 ml was then added to 1.5 ml of THF, therefore dissolving the particles completely in a 75:25 THF:water mix; 0.025 mg ml−1 (0.00612 mM) oil red content. The solution was sonicated for 2 minutes to ensure complete nanoparticle dissolution. The calibration was then recorded in the same manner as for the previous oil red calibration, yielding eqn (2) (see Fig. S5, ESI†).
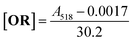 | (2) |
The similarity in the equations show the technique to be trustworthy in determining nanoparticle solution oil red content.
Fe3O4 content estimation
The method follows that of the blank calibration above. POFe nanocomposite particles were dispersed in 1 ml water, therefore oil red content was 0.01 mg ml−1 (0.02448 mM). A neodymium–iron–boron rare earth magnet was coated in parafilm and dropped inside the dispersion liquid and left for 60 hours to ensure all magnetised nanoparticles had aggregated to the magnet surface. The remaining dispersant became more transparent as magnetised nanoparticles were removed. It can therefore be inferred that the remaining oil red content relates to nanoparticles that do not bear Fe3O4. 0.5 ml of the remaining dispersion was added to 1.5 ml THF (forming a 25%:75% solution) and sonicated ((Covaris S2x with a duty cycle of 20, an intensity of 10 and 500 cycles per burst in frequency sweeping mode) to ensure complete nanoparticle dissolution. UV-vis spectra were then recorded and applied to the oil red calibration above, used to determine the concentration of the dissolved oil red. The difference in oil red content, i.e., from the initial 0.01 mg, shows the amount of removed particles due to magnetic interaction, and therefore shows the percentage of nanoparticles that contain Fe3O4.
Ibuprofen release studies
PCIFe and PBMIFe nanocomposite particles were dispersed in water at 2 mg ml−1 with respect to hydrophobic mass. 6 ml samples of each nanocomposite dispersion were added to dialysis tubing (MW cutoff 50000 Da) and placed in 30 ml water. Release studies were carried out at 20 °C and at 42 °C with constant stirring throughout. 1 ml volumes were removed at 30, 60, 90 and 120 minutes for UV-vis measurements, monitoring the IB peak at 272 nm. Following UV-vis analysis each solution was returned to the dialysis mixture.34
Results and discussion
Synthesis and characterisation
Previously we have utilised a combination of emulsion templating and freeze-drying to prepare organic nanoparticles of hydrophobic biocidal ingredients35 and dual-component hydrophobic fluorescence resonance energy transfer, FRET, dye nanoparticles.36 Herein, this method has been adapted further to prepare triple-component nanocomposite particles bearing three water-insoluble and commercially available ingredients: a polymer (PS), inorganic magnetic nanoparticles (oleic acid coated Fe3O4 particles) and a hydrophobic dye (OR). This strategy avoids many of the complications of previously reported approaches, namely (a) the in situ polymerisation of toxic monomers, (b) chemical modification of the metal oxide surface, (c) the need for encapsulation during the polymerisation reaction, and (d) lengthy purification techniques.In brief, our method (shown schematically in Fig. 1a–c) involved the preparation of an oil-in-water (O/W) emulsion in which up to three hydrophobic compounds were present in the volatile organic oil phase (toluene). The continuous aqueous phase contained a mixture of commercial stabilisers, commonly used in pharmaceutical products; a poly(ethylene glycol-graft-vinyl alcohol) copolymer (Kollicoat Protect®) and a non-ionic poly(ethylene glycol)/12-hydroxystearic acid ester surfactant (Solutol HS® 15). Following emulsification via extremely mild sonication (12 seconds) (Fig. 1a), the mixture was rapidly cryogenically frozen. The volatile organic (dispersed) and aqueous (continuous) phases were subsequently removed by freeze-drying to produce a solid, stable, porous monolithic matrix, composed of the water-soluble polymer stabilisers, bearing the water-insoluble hydrophobic nanocomposites (Fig. 1b). Upon addition of water, and subsequent dissolution of the stabilisers, nanocomposite particles were released to form sub-micron aqueous dispersions (Fig. 1c) stabilised by the water-soluble polymer and surfactant. The potential for loss of OR through oxidation via sonication, was studied using samples containing all materials in toluene (i.e. Kollicoat Protect® and Solutol HS® 15 suspended in toluene). UV-vis monitoring of the absorption at λmax = 518 nm was recorded prior to and following sonication for 12 seconds. No loss of the oil red signal was observed (see Fig. S1, ESI†). The target composition (wt/wt) of the inorganic–organic nanocomposite particles was 89% PS, 1% OR and 10% Fe3O4 (Fig. 1c, (ii)), from herein referred to as POFe. Non-magnetic organic nanoparticles (comprising 99% PS and 1% OR (wt/wt)) were also produced, referred to as PO (Fig. 1c, (i)). Both the nanocomposite materials were characterised in detail by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) (Fig. 1d and e). While the structures of both the monoliths bearing PO (Fig. 1d, (i)) and POFe (Fig. 1d, (ii)) appeared comparable, differing morphologies of the resulting nanoparticles where observed. In the absence of Fe3O4, spherical PO nanoparticles were produced (Fig. 1e, (i)) whereas upon SPION introduction, more irregular nanocomposite particles were formed (Fig. 1e, (ii)). This appears to arise from the additional inorganic material effecting polystyrene nucleation. TEM analysis showed the Fe3O4 embedded within the predominately polystyrene nanocomposite structures (Fig. 1e(ii)).
 | ||
| Fig. 1 Schematic representation of the emulsion templating, freeze drying process to prepare organic and inorganic–organic nanocomposite particles containing polystyrene (P), oil red (O) and Fe3O4 (Fe). (a) O/W emulsions bearing (i) toluene/P/O dispersed phase (ii) toluene/P/O dispersed phase with Fe nanoparticles; (b) post freeze drying monolithic structures bearing (i) PO and (ii) POFe; (c) aqueous nanodispersions bearing (i) PO and (ii) POFe; (d) SEM images (adhesive carbon disk covered aluminum stubs) of porous monoliths containing (i) PO and (ii) POFe; (e) TEM images (graphene oxide coated holey carbon films on 400 mesh) after drying of aqueous dispersions of (i) PO nanoparticles and (ii) POFe nanocomposites. | ||
The nanocomposite particles were analysed further by dynamic light scattering (DLS). The z-average diameter, polydispersity index (PdI) and zeta potential of POFe and PO were recorded as; POFe = z-average 779 nm, PdI 0.53 and zeta potential −10.8 mV, and PO = z-average 774 nm, PdI 0.49 and zeta potential −9.1 mV, therefore showing comparable particle characteristics.
The super-paramagnetism of the POFe nanocomposites was characterised via superconducting quantum interference, or SQUID, magnetometry (Fig. 2). SQUID analysis showed no appreciable effect of processing on the super-paramagnetic properties of the Fe3O4 contained within POFe, suggesting no oxidation to Fe2O3 as a result of the mild conditions employed here. The magnetisation saturation was measured as 38 emu g−1, which corresponds within error to that of the commercially bought Fe3O4 (42 emu g−1) employed during synthesis (See Fig. S6, ESI†) and represents a >90% retention of super-paramagnetism. As mentioned previously, lengthy sonication and in situ polymerisation has been shown to cause up to 40% reduction of super-paramagnetism, thought to be due to oxidation during processing.30,37
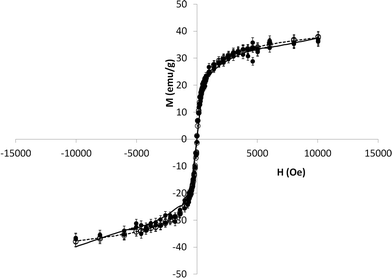 | ||
| Fig. 2 SQUID analysis of Fe3O4 within POFe (solid line, filled circles) and commercial Fe3O4 (dotted line, open circles). Error bars = 10%. | ||
Magnetic nanocomposites: static magnetic field interaction
The inclusion of OR within the magnetic nanocomposite particles gave rise to coloured dispersions that could be visually studied (Fig. 3). Response to an imposed magnetic field could therefore be readily observed by loss of colour as magnetic nanocomposite particles were removed from the bulk dispersion (Fig. 3(i) and (ii)). The facile synthetic process also allows for production of other inorganic–organic nanocomposites. Two similar dual-component particles were also prepared, comprising (wt/wt) either 90% PS/10% Fe3O4 (PFe) or 75% OR/25% Fe3O4 (OFe), both showing a visual response to the imposed magnetic field (Fig. 3(iii) and (iv) respectively). | ||
| Fig. 3 Photographs of inorganic–organic nanocomposite dispersions without exposure to magnetic field (left) and after 50 hours exposure (right): (i) POFe, (ii) POFe – cuvette turned through 90° to show particle accumulation, (iii) PFe and (iv) OFe. | ||
Response of POFe to the magnetic field was observed in detail by DLS. Measured z-average diameters were recorded every 10 seconds for a total of 80 minutes (Fig. 4a) with a magnet suspended above the cuvette, just touching the top of the liquid. A decrease in the observed z-average from approximately 780 nm to 300 nm was observed for POFe in the presence of the magnet however no discernible change was observed in the absence of the magnetic field (Table 1). The removal of coloured magnetic nanocomposite particles also led to a subsequent increase in POFe dispersion clarity, quantifiable as an increase in percentage transmittance at a wavelength of 700 nm (T700nm) using ultraviolet-visible (UV-vis) spectroscopy. Measurements every 30 minutes were taken until T700nm reached a plateau. A control dispersion (POFe with no magnetic field) was measured at the same intervals and for the same length of time. Fig. 4b (and Fig. S3, ESI†) shows a large T700nm increase from ∼0.2% to ∼45% after 50 hours due to removal of the coloured magnetic nanocomposites from the bulk dispersion; only a very slight increase in T700nm (∼0.25% to ∼1.0%) was observed for the control dispersion, likely arising from low levels of sedimentation.
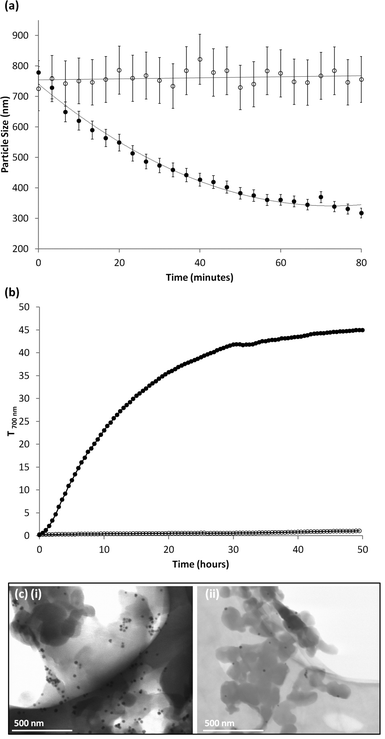 | ||
| Fig. 4 (a) Temporal DLS measurements of varying z-average diameter of POFe in the presence (closed circles) and absence (open circles) of a magnetic field. (b) Temporal UV-vis transmittance (700 nm) measurements of POFe in the presence (closed circles) and absence (open circles) of a magnetic field magnetic field. (c) TEM micrograph of POFe dispersion (i) before and (ii) after 80 minutes exposure to a magnetic field. | ||
| Z-average diameter [nm] | PdI | Number average [nm] | Volume average [nm] | |
|---|---|---|---|---|
| a Prior to interaction with magnetic field. b 80 minutes after application of the magnetic field. | ||||
| PO | 774 | 0.49 | 250 | 1340 |
| POFe a | 779 | 0.53 | 244 | 1412 |
| POFe b | 318 | 0.34 | 166 | 912 |
| PFe a | 780 | 0.66 | 358 | 3338 |
| PFe b | 292 | 0.33 | 171 | 1236 |
| OFe a | 724 | 0.53 | 223 | 3426 |
| OFe b | 239 | 0.35 | 154 | 2352 |
TEM microscopy of dried dispersions allowed a comparison (Fig. 4c) of the particles remaining in dispersion, after prolonged exposure to the magnetic field, with the initial dispersion prior to exposure. After 80 minutes interacting with the magnetic field, the particles that remain (were not attracted to the magnet) appear smaller, more spherical and contain relatively low levels of Fe3O4. Prior to magnetic interaction the TEM depicted a higher concentration of nanocomposite particles that aggregate on the TEM grid, most likely due to a drying effect, and display a higher abundance of embedded Fe3O4.
The dramatic decrease in z-average and increase in T700nm suggests the larger nanocomposite particles within the distribution contain a high Fe3O4 content and are removed more rapidly from the dispersion, leaving a distribution populated by smaller particles. This was further demonstrated where the number average, volume average and PdI are all reduced over time as a result of the loss of larger particles from the dispersion (Table 1 and Fig. S7–9, ESI†).
Magnetic nanocomposites: flow-through experiments
The magnetic properties of the particles offers the potential for controlling spatial accumulation using a magnetic field; the removal and localisation of POFe nanocomposites were investigated under dynamic flow conditions.38 The dispersions (1 ml) were passed through a glass capillary (1 mm internal diameter) at two flow rates (1 ml h−1 and 5 ml h−1) in the presence of magnets, arranged in contact with the glass capillary in several orientations depicted in Fig. 5a. Normalisation of T700nm, before and after flow past the magnet, allowed comparative studies of the effect of various magnet orientations and flow rates. | ||
| Fig. 5 (a) Schematic representation of magnet orientation along glass capillary tube. (i) Single magnet (horizontal poles), (ii) single magnet (perpendicular poles), (iii) two opposite magnets (perpendicular poles) and (iv) two adjacent magnets (perpendicular poles). Note; numbering system same for the following (b) and (c). (b) Optical images (flow rate = 5 ml h−1). (c) Magnetic nanocomposite aggregation under flow conditions (10 mg ml−1 total solid mass dispersed in water). Normalised % increase in T700nm after flow past magnets with different orientations at flow rates of 1 ml h−1 (black bar) and 5 ml h−1 (grey bar). | ||
As the dispersions flowed through the magnetic field, a gradual increase in accumulation occurred in the areas of maximum magnetic field strength (Fig. 5b). When a single magnet was aligned horizontally, nanocomposite aggregation occurred at the first pole of the magnet to a greater visual extent than the second, downstream pole (Fig. 5b, (i)) and little difference was observed when varying flow rate (Fig. 5c, (i)). If the single magnet was aligned perpendicular to the flow (one pole in contact with the capillary, Fig. 5a, (ii)), accumulation of a more dense layer was observed along the whole contact area (Fig. 5b, (ii)). A subsequent greater increase in transmittance was also measured for both flow rates, however this was more noticeable at 1 ml h−1 (Fig. 5c, (ii)). Addition of a second magnet, aligned perpendicular to the flow and on the opposite side of the capillary (Fig. 5a(iii) and b(iii)) showed accumulation across the internal diameter of the capillary, within the magnetic field generated between the magnets. Very little difference in removal of nanocomposite was seen compared to a single magnet and as measured by the normalised increase in transmittance at both flow rates (Fig. 5c, (iii)). When the second magnet was placed further along the capillary, with poles perpendicular to the direction of flow, (Fig. 5a, (iv)) accumulation was seen at both contact points, with less at the downstream magnet (Fig. 5c, (iv), see Fig. S10, ESI†). A considerable overall increase in normalised T700nm was also achieved; approximately 14-fold increase at 1 ml h−1 and approximately 7-fold increase at 5 ml h−1 over the single horizontal magnet.
% Fe3O4 content estimation
In order to estimate the percentage of nanocomposite particles that contained embedded Fe3O4, the OR-containing, three-component nanocomposites were exposed to a magnetic field for extended periods of time (60 hours) in an attempt to completely remove the fraction of the particle distribution containing Fe3O4. The removal of magnetic nanoparticles results in a more transparent and less coloured dispersion, therefore any nanocomposite particles remaining in the dispersion after extended exposure are inferred to not bear Fe3O4 and quantification of OR content would directly relate to the fraction of the dispersion without magnetic particle inclusion.Previous DLS and UV-vis experiments were each conducted for different timescales due to the sensitivity of the two techniques. Light scattering scales as the cube of the particle radius and therefore is highly affected by the loss of the lower fraction of larger particles within the distribution, allowing differences to be observed rapidly. The dispersions do not become clear or colourless within the DLS experiment timescale suggesting a high concentration of dispersed particles remain after 80 minutes application of the magnetic field. To measure large changes in transmittance using UV-vis techniques, considerably longer experiments (50 hours) were employed (Fig. 4).
To completely remove the fraction of magnetic nanocomposite particles, an exposure time of 60 hours was utilised in the presence of a submerged magnet (i.e. the magnet placed within the dispersion rather than suspended above). Absolute dispersion clarity was not achieved and as such it is appropriate to consider part of the distribution of particles formed as containing either PS alone, OR alone or both PS and OR, none of which will be attracted towards the applied magnetic field. Estimations of the percentage of nanocomposites containing all three hydrophobic compounds were accomplished through the quantification of remaining OR using UV-vis after removal of the submerged magnet and dissolution of all dispersed particles through addition of THF. The absorbance at λ = 518 nm (typical for OR in THF/water) decreased significantly, suggesting that approximately 80% of the coloured nanocomposite particles contain Fe3O4. (Determined from oil red calibration eqn (1) and calibration plot Fig. S3, ESI.†)
Drug release model studies
Model triple component nanocomposite particles were produced by substituting the hydrophobic drug ibuprofen (IB) for OR, and polycaprolactone (PC) or poly(n-butyl methacrylate) (PBM) for PS. PC and PBM were chosen to provide diversity of glass transition temperature (Tg) and allow studies of drug release and thermal response. The temperature of Fe3O4 SPIONS, following magnetic heating by applied oscillating magnetic fields (e.g. magnetic fluid hyperthermia therapy), is reported as locally reaching 42–45 °C,39 therefore release was studied at 20 °C and 42 °C. As previously, Kollicoat Protect® and Solutol HS® 15 stabilisers were employed and the same particle synthesis conditions used for POFe were adopted. The target composition of the nanocomposite particles was 63% polymer, 27% IB and 10% Fe3O4 (wt/wt), to improve the relative content of drug to polymer. After freeze-drying and redispersion, DLS measurements of polycaprolactone containing nanocomposites (PCIFe) showed z-average diameters of 458 nm, whilst poly(n-butyl methacrylate) nanocomposites (PBMIFe) were found to be 397 nm.The nanocomposite particles were dispersed at 2 mg ml−1 in water, were added to dialysis tubing, with a 50000 Da molecular weight cutoff and placed in water. Two samples were dialysed for each sample with one heated to 42 °C and the other controlled at 20 °C. Following previous reported procedures,34 UV-vis spectra were periodically recorded monitoring the peak at λ = 272 nm, corresponding to IB (see Fig. S11–14, ESI†).
Release of IB was observed (Fig. 6) from both PBMIFe and PCIFe nanocomposites, with dramatic increases achieved at elevated temperature for both materials. PBMIFe showed the highest response to elevated temperature (Fig. 6a and b), probably due to the Tg of poly(n-butyl methacrylate) being <42 °C (reported value, Tg = 20 °C)40 and polycaprolactone being above its Tg at all temperatures studied (reported value, Tg = −60 °C).40 At 20 °C, very little IB was released for PBMIFe and to ensure release at elevated temperature was not due to break-up or dissolution of the nanocomposites, DLS measurements were also recorded at 45 °C. Neither the particle size nor the derived count rate decreased over the 120 minute study period, therefore demonstrating the nanocomposite structures remain intact during drug release (see Fig. S15 and S16, ESI†).
 | ||
| Fig. 6 UV-Vis release study of IB from (a) PBMIFe at 42 °C, (b) PBMIFe at 20 °C, (c) PCIFe at 42 °C and (d) PCIFe at 20 °C. Error bars = 5%. | ||
Conclusions
In conclusion, we have demonstrated the synthesis of complex, three-component inorganic–organic nanocomposites using a generic emulsion templating, freeze-drying technique. Aqueous dispersions of magnetic nanocomposite particles bearing three hydrophobic ingredients (polymer, dye (or drug) and magnetic nanoparticles), were generated from the simple addition of water to the freeze-dried, porous solid monoliths. SEM and TEM showed irregular nanocomposite structures incorporating Fe3O4.Nanoparticle analysis demonstrated that 80% of the coloured nanocomposites contained Fe3O4 and were responsive to applied magnetic fields. Furthermore, the facile approach, which avoids lengthy sonication times and in situ polymerisation techniques, results in high retention of super-paramagnetism of the embedded Fe3O4. The high level of encapsulated Fe3O4 is comparable to other, more complex methods quoted in the literature.29 Model experiments to demonstrate drug release at varied temperature were also conducted using the hydrophobic drug ibuprofen, with polymers of varying glass transition temperatures. As such, differing drug release properties were observed at both 20 °C and 42 °C, mimicking the temperature of Fe3O4 under magnetic heating by applied oscillating magnetic fields.
This technique offers a platform to replace each of the hydrophobic ingredients with biologically active, hydrophobic compounds such as drugs or biocidal materials, thereby forming medically relevant nanocomposites. The demonstration of particle removal by a static magnet, and from flowing nanocomposite dispersions, also suggests in vivo or tracing/sensing applicability for targeted delivery of drugs or hyperthermia agents within tumours.
It is expected that long-term storage within the solid state, as freeze-dried monoliths, followed by redispersion at ambient temperature and administration, would offer considerable advantages with the potential to localise the particles using their magnetic properties and trigger cell-death and drug release through magnetic field manipulation. Release kinetics appears to be tuneable through choice of hydrophobic polymer employed.
Acknowledgements
The authors thank Research Councils UK and the EPSRC. We also acknowledge the Centre for Materials Discovery (University of Liverpool) and Pavel Borisov of the Rosseinsky group (University of Liverpool) for access to analytical equipment.Notes and references
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064 CrossRef CAS.
- F. M. Kievit and M. Zhang, Acc. Chem. Res., 2011, 44, 853 CrossRef CAS.
- D. K. Kim and J. Dobson, J. Mater. Chem., 2009, 19, 6294 RSC.
- Q. A. Pankhurst, N. K. T. Thanh, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2009, 42, 224001 CrossRef.
- K. M. Krishnan, IEEE Trans. Magn., 2010, 46, 2523 CrossRef CAS.
- Y. M. Wang, X. Cao, G. H. Liu, R. Y. Hong, Y. M. Chen, X. F. Chen, H. Z. Li, B. Xu and D. G. Wei, J. Magn. Magn. Mater., 2011, 323, 2953 CrossRef CAS.
- V. Salgueirino-Maceira and M. A. Correa-Duarte, Adv. Mater., 2007, 19, 4131 CrossRef CAS.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995 CrossRef CAS.
- K. Maier-Hauff, R. Rothe, R. Scholz, U. Gneveckow, P. Wust, B. Thiesen, A. Feussner, A. von Deimling, N. Waldoefner, R. Felix and A. Jordan, J. Neuro-Oncol., 2007, 81, 53 CrossRef CAS.
- M. Johannsen, U. Gneveckow, L. Eckelt, A. Feussner, N. Waldöfner, R. Scholz, S. Deger, P. Wust, S. A. Loening and A. Jordan, Int. J. Hyperthermia, 2005, 21, 637 CrossRef CAS.
- C. Alexiou, W. Arnold, R. J. Klein, F. G. Parak, P. Hulin, C. Bergemann, W. Erhardt, S. Wagenpfeil and A. S. Lubbe, Cancer Res., 2000, 60, 6641 CAS.
- P. Y. Chen, H. L. Liu, M. Y. Hua, H. W. Yang, C. Y. Huang, P. C. Chu, L. A. Lyu, I. C. Tseng, L. Y. Feng, H. C. Tsai, S. M. Chen, Y. J. Lu, J. J. Wang, T. C. Yen, Y. H. Ma, T. Wu, J. P. Chen, J. I. Chuang, J. W. Shin, C. Hsueh and K. C. Wei, Neuro-Oncology, 2010, 12, 1050 CrossRef CAS.
- S. Brule, M. Levy, C. Wilhelm, D. Letourneur, F. Gazeau, C. Menager and C. Le Visage, Adv. Mater., 2011, 23, 787 CrossRef CAS.
- K. Hayashi, K. Ono, H. Suzuki, M. Sawada, M. Moriya, W. Sakamoto and T. Yogo, ACS Appl. Mater. Interfaces, 2010, 2, 1903 CAS.
- K. Katagiri, Y. Imai and K. Koumoto, J. Colloid Interface Sci., 2011, 361, 109 CrossRef CAS.
- C. S. S. R. Kumar and F. Mohammad, Adv. Drug Delivery Rev., 2011, 63, 789 CrossRef CAS.
- S. Mornet, S. Vasseur, F. Grasset and E. Duguet, J. Mater. Chem., 2004, 14, 2161 RSC.
- A. M. Derfus, G. von Maltzahn, T. J. Harris, T. Duza, K. S. Vecchio, E. Ruoslahti and S. N. Bhatia, Adv. Mater., 2007, 19, 3932 CrossRef CAS.
- C. Fang and M. Q. Zhang, J. Mater. Chem., 2009, 19, 6258 RSC.
- D. K. Kim, M. Mikhaylova, F. H. Wang, J. Kehr, B. Bjelke, Y. Zhang, T. Tsakalakos and M. Muhammed, Chem. Mater., 2003, 15, 4343 CrossRef CAS.
- Z. R. Stephen, F. M. Kievit and M. Q. Zhang, Mater. Today, 2011, 14, 330 CrossRef CAS.
- J. H. Park, G. von Maltzahn, L. L. Zhang, M. P. Schwartz, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Adv. Mater., 2008, 20, 1630 CrossRef CAS.
- B. Gleich and R. Weizenecker, Nature, 2005, 435, 1214 CrossRef CAS.
- M. M. Lin, H. H. Kim, H. Kim, J. Dobson and D. K. Kim, Nanomedicine, 2010, 5, 109 CrossRef CAS.
- N. Tran and T. J. Webster, J. Mater. Chem., 2010, 20, 8760 RSC.
- W. H. Binder, D. Gloger, H. Weinstabl, G. Allmaier and E. Pittenauer, Macromolecules, 2007, 40, 3097 CrossRef CAS.
- M. Lattuada and T. A. Hatton, Langmuir, 2007, 23, 2158–2168 CrossRef CAS.
- Q. L. Fan, K. G. Neoh, E. T. Kang, B. Shuter and S. C. Wang, Biomaterials, 2007, 28, 5426 CrossRef CAS.
- C. K. Weiss and K. Landfester, in Hybrid Latex Particles: Preparation With (Mini)emulsion Polymerization, ed. A. M. van Herk & K. Landfester, Springer-Verlag, Berlin, 2010, p. 185 Search PubMed.
- F. Lan, K.-X. Liu, W. Jiang, X.-B. Zeng, Y. Wu and Z.-W. Gu, Nanotechnology, 2011, 22, 225604 CrossRef.
- J. Ban, K. Kim, H. Jung and S. Choe, J. Ind. Eng. Chem., 2010, 16, 1040 CrossRef CAS.
- J. P. Patterson, A. M. Sanchez, N. Petzetakis, T. P. Smart, T. H. Epps, III, I. Portman, N. R. Wilson and R. K. O'Reilly, Soft Matter, 2012, 8, 3322–3328 RSC.
- S. Hashimoto, T. Oda, K. Yamada, M. Takagi, T. Enomoto, N. Ohkohchi, T. Takagi, T. Kanamori, H. Ikeda, H. Yanagihara, E. Kita and A. Tasaki, Phys. Med. Biol., 2009, 54, 2571 CrossRef CAS.
- D. C. Niu, Z. Ma, Y. S. Li and J. L. Shi, J. Am. Chem. Soc., 2010, 132, 15144 CrossRef CAS.
- H. Zhang, D. Wang, R. Butler, N. L. Campbell, J. Long, B. Tan, D. J. Duncalf, A. J. Foster, A. Hopkinson, D. Taylor, D. Angus, A. I. Cooper and S. P. Rannard, Nat. Nanotechnol., 2008, 3, 506 CrossRef CAS.
- T. O. McDonald, P. Martin, J. P. Patterson, D. Smith, M. Giardiello, M. Marcello, V. See, R. K. O'Reilly and S. P. Rannard, Adv. Funct. Mater., 2012, 22, 2469 CrossRef CAS.
- L. P. Ramirez and K. Landfester, Macromol. Chem. Phys., 2003, 204, 22 CrossRef CAS.
- P. del Pino, A. Munoz-Javier, D. Vlaskou, P. Rivera Gil, C. Plank and W. J. Parak, Nano Lett., 2010, 10, 3914 CrossRef CAS.
- S. Laurent, S. Dutz, U. O. Hafeli and M. Mahmoudi, Adv. Colloid Interface Sci., 2011, 166, 8 CAS.
- B. E. Mattioni and P. C. Jurs, J. Chem. Inf. Comput. Sci., 2002, 42, 232 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TGA analysis; normalised SQID data; DLS data; UV-Vis data; optical microscope images. See DOI: 10.1039/c2jm34974d |
| This journal is © The Royal Society of Chemistry 2012 |