
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of bovine serum albumin (BSA)–dextran bio-nanoconjugate: structural, antioxidant and in vitro wound healing studies
Sonali S. Rohiwalab,
Z. Ellederovab,
Arpita P. Tiwarig,
Mohammed Alqarni c,
Sara T. Elazabd,
Gaber El-Saber Batihad,
Shivaji H. Pawarae and
Nanasaheb D. Thorat*f
c,
Sara T. Elazabd,
Gaber El-Saber Batihad,
Shivaji H. Pawarae and
Nanasaheb D. Thorat*f
aCentre for Interdisciplinary Research, D. Y. Patil University, Kolhapur – 416006, MH, India
bThe PIGMOD Center, Institute of Animal Physiology and Genetics, v. v. i., The Czech Academy of Sciences, Libechov, Czech Republic
cDepartment of Pharmaceutical Chemistry, College of Pharmacy, Taif University, P.O. Box 11099, Taif 21944, Saudi Arabia
dDepartment of Pharmacology, Faculty of Veterinary Medicine, Mansoura University, Mansoura 35516, Egypt
eCentre for Innovative and Applied Research, Anekant Education Society, T. C. College Campus, Baramati, MH, India
fDivision of Medical Science, Nuffield Department of Women's & Reproductive Health, University of Oxford, Oxford, UK. E-mail: thoratnd@gmail.com
gDepartment of Stem Cell and Regenerative Medicine, D.Y. Patil Education Society, Deemed to be University, Kolhapur, India
First published on 21st January 2021
Abstract
Glycation of proteins is often considered as a method to improve their functional properties for promising applications in wound healing. Furthermore, a marked increase in percentage of radical scavenging activity of the conjugates makes it an effective antioxidant synthetic strategy. A simple conjugation process was employed to develop bovine serum albumin–dextran conjugates (BSA–dextran) using Maillard reaction. Higher electrophoretic mobility and surface charge in the prepared conjugates was observed in native PAGE electrophoresis and zeta potential. Moreover, the fluorescence, FTIR and Raman analysis of the BSA–dextran conjugates shows significant shift in the fluorescence and wavelength as a consequence of conjugate formation. In vitro wound healing assay showed increased cell proliferation and migration effect. These finding suggests that BSA–dextran conjugate could open up a new practical way for exploration in the area of wound healing.
1. Introduction
Protein–polysaccharide conjugates, in which proteins are covalently bound to polysaccharides, have attracted substantial interest in the areas of wound healing, molecular diagnostics, protein therapeutics and tissue engineering. The increased understanding and synthetic control over polysaccharide and glycopolymer structures enhances their structural properties to be used in the biomedical field.1–3 The formation of protein conjugates is attributed to the ubiquitous nature of protein glycation reactions and the modification of some functional properties of proteins after their conjugation with carbohydrates.4 The glycation of proteins with polysaccharides by controlled dry heating involving the Maillard reaction is an effective way to improve certain functional properties of proteins; including their solubility, emulsifying properties, gel properties, and heat stability.5 Moreover, this approach also leads to improve other properties such as enhancement of their antioxidant effects and broadened their bactericidal effects along with its anticarcinogenic, antimutagenic and antihypertensive characteristics.6,7 At an early stage of the Maillard reaction, the protein BSA containing free amino groups, such as the ε-NH2 groups of lysine, terminal amino and guanido group of arginine, reacts with the carbonyl groups of dextran which after rearrangement step forms irreversible Maillard reaction products or Amadori products. In the advanced phase of the reaction, Amadori products undergo further transformation to fluorescent, colored substances, and cross-linked polymers.8BSA is prominent and most widely utilized serum protein structural/functional similarity to human serum albumin having 76% sequence homology and nearly identical pH-dependent conformational transitions. BSA is a water-soluble monomeric protein with a primary structure containing a single polypeptide chain with 583 amino acid residues and a molar mass of 66.4 kDa.9–11 The widespread use of polysaccharides along with proteins in various biological applications due to its low immunological response.12
Dextran is one of the widely used natural polysaccharide consisting of linear chains of α – 1,6 linked D-glucopyranose residues. Dextran has three hydroxyl groups on each glucopyranose repeat unit that are available for chemical modification with either cross-linking agents for hydrogel formation13,14 or bioactive molecules.15–19 Chevalier et al. had reported that the proteins modified with ribose and arabinose showed stronger radical-scavenging activity and suggested that the glycated protein could be used as an antioxidant in formulated foods as a functional ingredient due to its radical-scavenging activity and capacity to delay oxidative deterioration.20–22
The antioxidant protective system which involves enzymatic and non-enzymatic components accelerates wound healing. The wound healing process might be delayed or severe tissue damage can occur without proper balance of antioxidant activity.23 As well, it was noticed that the high oxidative stress leads to the depletion of non enzymatic antioxidants. This effect is more pronounced in chronic wounds than in acute wounds. Thus, supplementation for wounds with antioxidants should help to prevent oxidative damage of cells and enhance healing.24
In the present work, we prepare the BSA–dextran conjugate through simple conjugation process via Maillard reaction. The antioxidant properties of the conjugates were studied which is capable for effective wound healing process. Native PAGE electrophoresis and zeta potential displays the differential mobility of the prepared BSA–dextran conjugates. The spectroscopic methods such as UV-visible, fluorescence, FTIR and FT-Raman and circular dichronism (CD) spectroscopies provide an orthogonal comparison to these physical characterization approaches. Additionally, it provides the chemical specificity that allows the study of multiple bonds which are taking part in bonding and to quantify the change in the BSA conformation following conjugation. Further in vitro wound healing assay provides information regarding effective cell proliferation and migration. This report expands the horizon of using BSA–dextran conjugates as a potential carrier for enhancement of the wound healing activity with the help of its unique antioxidant property.
2. Experimental details
2.1 Reagent and chemicals
BSA (65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da), dextran (60000–90000 Da), acrylaminde and bis acrylamide, were procured from Sigma, India and were used without further purification. All the procured chemicals were of analytical grade. All aqueous solutions were prepared in double distilled water.
000 Da), dextran (60000–90000 Da), acrylaminde and bis acrylamide, were procured from Sigma, India and were used without further purification. All the procured chemicals were of analytical grade. All aqueous solutions were prepared in double distilled water.
2.2 Preparation of the BSA–dextran conjugates by dry heating
The BSA–dextran conjugates were prepared by the Maillard reaction with some modifications. The BSA–dextran mixtures in molar ratios of 1:1, 1:2, 1:3, and 1:4 were dissolved in distilled water and freeze-dried as reported previously.5 The lyophilized mixtures were stored in desiccators for seven days in the presence of saturated KBr solution at 60 °C and 79% relative humidity.
2.3 Characterization
To purify the BSA–dextran conjugates, the products were dissolved in deionized distilled water to a concentration of 0.25% (w/v) and passed through a 0.45 μm pore size membrane filter. Ultra-filtration was then performed with an YM-100 membrane (molecular weight cut-off of 100000; model HF-1, Ecoyeth Vision, Kharagpur, Kolkata, India) until half of the initial volume remained. The deionized distilled water was then added till the original volume. And the procedure was repeated three times. The retentates were freeze-dried and stored at 4 °C. The UV spectra of BSA–dextran conjugates and native BSA were measured in the wavelength range of 250–400 nm at room temperature with UV spectrophotometer (UV-3600 UV-visible-NIR spectrophotometer by SHIMADZU). The molecular structure of BSA–dextran conjugates was determined by FTIR spectroscopy and scanned at range (500–4000 cm−1) with Alpha ATR Bruker (Eco Model). The samples for FTIR analysis were prepared by grinding 98.99% KBr with 1–2% of the conjugates and pressing the mix to form a tablet.
For the FT-Raman spectra of the films were prepared and recorded in the spectral range of 39–3600 cm−1 using Fourier Transform Raman spectrometer (Bruker Multi-RAM, Germany Make) Nd:YAG laser source with excitation wavelength of 1064 nm and resolution 4 cm−1 at 336 mW laser power. Fluorescence spectra were measured after excitation at 280 nm, scanned at an emission wavelength range between 300 nm to 500 nm using a fluorescence spectrophotometer (Model no FP 8300, Spectrofluorimeter JASCO, Japan) at room temperature; using a quartz cuvette. The analysis was carried out at an excitation and emission slit wavelength of 3 to 5 nm, respectively. For zeta potential measurements the ultra-filtered samples of BSA–dextran conjugates were dispersed in 10 mM PBS (pH 8) and the dispersions were sonicated for 10 minutes at 25 °C, samples were scanned continuously. Each result was calculated from the average of 10 runs.
2.4 Native PAGE electrophoresis
Native polyacrylamide gel electrophoresis was performed25 with some modification using 4% stacking gel and 10% separation gel. One aliquot of glycated BSA solution was mixed with an equal volume of sample buffer (30% glycerol, 0.25 M Tris–HCl buffer, pH 6.8). Each lane was loaded with 20 μg of protein. Electrophoresis was carried out at constant current of 20 mA for about 3 h. Tris-glycine buffer (25 mM Tris and 250 mM glycine, pH 8.3) was used as the tank buffer. Gels were stained with Coomassie Brillant Blue R-250. Electrophoretic mobilities were obtained and calculated by the following formula.26where, d is distance travelled by protein band (in meter), l is length of the gel (in meter), t is time of electrophoresis (in sec) and U is potential difference applied (in V).

CD spectra were acquired with a J-815 spectropolarimeter (Jasco, Tokyo, Japan) to investigate differences in the secondary structures of dextran-conjugated BSA and native BSA. The spectral bandwidth was 2 nm and the time constant was 2 s at 25 °C. The concentration of the conjugate was 0.1 mg mL−1 of BSA in a 0.01 M phosphate buffer (pH 7.5), and the light path length of the quartz cell was 0.1 cm. For further analysis serial dilutions of native BSA as well as for BSA–dextran conjugates were made. CD measurement was performed in the range of 190–250 nm. The results were expressed in terms of mili ellipticity θ in deg cm2 dmol−1 and the relative α-helix content based on the relative intensity at 222 nm.
2.5 Determination of DPPH radical-scavenging activity
DPPH radical-scavenging activity of the MRPs was determined according to Gu et al. with modifications.27 An aliquot of BSA, dextran and MRPs (0.2 mL) was added to 4.8 mL of 0.25 mM DPPH in methanol. The solution was then mixed vigorously and allowed to stand at room temperature in the dark for 30 min. The mixture was centrifuged at 750 g for 10 min. The absorbance of supernatant was measured at 517 nm. These results were the average of three measurements and expressed as radical-scavenging activity (%). The percentage of DPPH radical-scavenging activity was calculated as follows:| Radical-scavenging activity (%) = [1 − (A517 nmblank − A517 nmtest)/A517 nmblank] × 100 |
2.6 Cell proliferation assay
For evaluation of cytotoxicity the AlamarBlue cell viability assay (Thermo Fisher Scientific) was performed. 3T3 fibroblasts cells were seeded on 12 well TPP® tissue culture plates (Sigma Aldrich), until the cell confluency reached up to 70–80%. The cells were incubated with all four conjugates 1:1, 1:2, 1:3 and 1:4 and control including only medium. Then 100 μL of AlamarBlue reagent (10×) was added to the cells, and incubated at 37 °C for 2 h before measuring the absorbance at 570 nm.
2.7 Scratch wound healing assay
The stimulatory effect of conjugate on migration of 3T3 fibroblasts cells was determined. The 3T3 fibroblast cells were seeded at a density of 5 × 105 cells per well into a 24-well TPP plate containing DMEM culture medium supplemented with 10% FBS and incubated overnight at 37 °C, in a humidified 5% CO2 atmosphere. After incubation, DMEM was completely removed and the adherent cell layer was scratched with a sterile pipette tip. Cellular debris was removed by washing off with phosphate buffer saline (PBS). The cells were treated with DMEM with 10% FBS containing 0.1 g mL−1 (dry weight) of conjugate, controls received only fresh DMEM. The cells were incubated (at 37 °C in humidified 5% CO2 atmosphere for 0 h, 24 h and 48 h) and then the recording of images of the scratch area was carried out in three different points, using a built-in camera in the microscope (10× magnification) at 0 h (just after scratching cells) and at 24 h and 48 h after incubation with 1:4 conjugate and control. Data were analyzed with TScratch wound healing analyzing tool in order to determine the width of the scratch and thus to calculate the rate of cells migration.
3. Results and discussion
3.1 Preparation of BSA–dextran bionano-conjugate with Maillard reaction
Maillard reaction is a useful tool to synthesize glycoconjugates of proteins. Such conjugates may help in wound healing by virtue of their antioxidant activity. Maillard reaction is influenced by reaction conditions like pH, temperature, time and relative humidity.28 The reaction rate was moderate and the side reactions were not obvious in the condition of the solid phase Maillard reaction at 60 °C and a relative humidity of 78.9% and neutral pH. The detail schematic representation of BSA–dextran conjugates prepared through Maillard reaction is shown in Fig. 1. The Maillard reaction was controlled in the very early stage to obtain early stage Maillard reaction product. After Maillard reaction native PAGE was performed to confirm the attachment. The higher mobility of the BSA–dextran conjugates proves that BSA has attached to higher molecular weight dextran. It also indicates that longer the reaction time more dextran molecules is attached to BSA and larger molecular weight of copolymer is formed. | ||
| Fig. 1 Schematic representation of BSA–dextran conjugates prepared through Maillard reaction. | ||
3.2 Native PAGE electrophoresis
To reveal the attachment of BSA with dextran the native PAGE was carried out as shown in Fig. 2 and the change in the electrophoretic mobilities of protein was calculated. | ||
| Fig. 2 Native polyacrylamide gel electrophoresis native PAGE profile of BSA–dextran conjugates after 7 days of Maillard reaction. Lane 1, native BSA; lane 2, native BSA + 250 mM glucose; lane 3, 1:1 conjugate, lane 4, 1:2 conjugate, lane 5, 1:3 conjugate, lane 6, 1:4 conjugate. | ||
The BSA attached dextran migrated with higher electrophoretic mobility than native BSA. The attachment of dextran affects the surface charge of proteins which attributes to the oxidative modification of surface amino acids. The pI of the glycated BSA has higher acidic value than native BSA. These observations were corroborated by zeta potential (ζ) measurements as shown in Table 1. It was observed that as the concentration of dextran increases with the increase in its zeta potential value. The increase in dextran concentration is the factor influences the electrophoresis mobility and the shape of the protein molecule. The glycation with high molecular weight dextran causes the folding of BSA molecule; ultimately causes a gain in the electrophoresis mobility.
| Parameter | Native BSA | Glu-BSA | 1:1 |
1:2 |
1:3 |
1:4 |
|---|---|---|---|---|---|---|
| Electrophoretic mobility μ (10 −8 m2 s−1 V−1) | 1.14 | 1.26 | 1.21 | 1.22 | 1.23 | 1.23 |
| Zeta potential ζ (mV) | −11.8 | −15.1 | −17 | −22 | −40 | −48 |
3.3 Zeta potential measurement
In this technique, voltage was applied across a pair of electrodes which is placed at both ends of a cell containing the particle dispersion of the BSA–dextran conjugate. There is the presence of fixed charge associated with any particle and is always surrounded by counter ions.29 Therefore, as positive counter ionic potential increases bound negative charge also increases. Charged particles are attracted towards the oppositely charged electrode. Since, the zeta potential is pH dependent, the measurements were performed at pH 8.0.In agreement with the native electrophoresis the zeta potential of the glycated BSA was shown to be higher than that of native protein. It was observed that as the concentration of dextran increases the zeta potential value increases. The zeta potential value for 1:1, 1:2, 1:3 and 1:4 were −17 mV, −22 mV, −40 mV and −48 mV respectively (Table 1), which is higher than that of the native BSA (−11.8 mV), Glu–BSA (−15.1) and dextran (−4 mV).
3.4 UV absorption spectroscopy of BSA–dextran conjugates
To study the effect of dextran conjugation on absorption properties of BSA, the absorption spectra of native BSA and BSA–dextran conjugates (1:1, 1:2, 1:3 and 1:4) were recorded as shown in Fig. 3(A). There is presence of aromatic amino acids like tyrosine, tryptophan, phenylalanine and disulphide bonds in native BSA which exhibits an absorption maximum peak at 282 nm. The BSA–dextran conjugates have the maximum absorption peak at around 279 nm with slight peak shifting. The shift in the wavelength to the lower value is called blue shift. This signifies that, there is a folding of native structure of protein and might be due to the shifting of tryptophanyl and tyrosyl residues from their values in aqueous solutions when they are either buried in the interior or lying on the surface of the protein.30 After conjugation the native structure of BSA is changed showing some conformational transitions. Moreover, the change in intensity of BSA (1.21 a.u.) and BSA–dextran conjugates were observed to be 0.422, 0.758, 0.525 and 0.727 a.u. for 1:1, 1:2, 1:3 and 1:4, respectively displaying the possibility of denaturation of some BSA molecules. Additionally, the change in the intensity of BSA in conjugates is due to the gradual concealment of aromatic amino acids i.e. tyrosine and tryptophan which plays a major role in absorbance intensity. Thus, it reveals that there is masking a of BSA surface with high molecular weight polymer dextran.
 | ||
| Fig. 3 (A) UV-visible absorption and (B) fluorescence spectroscopy spectrum for (a) native BSA, BSA–dextran conjugate of (b) 1:1 (c) 1:2 (d) 1:3 and (e) 1:4 molar ratios prepared at pH 7.0. | ||
3.5 Fluorescence spectroscopy
The intrinsic fluorescence spectroscopies of the BSA–dextran conjugates were analyzed to evaluate the conformational change in tryptophan. The fluorescence analysis of the BSA–dextran conjugates shows significant shift in the wavelength of the maximum of the intrinsic fluorescent emission with quenching of fluorescence, it clearly indicates that the protein conformation remained changed as a consequence of the conjugate formation. Fluorescence is usually dominated by the contribution of the tryptophan residues out of three aromatic amino acids i.e. tyrosine, tryptophan and phenylalanine. For both tyrosine and phenylalanine the absorbency at the wavelength of excitation and their quantum yield of emission are considerably lower than tryptophan. The tryptophan emission of a native protein and conjugates can be larger or smaller than the emission of free tryptophan in aqueous solution. Fluorescence emission spectra showed that, when excited at 280 nm, the maximum emission intensity appears at the wavelength of 328 nm for native BSA (Fig. 3(B)), and at 306 nm, 310 nm, 309 nm and 308 nm for the 1:1, 1:2, 1:3 and 1:4 conjugates respectively. Since considerable shift is observed in the conjugates, the microenvironment around the Trp residues of the BSA–dextran conjugates likely has been dissimilar to that of native BSA. Additionally, it was observed that as dextran concentration increases there is a decrease in the fluorescence intensity.31 Thus, a reduction in the fluorescence intensity suggests that the attachment of dextran to BSA affects partially the side chains of proteins in tertiary structure, without great disruption of native structure of BSA. It also suggests that binding of dextran to BSA in each conjugate occurred at the surface of the BSA molecule and fluorescence from the protein was shielded.
In the structure of BSA there is presence of two tryptophan groups which are embedded in two different domains: Trp134, located in proximity of the protein surface, but it is obscured in a hydrophobic pocket domain I, and Try214, located in an internal part of domain II.32 The Trp134 has larger spectral contribution, and Trp214 possesses a smaller fluorescence emission. This suggests that the main contribution to fluorescence is related to Trp134 which is located in the external part of domain I, which is more accessible to solvent.
3.6 Fourier-transform infrared (FTIR) spectroscopy
FTIR spectroscopy was used to investigate the nature of interactions between BSA and dextran as shown in Fig. 4(A). The secondary structure of proteins are determined by analyzing the structure of amide bonds which are centered at 3385 cm−1, 3113 cm−1, 1707 cm−1, 1533 cm−1 and 1242 cm−1, assigned to the stretching vibration of O–H, amide A (NH stretching vibration), amide I (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations), amide II (the coupling out phase of bending vibration of N–H and stretching vibration of C–N bands) and amide III is in the phase combination of N–H in plane bending and C–N stretching. Similarly, the spectrum of dextran exhibits polysaccharide characteristics absorption bands located in the region of 1180 to 953 cm−1 from vibration modes such as the stretching of C–C and C–O and the bending mode of C–H bonds.33 These are referred as the “saccharide” bands which are having more intensity at the mid-infrared spectrum region.34 The bands present in the dextran at 3521 cm−1, 1642 cm−1, 1146 cm−1 and 1084 cm−1 are due to the O–H stretching; water molecule bending, C–O vibrations respectively. While the bands at 913 cm−1, 845 cm−1 and 762 cm−1 are corresponding to α-glucopyranose ring deformation modes.
O stretching vibrations), amide II (the coupling out phase of bending vibration of N–H and stretching vibration of C–N bands) and amide III is in the phase combination of N–H in plane bending and C–N stretching. Similarly, the spectrum of dextran exhibits polysaccharide characteristics absorption bands located in the region of 1180 to 953 cm−1 from vibration modes such as the stretching of C–C and C–O and the bending mode of C–H bonds.33 These are referred as the “saccharide” bands which are having more intensity at the mid-infrared spectrum region.34 The bands present in the dextran at 3521 cm−1, 1642 cm−1, 1146 cm−1 and 1084 cm−1 are due to the O–H stretching; water molecule bending, C–O vibrations respectively. While the bands at 913 cm−1, 845 cm−1 and 762 cm−1 are corresponding to α-glucopyranose ring deformation modes.
 | ||
| Fig. 4 (A) FTIR and (B) Raman spectra for (a) native BSA, (b) dextran, and BSA–dextran conjugate of (c) 1:1, (d) 1:2, (e) 1:3 and (f) 1:4 molar ratios prepared at pH 7.0. | ||
The entire spectral features of the amide III of the conjugates showed decrease in the intensity. The partial shifting in the wavelength was observed with slight change in the native structure of BSA and dextran which might be due the chemical changes accompanying the Maillard reaction. Some functional groups including NH2 especially from lysine may be modified and the other Maillard products such as the Amadori compound (CO), Schiff base (CN) and pyrazines (C–N) may be increased by the Maillard reaction,35 which results in the substantial increase in the intensity of all the bands in the conjugates as compared to the native BSA and dextran. The partial changes in the wavelength are shown in the Table 2. The detail structural conformations were not visible in FTIR, so FT-Raman spectroscopy is acquired to investigate the structural interaction between protein and polysaccharide.
:1, 1:2, 1:3 and 1:4
| Assignationa unit: cm−1 | BSA | Dextran | Sample 1 | Sample 2 | Sample 3 | Sample 4 |
|---|---|---|---|---|---|---|
| a The band positions are in (cm−1) unit. | ||||||
| 1:1 |
1:2 |
1:3 |
1:4 |
|||
| O–H stretching | 3385 | — | 3392 | 3394 | 3396 | 3390 |
| Amide A (mainly NH stretching vibration) | 3113 | — | 3111 | 3111 | 3115 | 3117 |
| Amide I (mainly CO stretching vibrations) |
1707 | — | 1697 | 1683 | 1690 | 1693 |
| Amide II (the coupling out phase of bending vibration of N–H and stretching vibration of C–N bands) | 1533 | — | 1514 | 1529 | 1518 | 1516 |
| Amide III (is in the phase combination of N–H in plane bending and C–N stretching) | 1242 | — | 1274 | 1271 | 1336 | 1272 |
| O–H stretching | — | 3521 | 3608 | 3566 | 3553 | 3531 |
| Water molecule bending | — | 1642 | — | 1648 | — | 1645 |
| C–O vibrations | — | 1146 | 1147 | 1144 | 1146 | 1145 |
| 1084 | 997 | 992 | 1007 | 991 | ||
| α-Glucopyranose ring deformation modes | — | 913 | 901 | 902 | 794 | 899 |
| 845 | 790 | 749 | 750 | 753 | ||
| 762 | 752 | 649 | 696 | 697 | ||
3.7 FT-Raman spectroscopy
FT-Raman spectroscopy is a useful tool to investigate the structural and conformational characteristics of protein and polysaccharide. It was observed from the results that both FTIR and FT-Raman exhibit almost similar bands. Fig. 4(B) displays the FT-Raman spectra of native BSA, dextran and BSA–dextran conjugates (1:1, 1:2, 1:3 and 1:4) in the 250–3600 cm−1 region. Interpretation of the spectrum begins with those lines that are characteristic of the peptide backbone which was observed from the frequencies and intensities of the amide I band (CO stretching vibration) at 1661 cm−1. The amide III bands (mixture of C–N bond stretching and in-plane N–H bond bending) at 1277 cm−1 and 1246 cm−1 bands are associated with α-helix and random coil conformations. The band centered at 1551 cm−1 is contributed for amide II band. Along with all these bands the FT-Raman spectra also displays the bands for the aromatic ring modes. There is the presence of Tyr Fermi doublet at 824 cm−1 and 856 cm−1 and the 900–1000 cm−1 skeletal vibration regions.
Similarly, for saccharide some characteristic bands are attributed in the region at 409 cm−1 for skeletal modes of pyranose ring. The vibrations originating from the C–O–C of α-1,4a glycosidic linkage was observed as strong FT-Raman band vicinity at 913 cm−1. The band at 1132 cm−1 contributes to the two main vibrational modes, C–O stretching and C–O–H deformation and the band which is present at 1094 cm−1 attributed for the C–O–H deformation. The 1460 cm−1 band is associated with the CH2 deformation and 1356 cm−1 band in FT-Raman spectra is probably due to bending modes of C–H. The absorption band at 1272 cm−1 is a characteristic for CH2OH as well as for the C–O–H. Vibrational band originated due to the presence of carbon and hydrogen atoms is present in the region 1500–1300 cm−1. The coupling modes of C–O and C–C assigned is at 1040 cm−1. The observed wide band at 3100–3600 cm−1 shows the presence of O–H stretching.
It was observed that for all the BSA–dextran conjugates 1:1, 1:2, 1:3 and 1:4 there is slight difference in bonding with slight distortion of the characteristics bands of BSA (amide I, amide II, amide III and Fermi bands of tyrosine) and dextran (α-glucopyranose ring and C–O–C of α-1,4 glycosidic linkages). Decrease in the intensity of all characteristic bands was observed in the conjugates (1:1, 1:2, 1:3 and 1:4) as compared to that of control.
3.8 Secondary structural analysis of BSA and BSA–dextran conjugates by Far-UV circular dichroism (CD)
The secondary structure of BSA was also examined with help of Far-UV CD in the spectral region (190–250 nm). The CD spectrum from native BSA and BSA–dextran conjugates at pH 7.0 exhibited two negative minima at 208 nm and 222 nm and a maximum at 195–196 nm, indicating the presence of high α-helical content. The secondary structure of BSA contains about 50–68% α-helices and 16–21% β-structures.36 Interestingly, the increase in dextran concentration i.e. from (1:1 to 1:4) leads to gradual decrease in the negative band at 222 nm and 209 nm towards the baseline (less negative values) as shown in Fig. 5(A). These results indicate that there is slight decrease in the α-helical content of BSA in BSA–dextran conjugates, signifying that some conformational changes have occurred in BSA during conjugate formation.
 | ||
| Fig. 5 (A) Far-UV and (B) near-UV circular dichroism (CD) for (a) native BSA, and BSA–dextran conjugates (b) 1:1, (c) 1:2, (d) 1:3 and (e) 1:4 molar ratios prepared at pH 7.0. | ||
3.9 Tertiary structural analysis of BSA and BSA–dextran conjugates by near-UV circular dichroism (CD)
Alteration in the tertiary structure of BSA upon conjugate formation was examined by near-UV CD spectroscopy, which is sensitive to polarity, and thus the solvent exposure of the local environment of aromatic residues (27 Phe, 20 Tyr and 2 Trp) and 35 Cys that constitute 17 disulphide bridges and one free thiol of cysteine. Any transformations around these aromatic residues provide the useful information about the fluctuation in the tertiary structure of BSA in BSA–dextran conjugates. The near-UV CD spectrum of BSA and BSA–dextran conjugates is shown in Fig. 5, displayed two maxima 260 nm and 268 nm which are ascribed to phenylalanine side chains as studied previously.37 There is remarkable decrease in the ellipticity of both the peaks (260 nm and 268 nm) as the concentration of dextran is increasing i.e. from 1:1 to 1:4 (as shown in Fig. 5(B)). This indicates that there is gradual concealment of the aromatic amino acids from hydrophobic core of BSA molecule to the less polar environment upon the conjugation, suggesting that some changes might have occurred in tertiary structure.
3.10 Second-derivative UV
The ellipticity of the aromatic residues has a characteristic contribution at wavelength Trp (290–305 nm), Tyr (275–282 nm) and Phe (270–255 nm). The signals at 250 nm and 320 nm are attributed for the ellipticity of disulphide bridges. The Tyr showed maxima wavelength at 278 nm with the significant ellipticity at −3.05 for BSA molecules. For BSA–dextran conjugates slight blue shift at 277.22 nm, 277.83 nm, 276.12 nm and 276 nm was observed, with decrease in ellipticity (−4.91, −11.30, −6.35 and −11.91). The results point out that Tyr has become more exposed to solvent after conjugation which is also confirmed by fluorescence spectroscopy and exhibiting the same.3.11 Radical-scavenging activity
DPPH has been widely used for the determination of primary antioxidant activity i.e. the free radical scavenging activities of pure antioxidant compounds.38 DPPH radical was scavenged by MRPs by donation of hydrogen to form a stable DPPH-H molecule. The color of DPPH reagent changes from purple to yellow by acceptance of hydrogen radical from MRPs and a stable diamagnetic molecule is formed. The radical scavenging activity was in the decreasing order as follows: 1:4 > 1:3 > 1:2 > 1:1. This result could be explained by the highest MRPs formation during heating of BSA–dextran conjugate in 1:4 molar ratios, as the molar ratio of dextran was 4 times greater. There are more possibilities of dextran molecules to attach with BSA molecules in order to form MRPs; these results are in accordance with other report.27
Guérard et al. described that the MRPs obtained from protein hydrolysates showed darker color formation leads to stronger antioxidant activity, similar results were obtained in our case.39 The 1:4 BSA–dextran conjugate had shown the maximum radical scavenging activity as compared to other conjugates (1:1, 1:2 and 1:3). Similarly, Dong et al. has studied the antioxidant activity of hydrolyzed β-lactoglobulin–glucose MRPs with respect to heating time, maximum% inhibition was observed at heating time at and above 18 h.40 Herein, the heating time was 168 h showing 93% of inhibition. The changes in the DPPH radical scavenging activity of MRPs derived from all BSA–dextran conjugates are 74.39%, 88.53%, 91.21% and 93.88% for conjugate 1:1, 1:2, 1:3 and 1:4 respectively as shown in Fig. 6.
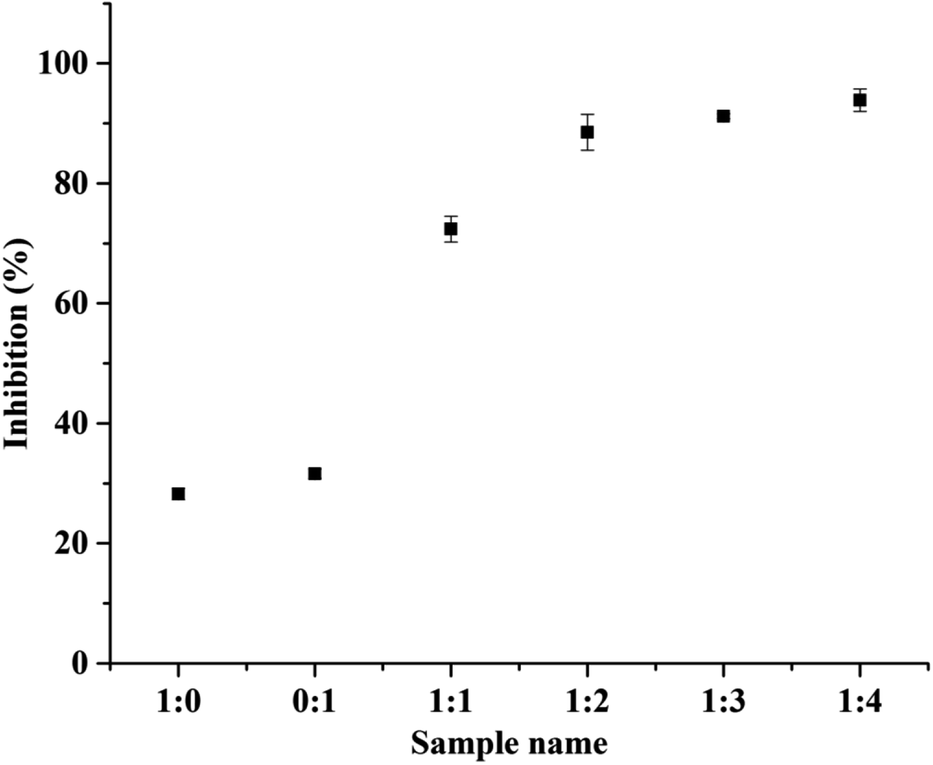 | ||
| Fig. 6 DPPH radical scavenging activity of controls BSA (1:0), dextran (0:1) and BSA–dextran conjugates of molar ratio 1:1, 1:2, 1:3 and 1:4. Data represent mean values ± SE of three independent experiments performed in triplicates. | ||
3.12 Cell viability assay
The cellular metabolic activity and population of live/dead cells relative to the untreated blank of 3T3 fibroblasts cells 24 h and 48 h post incubation with all the four conjugates is shown in Fig. 7(A). The viability of cells on conjugate sand control was evaluated by the AlamarBlue viability assay at different time points. The Alamar Blue assay incorporates a redox indicator that changes color or fluorescence in response to metabolic activity, and this is also strongly related to cell proliferation. The Alamar signal increased in cells growing in the presence of conjugate and then remained relatively constant as compare to the control. Similar results were observed for all four conjugates, the intensity increased with time and then remained constant. Thus, cells are able to grow and increase in number in the presence of all different conjugate. | ||
| Fig. 7 (A) Proliferation of 3T3 cells incubated with 0.1 g mL−1 of conjugates having molar ratio 1:1, 1:2, 1:3 and 1:4 in presence of medium. The control means cells growing in medium without any addition of the conjugate. (*P > 0.05, non-significant). Proliferation is presented as the absorbance in arbitrary units (Alamar Blue), (B) microscopy images of 3T3 fibroblast cells migration after scratch (i) control without treatment at 0 h, 24 h and 48 h and (ii) after treatment with 1:4 molar ratio conjugate at 0 h, 24 h and 48 h of 0.1 g mL−1 concentration. The scale bar is 10 mm. Data represent mean values ± SE of three independent experiments. Statistical analysis: paired t test ***P < 0.0005. | ||
3.13 Scratch wound healing assay
The evaluation of the effectiveness of BSA–dextran conjugate in wound healing process was performed in vitro using 3T3 fibroblasts cell line. Currently, cell culture is a popular and effective method to test the sensitivity of cells to selected groups present in the microenvironment. 3T3 fibroblasts cell have been proposed as a method for testing wound healing activity in vitro.41 Cell proliferation and migration are two extremely important features during the tissue formation phase in the wound healing. Scratch assay is a form to mimic a wound healing in in vitro. After disruption of cell monolayer there is a loss of cell to cell interaction which results in increase in the concentration of growth factors and cytokines at the edge of the wound resulting in initiation of migration and proliferation of cells.Remarkably, the extracts of 1:4 BSA–dextran conjugate (1:4 conjugate is used to perform scratch wound healing assay because it showed the best antioxidant activity) was able to promote fibroblasts proliferation and migration as compare to control (Fig. 7(B)(ii)). After 24 h of incubation with the extract of conjugate there was 26% ± 0.27 of open wounded area whereas in control it showed up to 53% ± 0.16 opened wounded area. After 48 h of incubation only 15% ± 0.41 of wounded area was open as compared to control in which wounded area was 46% ± 0.44. These results are in accordance with the cell proliferation assay showing increase in rate of metabolic activity as compared to control (Fig. 7(A)). This improved cell migratory effect evidence could be due to the ROS scavenging activity of BSA–dextran conjugate. There are several other polymeric conjugates are developed especially for wound healing like Zhu et al. have successfully fabricated a dextran–hyaluronic acid hydrogel enriched with sanguinarine (SA) incorporated into gelatin microspheres, which had high porosity, good swelling ratio, enhanced NIH-3T3 fibroblast cell proliferation, and sustained SA release profile.41 Similarly, Sun et al. have synthesized dextran-based polymers and scaffolds for controlled release and tissue engineering. They developed dextran-based hydrogels that are precisely manipulated with desired structural properties and encapsulated with angiogenic growth factors for therapeutic neovascularization, as well as their potential for wound repair.42 Wang et al. reported a novel dual-functional biodegradable dextran–poly(ethylene glycol) (PEG) hydrogel covalently conjugated with antibacterial Polymyxin B and Vancomycin (Vanco). This hydrogel is designed as a specific wound dressing material that eliminates prevailing bacteria and prevents further bacteria growth, while, enriching the side effects of antibiotics and accelerating tissue repair and regeneration.43
4. Conclusion
This study demonstrates that the synthesis of BSA–dextran conjugate through a green dry-heat process i.e. Maillard reaction provides potential bioactive material in wound healing. The BSA–dextran conjugate was interrogated both qualitatively and quantitatively via physiochemical characterization methods. The native PAGE and zeta potential were used to verify BSA–dextran conjugation and to identify the surface charge of the conjugates. Under the basic conditions the BSA attached dextran migrated with higher electrophoretic mobility i.e. 1.21, 1.22, 1.23 and 1.23 μ (10 −8 m2 s−1 V−1) than native BSA (1.14 μ (10 −8 m2 s−1 V−1)). These results were in accordance with zeta potential values of the conjugates (−17, −22, −40 and −48 mV), showing the influence of dextran concentration on mobilities. The structural evaluation was carried out by FTIR, FT-Raman, and fluorescence, far and near CD spectroscopy. The conjugates showed 93.88% inhibition in the radical scavenging activity which serves as novel material as it showed the best antioxidant activity as compared to the other conjugates. The scratch in vitro wound healing activity showed the effective cell migration and proliferation which is important for enhancement of the wound healing process. The strategy used in this work represents a simple method to obtain glycol-conjugates and its antioxidant and wound healing property is competent to be used as a bio-conjugate for wound healing application.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Indian Institute of Science, Bangalore, Center for Cellular and Molecular Biology, Hyderabad, Department of Biotechnology, Pune University, Pune and Department of Chemistry, Shivaji University, Kolhapur, MS, India for supporting our research. This work has been financially supported by the 0177-18 Seed Fund 940 through the European Huntington Disease Network and the National Sustainability Program of the Czech Ministry of Education, Youth and Sports (Project Number LO1609). The authors would also like to thank M. Vaskovicova for providing the 3T3 fibroblasts cells, the PIGMOD center, Institute of Animal Physiology and Genetics, v. v. i., the Czech Academy of Sciences, Libechov, Czech Republic.References
- D. Grande, S. Baskaran, C. Baskaran, Y. Gnanou and E. L. Chaikof, Macromolecules, 2000, 33, 1123–1125 CrossRef CAS.
- X. Z. Shu, Y. Liu, F. S. Palumbo, Y. Luo and G. D. Prestwich, Biomaterials, 2004, 25, 1339–1348 CrossRef CAS.
- D. Grande, S. Baskaran and E. L. Chaikof, Macromolecules, 2001, 34, 1640–1646 CrossRef CAS.
- F. K. Yeboah, I. Alli and V. A. Yaylayan, J. Agric. Food Chem., 1999, 47, 3164–3172 CrossRef CAS.
- S. H. Jung, S. J. Choi, H. J. Kim and T. W. Moon, Biosci., Biotechnol., Biochem., 2006, 70, 2064–2070 CrossRef CAS.
- Y. Sun, S. Hayakawa, M. Chuamanochan, M. Fujimoto, A. Innun and K. Izumori, Biosci., Biotechnol., Biochem., 2006, 70, 598–605 CrossRef CAS.
- J. A. Rufián-Henares and F. J. Morales, J. Agric. Food Chem., 2007, 55, 1480–1485 CrossRef.
- H. Jing and D. D. Kitts, Food Chem. Toxicol., 2002, 40, 1007–1015 CrossRef CAS.
- T. Peters, Adv. Protein Chem., 1985, 37, 161–245 CrossRef CAS.
- K. Hirayama, S. Akashi, M. Furuya and K. ichi Fukuhara, Biochem. Biophys. Res. Commun., 1990, 173, 639–646 CrossRef CAS.
- S. Era and M. Sogami, J. Pept. Res., 2009, 52, 431–442 CrossRef.
- S. G. Lévesque and M. S. Shoichet, Bioconjugate Chem., 2007, 18, 874–885 CrossRef.
- W. N. E. Van Dijk-Wolthuis, S. K. Y. Tsang, J. J. Kettenes-Van Den Bosch and W. E. Hennink, Polymer, 1997, 38, 6235–6242 CrossRef CAS.
- S. G. Lévesque, R. M. Lim and M. S. Shoichet, Biomaterials, 2005, 26, 7436–7446 CrossRef.
- M. P. Ribeiro, P. I. Morgado, S. P. Miguel, P. Coutinho and I. J. Correia, Mater. Sci. Eng., C, 2013, 33, 2958–2966 CrossRef CAS.
- G. Sun, X. Zhang, Y. I. Shen, R. Sebastian, L. E. Dickinson, K. Fox-Talbot, M. Reinblatt, C. Steenbergen, J. W. Harmon and S. Gerecht, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20976–20981 CrossRef CAS.
- M. Baudyš, D. Letourneur, F. Liu, D. Mix, J. Jozefonvicz and S. W. Kim, Bioconjugate Chem., 1998, 9, 176–183 CrossRef.
- S. Sugahara, M. Kajiki, H. Kuriyama and T. Kobayashi, Biol. Pharm. Bull., 2002, 25, 632–641 CrossRef CAS.
- Y. Chau, F. E. Tan and R. Langer, Bioconjugate Chem., 2004, 15, 931–941 CrossRef CAS.
- F. Chevalier, J. M. Chobert, C. Genot and T. Haertlé, J. Agric. Food Chem., 2001, 49, 5031–5038 CrossRef CAS.
- J. C. Slaughter, Biol. Rev., 2007, 74, 259–276 CrossRef.
- N. Phisut and B. Jiraporn, International Food Research Journal, 2013, 20, 1077–1085 CAS.
- T. Ponrasu, M. Kannappan Subamekala, M. Ganeshkumar and L. Suguna, J. Pharmacogn. Phytochem., 2013, 2, 77–84 Search PubMed.
- A. Soneja, M. Drews and T. Malinski, Pharmacol. Rep., 2005, 57, 108–119 Search PubMed.
- U. K. Laemmli, Nature, 1970, 227, 680–685 CrossRef CAS.
- G. Férard, Pure Appl. Chem., 1994, 66, 891–896 Search PubMed.
- F. Gu, J. M. Kim, K. Hayat, S. Xia, B. Feng and X. Zhang, Food Chem., 2009, 117, 48–54 CrossRef CAS.
- M. Mu, X. Pan, P. Yao and M. Jiang, J. Colloid Interface Sci., 2006, 301, 98–106 CrossRef CAS.
- R. A. Bohara, N. D. Thorat and S. H. Pawar, Korean J. Chem. Eng., 2015, 33, 216–222 CrossRef.
- C. Scheich, Protein Sci., 2004, 13, 370–380 CrossRef CAS.
- L. Jiménez-Castaño, M. Villamiel and R. López-Fandiño, Food Hydrocolloids, 2007, 21, 433–443 CrossRef.
- V. Militello, V. Vetri and M. Leone, Biophys. Chem., 2003, 105, 133–141 CrossRef CAS.
- F. L. Gu, J. M. Kim, S. Abbas, X. M. Zhang, S. Q. Xia and Z. X. Chen, Food Chem., 2010, 120, 505–511 CrossRef CAS.
- J. C. Cardoso, R. L. C. Albuquerque, F. F. Padilha, F. O. Bittencourt, O. De Freitas, P. S. Nunes, N. L. Pereira, M. J. V. Fonseca and A. A. S. Araújo, J. Therm. Anal. Calorim., 2011, 104, 249–254 CrossRef CAS.
- I. A. Farhat, S. Orset, P. Moreau and J. M. V. Blanshard, J. Colloid Interface Sci., 1998, 207, 200–208 CrossRef CAS.
- Y. A. Antonov and B. A. Wolf, Biomacromolecules, 2005, 6, 2980–2989 CrossRef CAS.
- R. Su, W. Qi, Z. He, Y. Zhang and F. Jin, Food Hydrocolloids, 2008, 22, 995–1005 CrossRef CAS.
- S. P. Wong, L. P. Leong and J. H. William Koh, Food Chem., 2006, 99, 775–783 CrossRef CAS.
- F. Guérard and M.-T. Sumaya-Martinez, J. Am. Oil Chem. Soc., 2003, 80, 467–470 CrossRef.
- S. Dong, A. Panya, M. Zeng, B. Chen, D. J. McClements and E. A. Decker, Food Res. Int., 2012, 46, 55–61 CrossRef CAS.
- Y. Abe, K. Inagaki, A. Fujiwara and K. Kuriyama, Eur. J. Pharmacol., 2000, 408, 213–218 CrossRef CAS.
- G. Sun and J. J. Mao, Nanomedicine, 2012, 7(11), 1771–1784 CrossRef CAS.
- L. Wang, X. Li, T. Sun, Y. H. Tsou, H. Chen and X. Xu, Macromol. Biosci., 2017, 1700325 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |