
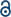 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic cross-linking of an alginate–acrylamide tough hydrogel system: time-resolved in situ mapping of gel self-assembly†
Akanksha Pragya‡
a,
Suhas Mutalik‡a,
Muhammad Waseem Younas‡a,
Siu-Kwong Pang‡a,
Pui-Kin Sob,
Faming Wangbc,
Zijian Zheng a and
Nuruzzaman Noor*a
a and
Nuruzzaman Noor*a
aThe Hong Kong Polytechnic University, Institute of Textiles and Clothing, Materials Synthesis and Processing Lab, Hung Hom, Kowloon, Hong Kong SAR, China. E-mail: nmnoor@polyu.edu.hk
bThe Hong Kong Polytechnic University, University Research Facility in Life Sciences, Hung Hom, Kowloon, Hong Kong SAR, China
cCentral South University, School of Architecture and Art, Changsha, China
First published on 12th March 2021
Abstract
Hydrogels are a popular class of biomaterial that are used in a number of commercial applications (e.g.; contact lenses, drug delivery, and prophylactics). Alginate-based tough hydrogel systems, interpenetrated with acrylamide, reportedly form both ionic and covalent cross-links, giving rise to their remarkable mechanical properties. In this work, we explore the nature, onset and extent of such hybrid bonding interactions between the complementary networks in a model double-network alginate–acrylamide system, using a host of characterisation techniques (e.g.; FTIR, Raman, UV-vis, and fluorescence spectroscopies), in a time-resolved manner. Further, due to the similarity of bonding effects across many such complementary, interpenetrating hydrogel networks, the broad bonding interactions and mechanisms observed during gelation in this model system, are thought to be commonly replicated across alginate-based and broader double-network hydrogels, where both physical and chemical bonding effects are present. Analytical techniques followed real-time bond formation, environmental changes and re-organisational processes that occurred. Experiments broadly identified two phases of reaction; phase I where covalent interaction and physical entanglements predominate, and; phase II where ionic cross-linking effects are dominant. Contrary to past reports, ionic cross-linking occurred more favourably via mannuronate blocks of the alginate chain, initially. Evolution of such bonding interactions was also correlated with the developing tensile and compressive properties. These structure–property findings provide mechanistic insights and future synthetic intervention routes to manipulate the chemo-physico-mechanical properties of dynamically-forming tough hydrogel structures according to need (i.e.; durability, biocompatibility, adhesion, etc.), allowing expansion to a broader range of more physically and/or environmentally demanding biomaterials applications.
1. Introduction
Hydrogels are water-encased gels (typically >90% H2O) composed of molecular chain networks, which find use, in part or whole, across various biomedical applications (e.g.; contact lenses, drug delivery carriers, tissue engineering scaffolds and prophylactics, in consumer products).1–11 Reports of tough hydrogels, especially Sun et al.'s 2012 account of double network (DN) tough hydrogels with remarkable mechanical properties (i.e.; DN of alginate–(poly)acrylamide(PAAm)), have further catalysed research into systems that can withstand (and recover) from various large mechanical forces, e.g.; for potential use in physiological load-bearing applications.12–14Alginates, amongst the most widely used gel-forming components, are algae-sourced polysaccharides possessing hydrocolloid properties, and are biocompatible, biodegradable, immunogenic, and non-toxic.13,15 Alginates are randomly 1-4-linked copolymers of repeating β-D-mannuronic acid (M-block) and α-L-guluronic acid (G-block) units – the acid block content, molecular weight and conformations, as well as the form and extent of cation-mediated ionic cross-linking, are crucial for alginates' gel-forming capacity and the resultant hydrogel chemo-physico-mechanical properties.16–18 However, these classic covalent single network alginate hydrogels are mechanically weak (e.g.; break at low strain (∼120% for alginate)), rendering them unsuitable for mechanical loading.12 DN approaches can markedly improve hydrogel toughness. Such DN tough hydrogels comprise two contrasting (i.e.; combinations of stiff/rigid but brittle networks with soft/ductile but mechanically weak networks) and interpenetrating block copolymer conjugated networks bound by myriad physico-chemical interactions over different length scales (i.e.; physical entanglement, ionic and covalent cross-linking), which also prevent dissolution of hydrophilic chains in hydrated polymer networks.1,12,19–23 These viscoelastic networks yield mechanical properties often orders of magnitude greater than their discrete components because the loosely cross-linked DN systems allow molecules to slightly pull apart over large areas efficiently distributing stress throughout the material bulk.1,24 Furthermore, the tough hydrogel materials properties (i.e.; permeability, stimuli responsivity, elastic modulus, fracture toughness and/or shear-thinning) are easily regulated through control of the preparation method and gel composition (i.e.; polymer volume fraction, temperature, and/or swelling agent).13,23,25–30 Thus, structure–function relationships can be gauged and tuned through variation in cross-link type and density within hydrogels.
Resilience (i.e.; ability to recover from elastic deformation), strength (i.e.; ability to bear a mechanical load) and toughness (i.e.; ability to resist fracture) are inherently contradictory material properties and so, hard to combine. High strength requires low mechanical dissipation (i.e.; suppression of dislocation and plastic deformation) while high toughness requires high mechanical dissipation during deformation (i.e.; large amounts of work before fracture).31 DN tough hydrogels (e.g.; alginate–PAAm networks) can offer both high toughness and resilience via delayed stiffening and mechanical dissipation due to broad physico-chemical bonding and varying but complementary physico-mechanical properties between the gel components.12,32
Physically cross-linked (i.e.; reversible) gels are bound by attractive non-covalent forces (i.e.; H-bonds, ionic cross-links and protein–ligand associations) between polymer chains.13 Thus, absent covalent cross-links, linear chains form networks via topological (i.e.; entanglement) interactions, so exhibiting viscoelastic rheology.21 Physical hydrogels, (e.g.; as formed by alginate), exhibit good toughness, but lack the creep resistance of covalent gels.13 Ionic cross-links (e.g.; via Ca2+) complexed with polyelectrolyte anions offer strong bonding and cross-link density, but also provide points of detachment/re-attachment, leading to, e.g.; self-healing activity and modified de-/swelling behaviour of hydrogels. For example, Ca2+ co-ordinates to M- and G-blocks on alginate during gelation, acting as junctions between blocks on adjacent chains (i.e.; egg-box model).33–36 Thus, the distribution of M and G units along the alginate chain, as well as the changing M/G ratio value of the alginate system, as preferential ionic cross-linking occurs to one uronic acid block over another, determines many physico-chemical properties of the gel structure that are closely related to their functionality, i.e.; high ratios results in a more elastic, flexible (although more fragile) gel whilst low M/G provide brittle, water-insoluble, more rigid gels.18,37,38 This is because the semi-rigid chains of G-rich alginates strongly electrostatically interact with Ca2+ via (G)–COO− groups, leading to either charge neutralisation of a single chain, or cross-linking across separate chains and so, possess greater durability due to the higher shear rate necessary to induce the chain orientation.39–42 Conversely, for M-rich alginates, the electrostatic interactions are less significant, especially for low molecular mass, and the viscosity decrease starts at a lower shear rate.43 However, there have been reports that low molecular weight and low M/G alginates produce the strongest, most well organised alginate structures.44
Chemically cross-linked (i.e.; permanent) gels are facilitated through various functional groups in the polymer backbone, (e.g.; hydroxyl, amine and hydrazide) often via specific chemical cross-linking agents (e.g.; N,N′-methylenebisacrylamide). The resultant permanent structure is usually more stable than for physically cross-linked, but may exhibit poor mechanical strength and toughness.13 The degree of covalent cross-linking is usually the most important factor in determining the resultant macroscopic properties (e.g.; mechanical strength, swelling and encapsulant release).45,46 Alternative chemical cross-linking methods include enzymatic linking and free-radical polymerisation.13,47
Both physically- and chemically-cross-linked hybrid hydrogel systems undergo entanglement as well as ionic and covalent cross-linking of multi-component polymer networks; the extent of cross-linking dependent on the polymer functional groups, as well as the size and type(s) of cross-linking agent used. Such systems are the focus of this paper. A systematic exploration into the dynamic formation processes of model DN alginate–PAAm tough hydrogels should yield a fuller understanding of the myriad dynamic bonding interactions that occur within the tough hydrogel polymer structure. While past (e.g.) NMR studies have reported on chemical structure, bonding and internal mobility of constituents, the nature of solution NMR processing, (i.e.; gel dissolution prior to analysis, as well as the fact that sample spectra need to be acquired at high-temperature to decrease the viscosity of the gel) confounds data outputs and may introduce inaccuracies into systems; the various required interventions may affect the bonding environment and so give erroneous interpretations of structural changes and relationships in cross-linking processes.16,48–51 Other methods (e.g.; mass spectrometry, XPS analysis etc.), also suffer from potential process inaccuracies for tough hydrogels.
This paper offers a detailed, time-resolved, investigation into tough hydrogel physico-chemical bonding effects and dynamic changes over a reaction, using myriad complementary analytical techniques (i.e.; spectroscopy (FTIR, Raman, UV-vis and fluorescence), microscopy (optical and video fluorescence), TGA-DSC, DLS, tensile- and compression-testing). Mechanistic insights into the formation routes and their correlations with resultant viscoelastic properties, are afforded using a non-intrusive continuous monitoring approach to give a clearer understanding of the structural changes over time, removing the need for guesswork or post-rationalisation with snapshot data, in order to highlight routes towards improved versatility and control over the resultant polymer physico-mechanical properties.48,51–55 We explored a model DN alginate–PAAm system, with good molecular affinity between the two networks, as aided by both ionic (CaSO4), and covalent (N,N′-methylenebisacrylamide) cross-linkers.
Past reports into alginate hydrogels (theoretical and experimental) have explained the favourability of uronic acid block co-ordination for ionic bonding, where G-block co-ordination has frequently reported as most favourable in accordance with the egg-box model. However, the same such in-depth reports have not yet been made for alginate-based tough hydrogel systems, to the best of our knowledge. We hypothesise that, in line with past reports, G-block alginate co-ordination will be favoured during ionic cross-linking over the course of a gelation process. In such a G-block co-ordination, progress in a dynamically evolving reaction will result in rising M/G (i.e.; a positive correlation of M/G with time).
2. Experimental
2.1 Double network (DN) tough hydrogel synthesis
Alginate polymer (Alg; from Laminaria hyperborea, Unichem), acrylamide (AAm; Unichem; 97%), N,N-methylenebisacrylamide (MBA; Alfa Aesar; >98%), N,N,N,N-tetramethylethylenediamine (TMEDA; Alfa Aesar; >99%), calcium sulfate (CaSO4; Unichem) and ammonium persulfate (APS; Alfa Aesar; (NH4)2S2O8; >98%) were used for synthesizing polyacrylamide. All materials were used as received without any further purification. [N.B.; It is recommended that a fresh source of TMEDA precursor is used for all polymerisation reactions.] Laser grade Rhodamine 6G (R6G) dye was procured from exciton and used without further purification. Deionized water (DI-H2O) was used for all experiments and spectroscopic studies.Double network hybrid alginate–polyacrylamide (Alg–PAAm) tough hydrogels were synthesised using known, free radical copolymerisation protocols.56–58 Sodium alginate polymer (6.76 g) and monomeric AAm (40.54 g) at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 ratio, were dissolved in DI-H2O (300 ml) then stirred for at least 2 h to remove all alginate lumps.1 All experimental characterisation (Raman, FTIR, UV-vis, and fluorescence spectroscopies) used 50 mL of this stock solution for free radical polymerisation. The free radical polymerisation was triggered through the introduction of: APS initiator (67.5 mg), TMEDA accelerator/catalyst (0.21 μL), MBA co-monomer/covalent cross-linker (4.05 mg) and CaSO4 ionic cross-linker (148.5 mg), which were homogenised on a vortex mixer immediately prior to loading and analysis.34,59 Reactions using R6G as tracker dye (i.e.; UV-vis and fluorimetry), used 0.04 M dye, which was fully and homogeneously dispersed in the stock solution, at the initial stage of stock solution preparation. All synthesis and analyses were carried out at room temperature and pressure (RTP), in air ambient. Beyond the initial reaction, no further treatments (e.g.; sealing, silicone oil, etc.) were conducted on samples.
:6 ratio, were dissolved in DI-H2O (300 ml) then stirred for at least 2 h to remove all alginate lumps.1 All experimental characterisation (Raman, FTIR, UV-vis, and fluorescence spectroscopies) used 50 mL of this stock solution for free radical polymerisation. The free radical polymerisation was triggered through the introduction of: APS initiator (67.5 mg), TMEDA accelerator/catalyst (0.21 μL), MBA co-monomer/covalent cross-linker (4.05 mg) and CaSO4 ionic cross-linker (148.5 mg), which were homogenised on a vortex mixer immediately prior to loading and analysis.34,59 Reactions using R6G as tracker dye (i.e.; UV-vis and fluorimetry), used 0.04 M dye, which was fully and homogeneously dispersed in the stock solution, at the initial stage of stock solution preparation. All synthesis and analyses were carried out at room temperature and pressure (RTP), in air ambient. Beyond the initial reaction, no further treatments (e.g.; sealing, silicone oil, etc.) were conducted on samples.
2.2 Tough hydrogel materials characterisation
Subsequent to initial gel preparation and loading, there was no further sample movement or exchange – all readings were taken on the same sample, in the same configuration, as the gelation evolved. Time-based readings were initiated as soon as the reaction components were mixed in a single vessel; the time codes corresponding to the time reading at which point each data acquisition was initiated. Tough hydrogel in situ structure-bonding evolution effects in real-time, at RTP, were examined by: Raman spectroscopy (BaySpec Nomadic) with a 532 nm laser excitation source (100% intensity), over 200–3200 cm−1, at 20 s integration, with any cosmic ray artefacts manually removed through data averaging; transmission ATR-FTIR (Perkin-Elmer Spectrum 100) over 4000–650 cm−1, at 4 cm−1 resolution and 16 averaged scans; UV-visible spectroscopy (UH5300 Hitachi) recorded at 1 nm step, across 200–800 nm, at 400 nm.min−1 against a DI-H2O reference standard; fluorescence spectroscopy (Edinburgh Instruments Spectrofluorometer FS5) excited at 470 nm, recorded over 500–700 nm, at a 1 nm step size and 1 s dwell time.2.3 Tough hydrogel mechanical properties characterisation
Mechanical properties were evaluated using uniaxial tensile and unconfined compression tests at RTP, to generate stress–strain curves. Compressive tests were done on an INSTRON 4411 with 5000 N (ASTM D1424-09) at constant crosshead velocity of 10 mm min−1 on cylindrical samples (18 × 35 mm; h × d).22 Tensile tests, on (size 75 × 40 × 2 mm; l × w × h) rectangular samples with notch and without notch, and (50 × 4 × 3 mm; l × w × h) dumbbell-shaped samples, were done on an INSTRON 5566 with a 500 N load cell (ASTM D1424-09), and 20 mm gauge length, at a constant extension rate of 100 mm.min-1.61 Because creep, high stretchability, water loss and time consumption were major concerns for data accuracy, a relatively high strain rate of 100 mm min−1 was used. For notched samples, an initial notch of ∼2 mm was cut using a razor blade and measured using calipers. Tensile strength was taken from the point of maximum stress. The modulus was taken from the average slope over 0–10% of strain ratio from stress–strain curves.34,613. Results
3.1 Reaction overview
A double-network (DN) tough hydrogel system was formed based on a 1:6 alginate(Alg)–acrylamide(AAM) ratio, in accordance with Sun et al.'s procedure (Fig. 1; deviations from conventional mechanisms are based on the experimental data from this paper). APS and TMEDA together combined to overcome the lack of experimental photo-activation, speeding up gelation as well as controlling the structure and uniformity of gels. The APS-initiator undergoes homolytic fission to produce SO4−. Radicals via accelerated conversion from sulfide ions in combination with TMEDA, which in turn also produce hydroxyl radicals in contact with water.62 These active initiator ions help convert cross-linkable C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C to C–C in AAm, as they co-ordinate to Alg, as a result of sulfate addition onto AAm, aiding polymerisation.62–64 Thus, the APS influences polymerisation speed as well as gel uniformity. The covalent cross-linker MBA, affords random bonding and co-polymerisation, eventually resulting in gel formation.
C to C–C in AAm, as they co-ordinate to Alg, as a result of sulfate addition onto AAm, aiding polymerisation.62–64 Thus, the APS influences polymerisation speed as well as gel uniformity. The covalent cross-linker MBA, affords random bonding and co-polymerisation, eventually resulting in gel formation.
 | ||
| Fig. 1 Schematic overview of the alginate–acrylamide double network tough hydrogel proposed reaction process according to the data from this study, including; covalent bond formation routes (A and B); ionic cross-linking routes (C), and; summary of the overall bonding effects involved in the overall hydrogel (D). | ||
The room-temperature, free-radical polymerisation process was analysed in situ, in a time-resolved manner, using a host of complementary characterisation techniques. Insights into the evolution of bonding interactions and their potential impacts on gel mechanical properties, were obtained as a result. The CaSO4 ionic cross-linked system was explored in detail, although similar trends were observed for equivalent molar ratio variations of alternative cationic cross-linker (i.e.; MgSO4, Na2SO4, CaCl2, and Ca–lignosulfonate); see ESI.†
Tough hydrogel properties (and gel properties in general), can be controlled via the onset and extent of gelation, with the subsequent mechanical properties modified via chemical precursor component presence (including the presence of metal (e.g.; calcium) salts), relative concentrations and relative ratio variation.65,66 The unstable colloidal system dynamically agglomerates into a non-ergodic gel over time, as facilitated by charged functional groups at the gel surface. In such out-of-equilibrium systems, the DN tough hydrogel undergoes extensive entanglement, increasing interpenetrating bond formation and cross-linking, which in turn increases the likelihood of further intermolecular associations, resulting in increased shear thickening, gel viscosity and elastic characteristics. In general, the macroscopic, rheological properties of polymers arise from microscopic entanglement of dynamic polymer chains and the constraints imposed by gelation, as determined by reptation dynamics, including the resultant elastic properties of polymeric gels.67,68
3.2 Fluorescence spectroscopy emission data
R6G fluorescent tracer dye was used as a sensitive stain that co-ordinates to the dynamically forming tough hydrogel, and allows for the reaction process to be accurately followed in real-time.69 During the formation process, a visually-detectable change occurs in the R6G from approximately pink-red to an orange coloured emission (Fig. 2).70 | ||
| Fig. 2 Spectrofluorescence spectra of a double network alginate–acrylamide tough hydrogel system over time, in the presence of R6G tracer dye, for the 0–60 minute period (A) and the 24–168 h period (B). | ||
In the time-evolved fluorescence emission spectra, from the first minute onwards, there are two clearly defined signals for the R6G; at λ560 and λ597. The band splitting and changes are indicative of varying monomer and dimer concentrations. The band at λ597 indicates H-type and J-type fluorescent dimer formation, depending on the specific configuration adopted upon interaction.69,71,72 The band at λ560 is due to fully solvated and relatively well-isolated monomeric dye molecules.
After ∼519 s, there is a fluorescence band shift from a doublet to a single, broad band at λ594, which is attributed to excimer formation and also signifies the onset of the main gelation process.73 Empirically, it arises due to the broad (and continued) conversion of the monomers and dimers of the precursors condensing and cross-linking into longer chain polymers that undergo further entanglement and aggregation, as gelation progresses.74,75 The high density of subsequent intermolecular interactions results in the formation of electronically excited states; excimer emission.76 This process is caused by enhancement of the polarisability in the dye-surrounding environment that results from the close packing of dye moieties in dimers and aggregates.71 The observed spectral band is generally broader because the fluorescence occurs between a bound upper state and an unbound ground state, meaning a broader range of vibrational states can be inhabited (due to fewer quantisation selection rule pressures).77
There is little further change in trace profile apart from decreasing band intensity (i.e.; a static fluorescence quenching process), indicating a stable tracer dye with little further change in the R6G-surrounding environment and that chains are broadly in the most condensed state, although further agglomeration and aggregation still continues over the first hour of reaction and during the course of a week.71,78–80
3.3 UV-vis absorption spectroscopy
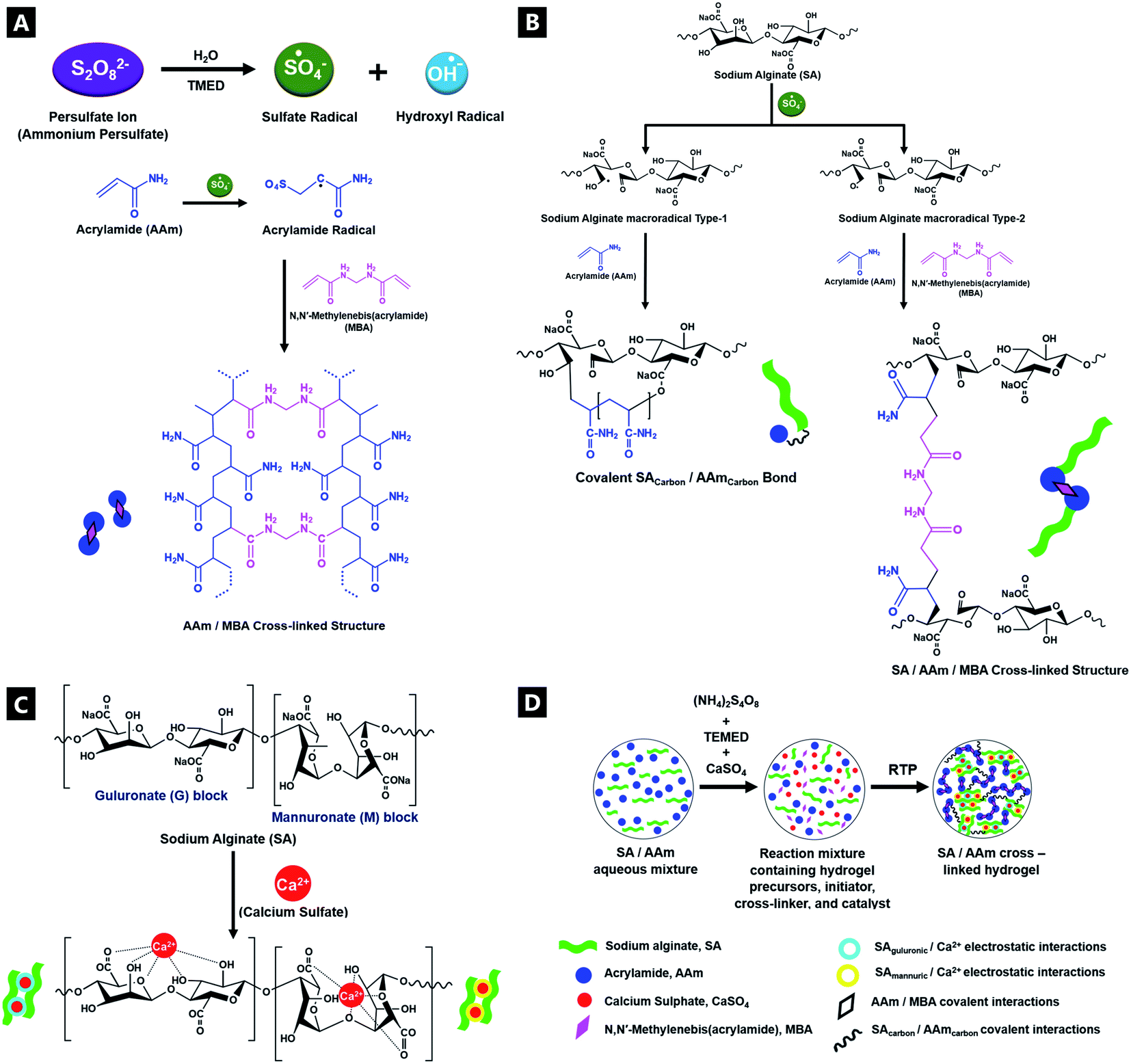
UV-vis analyses of the hydrogel formation process were also done in the presence of cationic R6G fluorescent tracer dye (Fig. 3A–F), to follow the increase of cross-link density in situ and in real time, as a corollary to the fluorimetry data.81 Seven regions of interest are identified at λ242; λ332; λ349; λ410; λ505; λ533, and; λ668. Analysis indicates differing intensity variation relationships, reflecting different reaction regimes – tentative assignments have been made. The bands at λ410 and λ668, are primarily attributed to sodium alginate (ESI Fig. S5†), in contrast to past reports, which asserted that there is no observable signal from sodium alginate.82,83 The λ411 shows little movement in λmax position, but diminishes in intensity during the reaction, indicating consumption of the sodium alginate precursor as it undergoes polymerisation. The strong signal at λ242, is predominantly attributable to the acrylamide component, although the APS, MBA and TMEDA, are all thought to also contribute. These all undergo a heavy intensity reduction, blue-shift and tailing, over time as the elements undergo conjugation; the largest decrease corresponding to consumption of acrylamide during cross-linking.84 The band profile change indicates a differential reaction rate for the different components. Two bands at λ349 and λ533 are attributed solely to the R6G dye and decrease in spectral intensity as cross-linking proceeds within the hydrogel. In such reactions, as reflected in the fluorimetry data, the positively charged dye co-ordinates to the forming hydrogel, likely via hydrogen bonds, such that the spectral features undergo change as the cross-linking reaction proceeds and the dye is consumed.85,86 The λ349 signal is attributed to π → π* transitions arising from dye co-ordination to the forming hydrogel.87,88 The λ533 profile arises due to the excellent co-ordination abilities of R6G to Ca2+.89,90 | ||
| Fig. 3 UV-vis spectra of a double network alginate–acrylamide tough hydrogel system over time, for the 0–60 min period in the presence of R6G tracer dye (full spectrum (A); baselined windows of interest (B–F)). | ||
3.4 FTIR and Raman spectroscopy M/G ratio
Alginates are randomly 1-4-linked copolymer salts of repeating β-D-mannuronic acid (M-block) and α-L-guluronic acid (G-block) units, in the form of homopolymeric (MM- or GG-blocks) and heteropolymeric sequences (MG- or GM-blocks).40,64,91 Ca2+ preferentially cross-links via certain binding sites (i.e.; M-block carboxyl, G-block carboxyls and hydroxyls), in accordance with different adsorption enthalpies.92,93 M-blocks form β-(1-4) linkages, resulting in linear and flexible conformations while the C5-epimer of G-blocks gives rise to α-(1-4)-linkages, yielding steric hindrance around the carboxyl groups, yielding folded and rigid structural conformations, responsible for the pronounced stiffness of the molecular chains.15 Thus, the ionic co-ordination route has marked effect on the resultant gel mechanical properties.Comparisons of M/G ratio allows a valuable semi-quantitative appraisal of the relative composition of alginate-based gels, whose initial composition is based on the original seaweed source, as well as any changes that occur during a reaction. The specific ratios depend on the presence, identity and concentration of the ionic cross-linker (i.e.; Ca2+) and the ability to readily ion-exchange.93,94 These M/G ratios will change over the course of a reaction. There are broadly three FTIR doublets from which M/G are empirically approximated, of which we utilised two; M∼1290 cm−1/G∼1320 cm−1, and; M∼1030 cm−1/G∼1080 cm−1 (no doublet was observed at M808 cm−1/G787 cm−1).95–97 For Raman spectroscopy, the characteristic bands were at M∼975 cm−1/G∼825 cm−1.98,99 Comparison between this complementary data is valuable as IR is more sensitive to side group vibrations and Raman is more sensitive to skeleton vibrations; any difference between the values is thought due to the inherent mechanism of the methods.98 A baseline method was used and maximum intensities compared, for all datasets (Fig. 4).41
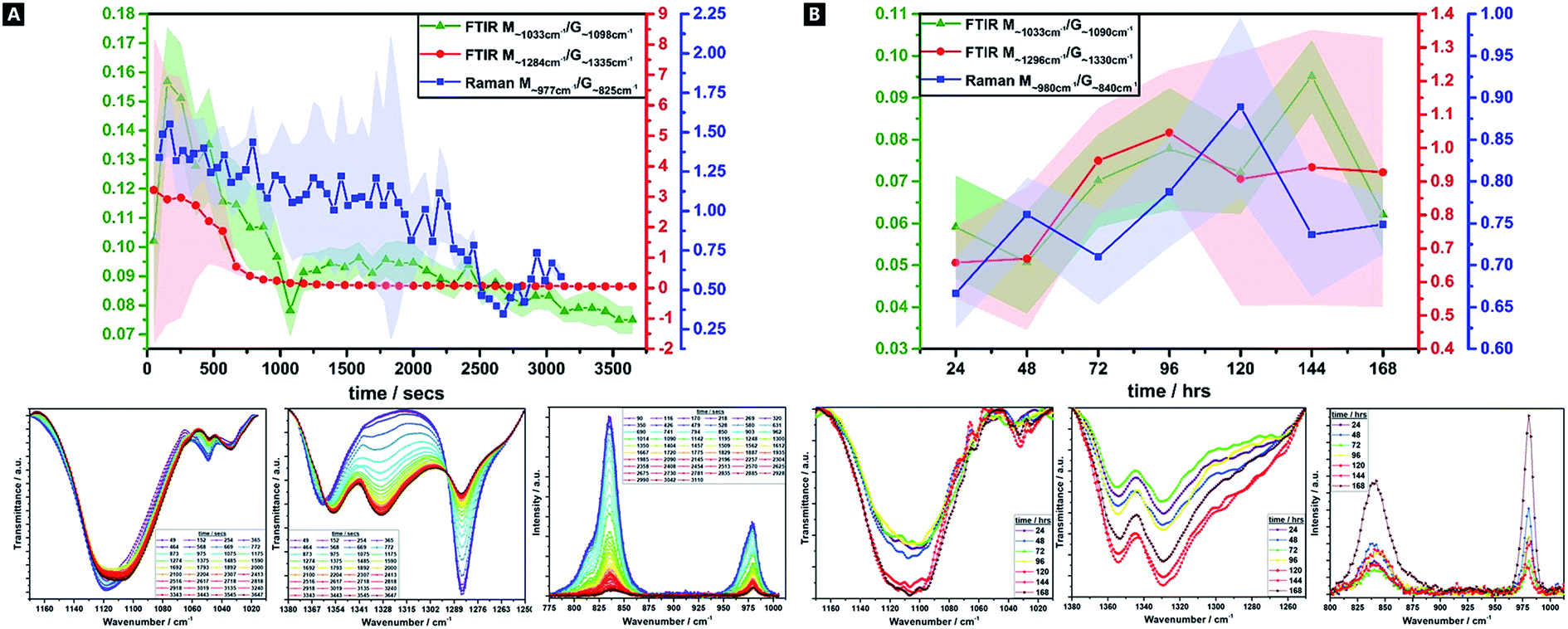 | ||
| Fig. 4 Alginate M/G ratio variations over time as calculated from FTIR (1033 cm−1/1098 cm−1 & 1284 cm−1/1335 cm−1) and Raman spectroscopy (977 cm−1/825 cm−1) data, over the 60 minute (A) and 24–168 h ranges (B). | ||
There is a decrease in M/G and a greater decrease in the intensity ratio of some of the most representative bands associated to M-units, over 60 min, which suggests that the di-equatorial, glycosidic-linked M-blocks form complexes with Ca2+ as cross-linking and the reaction proceeds; the predominance of bonds formed via such units, initially.94 Across the reaction, both M- and G-units are clearly involved in cross-linking, but the rate of change of M-block signal decrease indicates greater involvement. This change is broadly correlated across both Raman and FTIR M/G data, with similar onsets of change, rates of change and trends in M/G ratio, despite the difference in absolute values. It is only over the longer-term curing process (24–168 h), that G-block bonding becomes more favourable and this is thought to arise due to the decreasing number of freely available M-block units for Ca2+ to easily co-ordinate to. Thus, during the cross-linking analysis period in our tough hydrogel system, an acidic polysaccharide comprising a major fraction of G-block residues (i.e.; G-rich) is formed initially, which in turn explains the relatively high tensile strength of the experimentally obtained tough hydrogel.17,18,40,100
This unexpected result contradicts the “egg-box model”, since G-blocks have previously been considered to have more affinity to Ca2+ than M blocks, although it may be explained by the presence and relative concentration of the Ca2+, which is known to produce stronger gels in high-M alginate and/or above a critical cation concentration.42,44,101,102 Initially, there is little change in the ratio, as well as little change in the absolute intensities – the ionic interactions do not predominate, and where it occurs, it seems to occur relatively evenly between M and G units. From ∼8 min onwards, the bonding occurs more favourably via the M-units in Alg-PAAm DN tough hydrogels. The largest rate of change seemingly occurs within the first 10–15 min, after which a much slower rate of change is observed, (i.e.; both M- and G-block co-ordination are favoured thereafter).
3.5 ATR-FTIR spectroscopy
Fig. 5 displays the time-resolved ATR-FTIR spectra of the Alg–PAAM tough hydrogel during gelation, highlighting the evolving and changing bonding transformations, including ion exchange, across various functional groups (e.g.; hydrophilic carboxyl and hydroxyl groups) and cross-linkable CC bonds, that facilitate hydrogel formation.45 Peak shifts and profiles evolve mainly within the first ∼60 min, although further changes are observed over ∼168 h. Furthermore, there are seemingly two broad reaction stages within this first hour; phase I occurs within ∼10 min, and phase II spans the next ∼50 min and beyond. Due to the multiple components involved in the gel and the overlapping signals present, a tentative assignment is offered here, especially in areas where corroborating literature sources are absent. Unsurprisingly, the majority of FTIR peaks and Raman bands correspond to those of acrylamide and alginate.
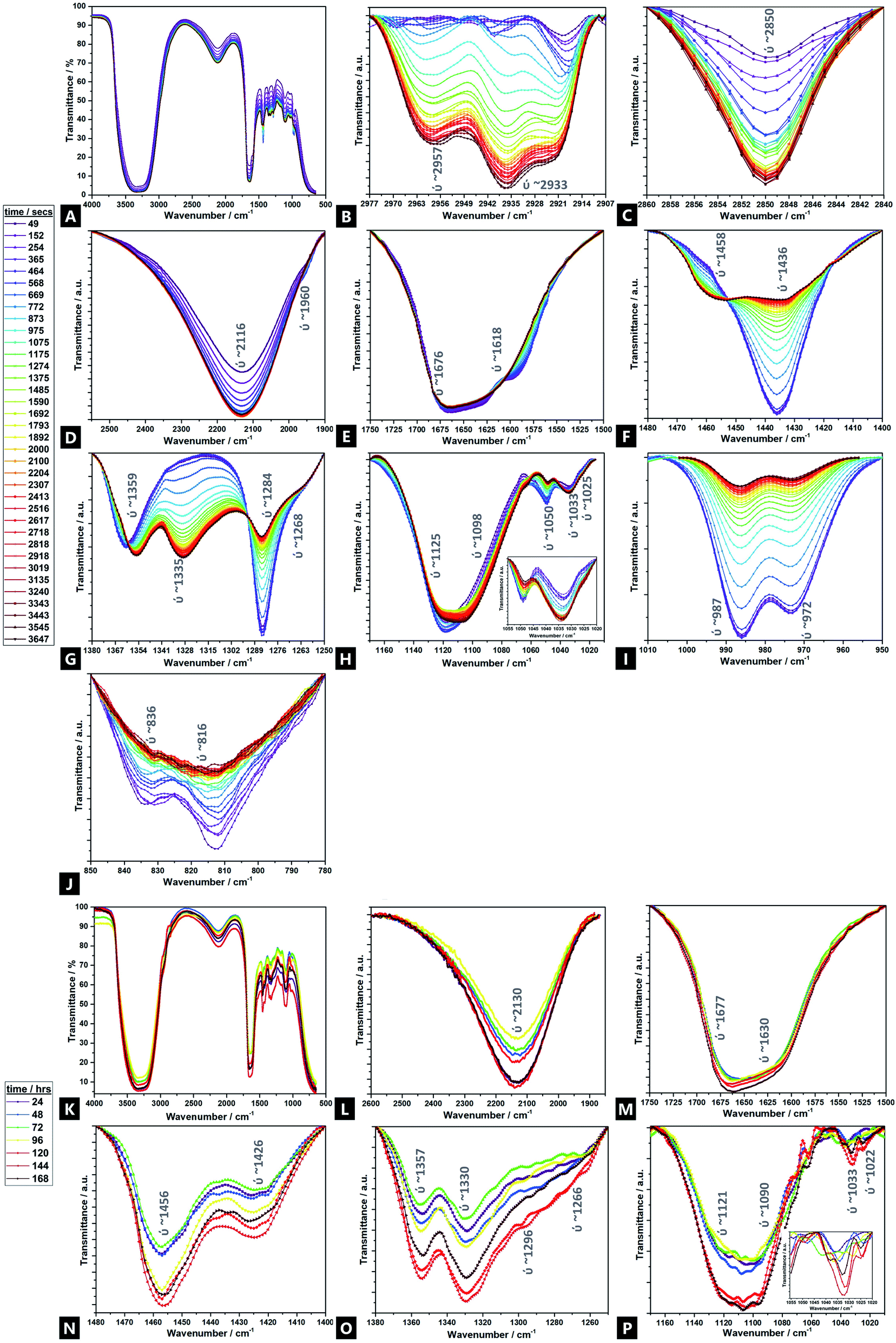 | ||
| Fig. 5 FTIR spectra of a double network alginate–acrylamide tough hydrogel system over time, for the 0–60 min period (full spectrum (A); baselined windows of interest (B–J)) and the 24–168 h period (full spectrum (K); baselined windows of interest (L–P)). | ||
For the transitions of interest in phase I, there is an increase in signals at 3380 cm−1 (ν(OH)), 2918 cm−1 (νs(CH3)), 2850 cm−1 (νas(CH3)), and 2116 cm−1 (ν(CN)).103 There is likewise, a speedy decrease in signal at 1676 cm−1 (νs(CO) amide I). In phase II, there is an increase in signal at 2957 cm−1 (ν(CH)), 2933 cm−1 (νas(CH2)), and 1033 cm−1 (Mn+–O), as well as new peaks that grow in intensity at 1458 cm−1 (νas(CH3)), 1359 cm−1 (νs(CH3)), and 1335 cm−1 (ω(CH)), all of which continue to evolve up to 168 h41,104–106 There is also a relative decrease of peaks at; 1618 cm−1 (νas(COO−)), 1436 cm−1 (ρ(CH)), 1420 cm−1 (νs(COO−)), 1284 cm−1 (ν(CO)), 1125 cm−1 (ν(CO)), 1050 cm−1 (νas(C–O–C)), 987 cm−1 (G-block δ(CC)), 972 cm−1 (G-block ν(CO)), 836 cm−1 (M-block δ(C1–H)) and 816 cm−1 (M-block residues). The signals at 1268 cm−1 (ν(CO)), 1098 cm−1 (νas(C–O–C)) and 1022 cm−1 (ν(CO)), broadly remain unchanged.107,108
Naturally, because of the precursors used and the nature of the hydrogel itself, all IR spectra are dominated by a strong, broad hydroxyl band spanning 3100–3600 cm−1.109 Possibly, there is an N–H stretch also present but drowned out by the strength of the overlapping –OH stretch ∼3380 cm−1. As the reaction proceeds, beyond the first hour and into 24–168 h, the –OH stretch broadens, accompanied by a slight increase in intensity. However, it is difficult to deconvolute from the contribution of the N–H shoulder (∼3250 cm−1) and so parse whether the contributions are from increased intramolecular and/or intermolecular bonding.106
Signals associated with alkyl groups increase during the reaction.15 Many of these changes occur within the first ∼10 min, indicating bonding involvement through unsaturated carbons, in the initial stages of reaction. Peaks at 2850 cm−1 and 2918 cm−1 are present and increase with time. There are also two new peaks, at 2933 cm−1 and 2957 cm−1, not present at the outset, that gradually emerge over the course of the reaction, with increasing speed of onset beyond the ∼600 seconds of reaction mark, i.e.; they are associated a different phase of the hydrogel formation to the 2850 cm−1 and 2918 cm−1 signals. Similarly, signals related to geminal methyl groups, at 1458 cm−1 and 1335 cm−1, also not present initially, become apparent in phase II, growing in intensity thereafter.15
In the double bond stretching region, four peaks can be clearly observed; those at 1618 cm−1 and 1436 cm−1 are attributed to asymmetric and symmetric stretching vibrations of CO from the –COO− groups of alginate respectively.15,104,110–112 As the reaction proceeds, the signal decreases, broadens and shifts slightly, to higher wavenumber, indicating exchange of cross-linking ions at the alginate and the procession of ionic bonding via Ca2+.113
Signals assigned to the bending vibrations of the NH(amide band); –COC– and –CO stretch; and –OH angular coupling, indicating the existence of free carboxyl groups, decrease over time109 In addition, another broad, medium-intensity signal is present throughout the reaction; a –CN stretch at 2116 cm−1, characteristic of plant gums like alginate that increases over the course of the reaction, which may in turn facilitate acylhydrazone bond formation to AAm.114,115 The 1033 cm−1 region increases with time – this signal is also associated with metal–oxygen bonds; the relative signal increase indicating greater Ca2+-binding involvement with reaction time, as calcium displaces sodium.106
Peaks at 1676 cm−1 and 1436 cm−1 broaden with time and increasing Ca2+ incorporation.106,116 As cross-linking proceeds, the characteristic amide I carboxylate group at 1676 cm−1, grows in prominence while the aldehyde vibrational band diminishes, indicating its critical role in cross-linking to form a conjugated amine system. The change in the singlet at 1436 cm−1, likely corresponding to a CO conjugated, symmetric stretch of COO groups, decreases in intensity and converts into a doublet; the new peak found at higher wavenumbers. This peak fission is likely due to the stretching vibrations of C–O and the O–H angular coupling, which are specific to ionic binding; the Ca2+ modifies the environment around the carbonyl group and also signifies that much of the cross-linking occurs via the carbonyl groups.117 The decrease of a M-block C–O–C pyranose stretch at 1050 cm−1 over the course of the reaction again shows the consumption and binding that occurs via such units, although the 1033 cm−1 peak does not undergo such a marked change.104,106 No peaks were observed at 1010 cm−1, which would otherwise be observed, as related to Ca2+-oxygen bonds to G-blocks; the absence again supporting the favourability of the M-block co-ordination.106 This is further supported by the decreasing intensity of two bands at 1284 cm−1 and 1098 cm−1, as well as the broad, weak signal at 1050 cm−1 which correspond to elongation in the C–O–C asymmetric stretch.109,110,118 All these peaks shift to lower wavenumbers as calcium content increases, indicating weakening of C–C and C–O bonds due to sharing with Ca2+.106
The weak doublet at 987 cm−1 and 972 cm−1 likely corresponds to α-(1-4)-glycosidic bonds, as observed in the original alginate precursor, and the decreased signal over time is related to the interaction of these binding sites to Ca2+.106 Additionally, a very weak doublet related to the MM bands at 816 cm−1 and 836 cm−1 decreases over the first 60 min (largely absent after ∼20 min), as ionic cross-linking proceeds, until completely absent by the latter stages of the reaction.113,119,120
3.6 Raman spectroscopy
Raman spectroscopy data are in broad agreement with those of FTIR spectroscopy and show that Raman bands shift and undergo changes in peak profile and intensity during gelation, indicating broader evolving structural changes.121 Tentative assignments of the multicomponent system are offered in Fig. 6 (individual precursor signals are in ESI Fig. S6†). | ||
| Fig. 6 Raman spectra of a double network alginate–acrylamide tough hydrogel system over time, for the 0–60 minute period (full spectrum (A); baselined windows of interest (B–D)) and the 24–168 h period (full spectrum (E); baselined windows of interest (F–H)). | ||
Large intensity changes were observed for the –OH stretching vibration, possibly due to consumption of the individual monomers during gelation (i.e.; covalent binding as well as hydrogen bond interactions). New Raman bands evolved in two regions over the reaction; ∼2930 cm−1, and ∼1460 cm−1, attributed to; acrylamide isopropyl –CH2 symmetric stretches, and acrylamide –CH2 bends respectively. The ∼2930 cm−1 band undergoes a large increase, from near non-existent, to high intensity, which indicates that polymer chains undergo bond saturation (i.e.; sp3-C–H stretch) as a result of covalent cross-linking.15,122,123 In addition, a shoulder starts to develop and increase in intensity after 24 h, at ∼2870 cm−1, which corresponds to aliphatic –CH2−.124,125 Likewise, the ∼1460 cm−1 signal also becomes increasingly present, as the reaction proceeds.
All other Raman bands diminished significantly during the reaction process. Bands above 1300 cm−1 are attributed to deformations of –CHx functional groups and stretching vibrations of the carboxylate functional group –COO–.126 Thus, unsaturated acrylamide carbon bands at ∼3114 cm−1 vanish over time; the ∼3040 cm−1 band greatly diminishes in intensity. Furthermore, a band corresponding to N2 reaction by-products, appears at ∼2330 cm−1; the intensity of the free N2 vibron increases over the course of an hour but disappears after 24 h127
The C–O and COO stretching vibrations present over 1800–1400 cm−1 and can be used to characterise the alginate and identify different cation binding states. The C–O stretch band does not occur near ∼1730 cm−1, as would be expected but rather, the asymmetric stretch bands of the carboxylate (COO) appear as a weak, broad peak at ∼1617 cm−1, possibly due to the increased mass of the Ca2+ counter ion, as compared to the proton or sodium. The triplet at ∼1600 cm−1/1636 cm−1/1676 cm−1 corresponds to carboxylate stretches, including the carbonyl of the non-self-associated amide (1636 cm−1) and the carbonyl asymmetric and symmetric stretches of the associated amide (1600 cm−1 and 1680 cm−1 respectively).93,128–130 All three diminish as cross-linking occurs, as well as broaden and undergo a slight change in their relative ratios, during gelation (i.e.; as a result of the difference between carboxylic acid (–COOH) and calcium carboxylate salt (–COO– Ca2+–OOC–)); the Ca2+ replacing protons effecting shifts to higher wavenumbers.15,94,131 In addition, a band at ∼1436 due to the symmetric vibration of ν(COO−); initially starting as a shoulder after 1 h and then becomes an overlapping doublet after 24 h (i.e.; 1430 cm−1 and 1457 cm−1).15,132 The changes arise from interactions of alginate with Ca2+ from the Na+ conformer, perhaps as a result of binding via both oxygen atoms of the carboxylate function; the relative increase in ∼1457 cm−1 due to the resultant conformational changes in the polyacrylamide chain.94,99,128,132,133
The 1400–1200 cm−1 region corresponds to –C–H deformation, as well as –N–H and –C–O stretching vibrations. More specifically, alginate vibrations of the polymer backbone are located at wave numbers <1300 cm−1.94 At 1300 cm−1 a weak CO vibration band appears.15 The band at 1287 cm−1 (AAm –CH2 vibrations) diminishes over 60 min, but then reappears as a stronger band at 1334 cm−1 (–N–H stretch) after 24 h, which corresponds to copolymerisation and imidisation.134–136 A small broad band at ∼1223 cm−1, corresponds to the –C–O–C– stretch, evolves beyond 24 h; although nothing is seen in the initial 1 h137
The 1200–1000 cm−1 region corresponds to C–O–H deformation, C–C–H deformation, C–O, and C–C (∼1123 cm−1) stretching vibrations, as well as symmetric and asymmetric vibration bands of COC bonds typical of polysaccharide rings.15 The –C–C– band undergoes broadening and a slight intensity decrease over time.138 The band at 1090 cm−1 is attributed to glycosidic ring breathing of the alginate.126 A band at ∼1056 exhibits a large intensity decrease in the initial hour and is completely absent after 24 h. New bands also arise at 1034–1016 cm−1 (and ∼850 cm−1) related to metal–oxygen–metal bonds, which could correspond to partial bonding between Na+ (and subsequently Ca2+) and oxygen atoms in the G-blocks of alginate (at ∼1025 cm−1).99,139
Skeletal stretching, deformation modes, and ring breathing are identified in the 1000–700 cm−1 region. Three sharp bands at ∼809 cm−1, ∼876 cm−1 and ∼977 cm−1 are assigned to skeletal –C–C∼ and –CO stretching; deformational –C–C–H, and; –C–CO bending modes, respectively. Their relative intensities change characteristically on going from M-rich content alginates to low-M content ones, which is also in agreement with the experimentally observed M/G changes (Fig. 4).94 The ∼876 cm−1 band decreases in intensity but there is no further shift, meaning there is no subsequent weakening of the –C–C– and –CO bonds, since there is no sharing of the bonds with the cation.94 Additionally, the ∼806 cm−1 spectral band confirms the presence of α-configuration G-units.99 Another influential spectral band centred at 955 cm−1 is thought to be a marker for alginate (it corresponds to G-rich, M-rich or mixed parts) and decreases in intensity as polymerisation proceeds through consumption of the alginate precursor, although there is no associated band shift.99 A weak, broad shoulder at ∼766 cm−1 arises after 24 h, although there is nothing present in the first 60 min of reaction. This is attributed to the υ4-asymmetric carbonate bending mode, which may correspond to metal carbonate formation.140,141
The region below 700 cm−1 is attributed to pyranosyl ring deformation, C–O–C glycosidic linkage vibrations, as well as co-ordination interactions of Ca2+, primarily from alginic acid.93 A band at ∼627 cm−1 (δ(CO, trans)) diminishes and almost disappears after 1 h, as polymerisation partly occurs through the carbonyl groups, and then re-develops at –642 cm−1 (δ(CO, cis)).142 Thus, there is a partial trans to cis transition at the carbonyl sites, perhaps facilitated by unzipping of the copolymer as the co-ordinating metal cation moves away, and hydrogen bonding comes to the fore. Such co-ordination conversions have previously been reported for proteinaceous polymers.143,144 The band at ∼502 cm−1 diminishes and almost disappears after 1 h and then later re-develops at ∼485 cm−1, possibly due to changing interactions with the phenyl groups and changes in the ring conformation.145–147 Finally, the diminishment of intensity in the low-frequency <200 cm−1 region, beyond ∼10 min, indicates increasing restriction placed on localized lattice vibrations as a result of the gelation and densification process.148,149
3.7 Tensile and compression testing
Bonding effects evolved over 168 h, as reflected in mechanical properties variation, e.g.; stress–strain curves show the non-linear viscoelastic behaviour of the tough hydrogel (Fig. 7). There was also a variation in materials properties under compression and tension. Mechanical property data could only be obtained for gels that had been aged a minimum of ∼4 h. All tensile curves, regardless of configuration, typically show four stages of deformation; (1) elastic behaviour up to ∼150–250% strain corresponding to the elastic peak stresses; (2) yield point; (3) ultimate tensile strength, and; (4) the failure point.12 Following application of strain, an instantaneous network of permanent and transient cross-links arise, thus causing the elastic peak stress. | ||
| Fig. 7 Tensile strain–stress curves of (A) rectangular notch-less, (B) rectangular notched, (C) dumbbell-shaped specimens, and; (D) time dependent variation in the mechanical properties of the alginate–acrylamide dumbbell-shaped tough hydrogel system. Compressive stress–strain curve (E–H) for 1–5 cycles. Variation of compressional resistance with time for different load-unload cycles (I). Compressive stress–strain curve at 50–80% strain (J–M). Variation of compressional resilience with time for different strain percentages (N). | ||
As time progresses, stress relaxation occurs due to unzipping of ionic cross-links, as well as disruption of transient physical crosslinks, enabling topological rearrangement of chains. This is thought to be facilitated by water migration and exudation; well-known to be the predominant mechanism by which stress-relaxation in hydrogels occurs.150,151 The subtle absence of yield-point in the 4 h aged sample shows that the hydrogel is still undergoing reversible, complex breaking and reformation processes with chain undulation, contributing to the initial stress application before the yield point. The high strength and toughness of traditional double gel networks are derived from the rigid yet brittle, ionic sacrificial network; the unzipping effect of Ca2+ cross-links exceed the process of softening commonly encountered in highly stretchable hydrogels.1,22,152,153 Thus, the increasing strain hardening affords increasing tensile properties with aging, namely, toughness (up to ∼650 kJ m−3; a ∼117% increase), ultimate tensile strength (up to ∼78 kPa; a ∼60% increase) and Young's modulus (up to ∼21 kPa; a ∼160% increase).154 Whilst toughness and tensile strength tend to increase and then plateau after ∼48 h of aging, the elastic modulus continues increasing with aging time, in correlation with increasing cross-link density.155 These properties are in line with the traditional double network gels where the high mechanical properties are derived from the strong entanglement and contrasting network structures.156,157 The high toughness is due to energy dissipation induced by the unzipping of physical cross-links across alginate networks, allowing high stresses to be shifted between adjacent alginate G-blocks, which have a stiffer configuration due to a greater hindered rotation about the glycosidic linkages.158 This also explains the improvement in experimental mechanical properties over the course of 168 h; there is a greater G-block co-ordination with time. The conventional trade-off between ultimate strength, stiffness (elastic modulus) and toughness is not encountered here; there is a simultaneous increase in all properties.60,156,159,160
The partially overlapping nature of the five-cycle, compressional loading–unloading curves, and presence of loops indicate a small degree of network structure damage in the conventional double network hydrogels.19,161 However, subsequent to the first cycle, there is also almost no decrease in maximum stress for the following four cycles, which confirms the highly resilient behaviour; the hydrogel dissipates energy effectively. Even at 80% strain, the hydrogel shows high recoverability of 65–70%; the loss of full recovery attributed to partial rupture of covalent cross-links and disruption in topological entanglements which decreases the overall cross-link density.162,163 Such disruption cannot be restored under ambient conditions, thus decreasing compressive stress and compressional resilience.164 Likewise, the maximum energy dissipation by the unzipping calcium cross-links occurs in the first cycle (Fig. 7C), with no prominent dissipation observed thereafter. During compression, ionic cross-links consume the dissipated energy, yielding good compressional resilience (compressive stress maximum of ∼145 kPa), and covalent crosslinks help preserve elasticity, simultaneously.152 This cyclic softening and a simultaneous strain hardening as the compression cycle proceeds, is in accordance with recent reports on sliding gels by Ito and coworkers.165–167
4. Discussion
The internal arrangement of the copolymer structure materially impacts the favourability of subsequent co-ordination to the alginate backbone and so, the favoured reaction path in a tough hydrogel formation process. Knowledge of such rearrangements is important, since pre-dominant co-ordination through one backbone unit-type preferentially over the other manifests marked differences in the resultant mechanical properties. Furthermore, the agglomeration route, as facilitated by the cross-linkers, and through electrostatic repulsion minimisation, indicates the mechanism by which aggregation is favoured.168–171Specifically, for the model DN Alg–PAAm tough hydrogel system explored in this study, time-resolved investigations identify two broad stages in tough hydrogel formation, where different bonding interactions seem to predominate across the macromolecular chains in solution, which has a critical impact on the resultant mechanical properties. Initially, during phase I, physical entanglement and covalent cross-linking are the major drivers, as monomers and dimers cross-link into longer polymer chains, signifying gelation onset.73 Covalent bonding is thought to proceed via both the unsaturated carbon bonds on the Alg–PAAm frameworks, as well as the Alg-carbonyl groups which bind to the AAm-amide groups, via a MBA bridge.14,107,114,172,173
After ∼8–12 min (and not appreciably earlier), in phase II, ionic cross-linking effects come to the fore, as Ca2+-mediated cross-linking initially proceeds via both M-block (slightly more favourable) and G-block units.15,110 M-block co-ordination is thought to proceed via a bidentate bridging co-ordination, with the Alg-carboxylate groups playing a key role.92,117,172 This also corresponds with increasing stiffness of the structure. The delayed onset of ionic contributions observed experimentally, may be partly due to the slower dissolution of the CaSO4 precursor, which in turn delays the ionic contributions to bonding. This seemingly results in a more effective tough hydrogel system with more beneficial mechanical properties. Past empirical reports indicate that when insufficient durations are spent at each cross-linking stage (e.g.; faster onset of ionic cross-linking contribution that bypasses the dimerization phase), there is incomplete diffusion and entanglement of molecular chains, non-ideal cross-linking effects, and void formation, resulting in weaker and unsatisfactory mechanical properties.34,74,75,174 Equivalent molar ratio variations of alternative cationic cross-linker (i.e.; CaCl2, MgSO4, Na2SO4, and Ca-lignosulfonate) were also explored, in place of CaSO4 (see ESI†). Thus, ideal mechanical properties (i.e.; ultimate strength and toughness), need optimisation of cross-link density and type in polymer systems, so as to improve (e.g.) the energy dissipation mechanism.175–178
5. Conclusions
This study explored mechanistic insights into the stages and factors involved in tough hydrogel formation and its correlation with the subsequent mechanical properties. A model double network alginate–acrylamide polymeric system was explored, in the presence of both ionic (CaSO4), and covalent (N,N′-methylenebisacrylamide) cross-linkers. The study offered an alternative reaction route to that predicted in the hypothesis; specific to ionic binding, and contrary to past reports, we observed a particular favouring of M-group co-ordination by Ca2+-linkers during gelation. More broadly, in the overall gelation reaction, two formation stages were identified spanning physical entanglement, dimerisation and covalent cross-linking in phase I, prior to the predominance of ionic cross-linking bonding interactions in phase II, which in turn seems to impact the exhibited mechanical properties.The detailed insights and broader findings offered in this paper may facilitate practical new synthetic routes to enhance the intrinsic gel chemo-physico-mechanical properties via direct intervention at various bonding stages, during the evolving cross-linking processes. We expect formation mechanisms in similar systems to be governed by the time-dependent bond formation, near-surface viscoelasticity of the bulk material, as well as the molecular architecture and composition of the gel relative to the crosslinking dynamics. These foundational insights will also aid understanding of tissue failure, tissue repair therapies, and design principles for future biomaterials and functional polymer gels. Such investigations will be the focus of future studies.
Author contributions
AP, SM and MWY carried out the lab syntheses and acquired data. AP organised and analysed data, made figures, and composed the mechanical properties analyses. SKP helped with all aspects of experimental data acquisition, feedback on analysis and edited the manuscript. PKS acquired MALDI-TOF MS and analysis. FW and ZZ helped with project planning, feedback on analysis and edited the manuscript. NN planned and funded the study, analysed data, made figures and composed the manuscript.Conflicts of interest
There are no known conflicts to declare.Acknowledgements
Nuruzzaman Noor would like to thank the Hong Kong UGC-RGC (25303318) as well as both the Institute of Textiles and Clothing and the Faculty of Applied Sciences and Textiles of The Hong Kong Polytechnic University (1-ZVK4 & 1-ZVLR), for funding. The authors would like to acknowledge support from the HK PolyU Materials Research Centre.References
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- Z. Zhang, Y. Chen and J. Guo, Phys. E, 2019, 105, 212–218 CrossRef CAS.
- J. Yi, K.-C. T. Nguyen, W. Wang, W. Yang, M. Pan, E. Lou, P. W. Major, L. H. Le and H. Zeng, J. Colloid Interface Sci., 2020, 578, 598–607 CrossRef CAS PubMed.
- Y. Yue, X. Wang, J. Han, L. Yu, J. Chen, Q. Wu and J. Jiang, Carbohydr. Polym., 2019, 206, 289–301 CrossRef CAS PubMed.
- Y. Liang, J. Xue, B. Du and J. Nie, ACS Appl. Mater. Interfaces, 2019, 11, 5441–5454 CrossRef CAS PubMed.
- X. Du, L. Wu, H. Yan, L. Qu, L. Wang, X. Wang, S. Ren, D. Kong and L. Wang, ACS Biomater. Sci. Eng., 2019, 5, 2610–2620 CrossRef CAS PubMed.
- Z. Cao, Y. Wang, H. Wang, C. Ma, H. Li, J. Zheng, J. Wu and G. Huang, Polym. Chem., 2019, 10, 3503–3513 RSC.
- D. Gan, W. Xing, L. Jiang, J. Fang, C. Zhao, F. Ren, L. Fang, K. Wang and X. Lu, Nat. Commun., 2019, 10, 1487 CrossRef PubMed.
- V. T. Tran, M. T. I. Mredha, S. K. Pathak, H. Yoon, J. Cui and I. Jeon, ACS Appl. Mater. Interfaces, 2019, 11, 24598–24608 CrossRef CAS PubMed.
- G. Qu, Y. Li, Y. Yu, Y. Huang, W. Zhang, H. Zhang, Z. Liu and T. Kong, Angew. Chem., Int. Ed., 2019, 58, 10951–10955 CrossRef CAS PubMed.
- X. Wang, H. Hu, Z. Yang, L. He, Y. Kong, B. Fei and J. H. Xin, Smart Mater. Struct., 2014, 23, 125027 CrossRef.
- Y. Liu, W. He, Z. Zhang and B. Lee, Gels, 2018, 4, 46 CrossRef PubMed.
- D. Bhatnagar, M. Simon and M. H. Rafailovich, in Recent Advances in Biopolymers, InTech, 2016 Search PubMed.
- J. Liu, S. Lin, X. Liu, Z. Qin, Y. Yang, J. Zang and X. Zhao, Nat. Commun., 2020, 11, 1071 CrossRef CAS PubMed.
- M. M. M. Soledad Lencina, Z. Iatridi, M. A. Villar and C. Tsitsilianis, Eur. Polym. J., 2014, 61, 33–44 CrossRef CAS.
- A. Boucelkha, E. Petit, R. Elboutachfaiti, R. Molinié, S. Amari and R. Z. Yahaoui, J. Appl. Phycol., 2017, 29, 509–519 CrossRef CAS.
- F. Hentati, C. Delattre, A. V. Ursu, J. Desbrières, D. Le Cerf, C. Gardarin, S. Abdelkafi, P. Michaud and G. Pierre, Carbohydr. Polym., 2018, 198, 589–600 CrossRef CAS PubMed.
- M. J. Costa, A. M. Marques, L. M. Pastrana, J. A. Teixeira, S. M. Sillankorva and M. A. Cerqueira, Food Hydrocolloids, 2018, 81, 442–448 CrossRef CAS.
- X. Lu, C. Y. Chan, K. I. Lee, P. F. Ng, B. Fei, J. H. Xin and J. Fu, J. Mater. Chem. B, 2014, 2, 7631–7638 RSC.
- A. Memic, H. A. Alhadrami, M. A. Hussain, M. Aldhahri, F. Al Nowaiser, F. Al-Hazmi, R. Oklu and A. Khademhosseini, Biomed. Mater., 2015, 11, 014104 CrossRef PubMed.
- S. R. Raghavan and J. F. Douglas, Soft Matter, 2012, 8, 8539 RSC.
- J. Li, Z. Suo and J. J. Vlassak, J. Mater. Chem. B, 2014, 2, 6708–6713 CAS.
- F. Wu, L. Chen, Y. Li, K. I. Lee and B. Fei, J. Mater. Sci., 2017, 52, 4421–4434 CrossRef CAS.
- Y. Zhu, H. Inada, A. Hartschuh, L. Shi, A. Della Pia, G. Costantini, A. L. Vázquez de Parga, R. Miranda, A. Barbier, C. Mocuta, R. Belkhou, B. Bhushan, J. H. Hoo, K. S. Park, R. Baskaran, K. F. Böhringer, W. Lu, M. Nosonovsky, M.-H. Ham, A. A. Boghossian, J. H. Choi, M. S. Strano, A. Lang, M. L. Habegger, P. Motta, B. Bhushan, T. Bachmann, H. Wagner, D. W. Brenner, J. Chen, N. Shakiba, Q. Tan, Y. Sun, J. R. Greer, M. Laver, S. M. Khaled, A. Parodi, E. Tasciotti, B. C. Dave, S. B. Lockwood, C. Musicanti, P. Gasco, F. Vollrath, A. Booth, A. C. McIntosh, N. Beheshti, R. Walker, L. U. Larsson, A. Copestake, H. Hwang, Y.-K. Cho, J. Chen, M. Chu, C. R. Gordijo, X. Y. Wu, Y. Sun, M. Kolle, U. Steiner, S.-W. Wang, F. Ceyssens, R. Puers, X. Han, S. Mao, Z. Zhang, L. Jiang, L. Lin, R. Ragan, V. Lughi, C. Drummond, M. Ruths, W. Mu, J. B. Ketterson, P. Berini, Y.-P. Zhao, F.-C. Wang, S. Prakash, S. J. Henley, J. V. Anguita, S. R. P. Silva, M. Chanana, C. Mateo, V. Salgueirino, M. A. Correa-Duarte, S. Kar, S. Talapatra, J. Calvo Fuentes, J. Rivas, M. A. López-Quintela and S. Tsuda, in Encyclopedia of Nanotechnology, Springer Netherlands, Dordrecht, 2012, pp. 2459–2470 Search PubMed.
- Y. Zhai, X. Meng, H. Duan, Z. Ding, Y. Liu and L. Lucia, Macromol. Chem. Phys., 2016, 217, 32–38 CrossRef CAS.
- R. Parhi, Adv. Pharm. Bull., 2017, 7, 515–530 CrossRef CAS PubMed.
- X. Peng, T. Liu, C. Shang, C. Jiao and H. Wang, Chin. J. Polym. Sci., 2017, 35, 1268–1275 CrossRef CAS.
- J. Hua, P. F. Ng and B. Fei, J. Polym. Sci., Part B: Polym. Phys., 2018, 56, 1325–1335 CrossRef CAS.
- F. Wu, L. Chen, Y. Wang and B. Fei, J. Mater. Sci., 2019, 54, 12131–12144 CrossRef CAS.
- B. Kumar, N. Noor, S. Thakur, N. Pan, H. Narayana, S. Yan, F. Wang and P. Shah, ACS Omega, 2019, 4, 15348–15358 CrossRef CAS PubMed.
- S. Lin, Y. Zhou and X. Zhao, Extreme Mech. Lett., 2014, 1, 70–75 CrossRef.
- X. Zhao, S. Lin and H. Yuk, Compliant yet tough hydrogel systems as ultrasound transmission agents, US Pat., US9878506B2, https://patents.google.com/patent/US9878506, (accessed 2 July 2020) Search PubMed.
- X. Ding and Y. Wang, J. Mater. Chem. B, 2017, 5, 887–906 RSC.
- M. Anurup, R. Monika, M. Rabibrata, B. Provas and C. Jyotirmoy, Front. Boeng. Biotechnol., 2016 DOI:10.3389/conf.FBIOE.2016.01.00171.
- H. Jiang, L. Fan, S. Yan, F. Li, H. Li and J. Tang, Nanoscale, 2019, 11, 2231–2237 RSC.
- R. V. Kulkarni and B. Sa, J. Drug Targeting, 2008, 16, 167–177 CrossRef CAS PubMed.
- J. M.-L. Kok and C.-L. Wong, Sustainable Chem. Pharm., 2018, 9, 87–94 CrossRef.
- C. Peteiro, in Alginates and Their Biomedical Applications, ed. B. Rehm and M. Moradali, Springer Singapore, 2018, pp. 27–66 Search PubMed.
- P. De Vos, Biomaterials, 1997, 18, 273–278 CrossRef CAS PubMed.
- M. A. Fawzy, M. Gomaa, A. F. Hifney and K. M. Abdel-Gawad, Carbohydr. Polym., 2017, 157, 1903–1912 CrossRef CAS PubMed.
- E. Gómez-Ordóñez and P. Rupérez, Food Hydrocolloids, 2011, 25, 1514–1520 CrossRef.
- C. K. Siew, P. A. Williams and N. W. G. Young, Biomacromolecules, 2005, 6, 963–969 CrossRef CAS PubMed.
- A. Dodero, S. Vicini, M. Alloisio and M. Castellano, J. Mater. Sci., 2019, 54, 8034–8046 CrossRef CAS.
- P. E. Ramos, P. Silva, M. M. Alario, L. M. Pastrana, J. A. Teixeira, M. A. Cerqueira and A. A. Vicente, Food Hydrocolloids, 2018, 77, 8–16 CrossRef CAS.
- H. Zhang, A. Patel, A. K. Gaharwar, S. M. Mihaila, G. Iviglia, S. Mukundan, H. Bae, H. Yang and A. Khademhosseini, Biomacromolecules, 2013, 14, 1299–1310 CrossRef CAS PubMed.
- I. Khalid, M. Ahmad, M. Usman Minhas, K. Barkat and M. Sohail, Adv. Polym. Technol., 2018, 37, 985–995 CrossRef CAS.
- H. Sojoudi, M. Wang, N. D. Boscher, G. H. McKinley and K. K. Gleason, Soft Matter, 2016, 12, 1938–1963 RSC.
- Y. E. Shapiro, Prog. Polym. Sci., 2011, 36, 1184–1253 CrossRef CAS.
- G. Ben Messaoud, P. Le Griel, D. Hermida-Merino, S. L. K. W. Roelants, W. Soetaert, C. V. Stevens and N. Baccile, Chem. Mater., 2019, 31, 4817–4830 CrossRef CAS.
- Y. Liu, K. Zhang, J. Ma and G. J. Vancso, ACS Appl. Mater. Interfaces, 2017, 9, 901–908 CrossRef CAS PubMed.
- L. Francis, K. V Greco, A. R. Boccaccini, J. J. Roether, N. R. English, H. Huang, R. Ploeg and T. Ansari, J. Biomater. Appl., 2018, 33, 447–465 CrossRef CAS PubMed.
- B. Ding, H. Gao, J. Song, Y. Li, L. Zhang, X. Cao, M. Xu and J. Cai, ACS Appl. Mater. Interfaces, 2016, 8, 19739–19746 CrossRef CAS PubMed.
- M. Nakamura, M. Okano and S. Watanabe, ACS Appl. Polym. Mater., 2019, 1, 3008–3016 CrossRef CAS.
- Y. Shmueli, J. Jiang, Y. Zhou, Y. Xue, C.-C. Chang, G. Yuan, S. K. Satija, S. Lee, C.-Y. Nam, T. Kim, G. Marom, D. Gersappe and M. H. Rafailovich, ACS Appl. Polym. Mater., 2019, 1, 1559–1567 CrossRef CAS.
- M. J. Van Vleet, T. Weng, X. Li and J. R. Schmidt, Chem. Rev., 2018, 118, 3681–3721 CrossRef CAS PubMed.
- X. P. Morelle, W. R. Illeperuma, K. Tian, R. Bai, Z. Suo and J. J. Vlassak, Adv. Mater., 2018, 30, 1801541 CrossRef PubMed.
- H. J. Naghash and O. Okay, J. Appl. Polym. Sci., 1996, 60, 971–979 CrossRef CAS.
- D. J. Beebe, J. S. Moore, J. M. Bauer, Q. Yu, R. H. Liu, C. Devadoss and B.-H. Jo, Nature, 2000, 404, 588–590 CrossRef CAS PubMed.
- C. H. Yang, M. X. Wang, H. Haider, J. H. Yang, J.-Y. Y. Sun, Y. M. Chen, J. Zhou and Z. Suo, ACS Appl. Mater. Interfaces, 2013, 5, 10418–10422 CrossRef CAS PubMed.
- Y. Xiao, E. A. Friis, S. H. Gehrke and M. S. Detamore, Tissue Eng., Part B, 2013, 19, 403–412 CrossRef CAS PubMed.
- Y. Wang, Y. Xue, J. Wang, Y. Zhu, Y. Zhu, X. Zhang, J. Liao, X. Li, X. Wu, Y.-X. Qin and W. Chen, Polymers, 2019, 11, 1112 CrossRef CAS PubMed.
- R. Tripathi and B. Mishra, AAPS PharmSciTech, 2012, 13, 1091–1102 CrossRef CAS PubMed.
- A. Giz, H. Çatalgil-Giz, A. Alb, J.-L. Brousseau and W. F. Reed, Macromolecules, 2001, 34, 1180–1191 CrossRef CAS.
- S. Pentlavalli, P. Chambers, B. N. Sathy, M. O'Doherty, M. Chalanqui, D. J. Kelly, T. Haut-Donahue, H. O. McCarthy and N. J. Dunne, Macromol. Biosci., 2017, 17, 1700118 CrossRef PubMed.
- P. Kujawa, A. Audibert-Hayet, J. Selb and F. Candau, Macromolecules, 2006, 39, 384–392 CrossRef CAS.
- J. R. Tse and A. J. Engler, Curr. Protoc. Cell Biol., 2010, 47, 10.16.1–10.16.16 Search PubMed.
- M. Abadi, M. F. Serag and S. Habuchi, Nat. Commun., 2018, 9, 5098 CrossRef PubMed.
- T. Annable, R. Buscall, R. Ettelaie and D. Whittlestone, J. Rheol., 1993, 37, 695–726 CrossRef CAS.
- M. L. Ferrer and F. del Monte, Langmuir, 2003, 19, 650–653 CrossRef CAS.
- S. Saha, M. U. Chhatbar, P. Mahato, L. Praveen, A. K. Siddhanta and A. Das, Chem. Commun., 2012, 48, 1659–1661 RSC.
- M. C. Gutiérrez, M. J. Hortigüela, M. L. Ferrer and F. del Monte, Langmuir, 2007, 23, 2175–2179 CrossRef PubMed.
- F. M. Zehentbauer, C. Moretto, R. Stephen, T. Thevar, J. R. Gilchrist, D. Pokrajac, K. L. Richard and J. Kiefer, Spectrochim. Acta, Part A, 2014, 121, 147–151 CrossRef CAS PubMed.
- A. Penzkofer and Y. Lu, Chem. Phys., 1986, 103, 399–405 CrossRef CAS.
- U. T. D. Huynh, A. Lerbret, F. Neiers, O. Chambin and A. Assifaoui, J. Phys. Chem. B, 2016, 120, 1021–1032 CrossRef CAS PubMed.
- I. Braccini and S. Pérez, Biomacromolecules, 2001, 2, 1089–1096 CrossRef CAS PubMed.
- M. A. Slifkin, Nature, 1963, 200, 766–767 CrossRef CAS.
- J. M. Steves, L. T. Tan, J. A. Gardella, R. Hard, W. L. Hicks, A. N. Cartwright, B. Koc and F. V. Bright, Appl. Spectrosc., 2008, 62, 290–294 CrossRef CAS PubMed.
- A. Srivastava, J. H. Waite, G. D. Stucky and A. Mikhailovsky, Macromolecules, 2009, 42, 2168–2176 CrossRef CAS PubMed.
- V. Martínez Martínez, F. López Arbeloa, J. Bañuelos Prieto and I. López Arbeloa, J. Phys. Chem. B, 2005, 109, 7443–7450 CrossRef PubMed.
- X. Cai, J. Yu, L. Xu, R. Liu and J. Yang, Food Chem., 2015, 174, 291–298 CrossRef CAS PubMed.
- X. Kang, Y. Yu, Y. Bao, W. Cai and S. Cui, Polym. Chem., 2015, 6, 4252–4257 RSC.
- Y. K. Verma, R. P. Tripathi and G. U. Gangenahalli, React. Funct. Polym., 2016, 102, 130–136 CrossRef CAS.
- I. Moreno-Villoslada, M. Jofré, V. Miranda, R. González, T. Sotelo, S. Hess and B. L. Rivas, J. Phys. Chem. B, 2006, 110, 11809–11812 CrossRef CAS PubMed.
- Y. Li, W. He, Q. Peng, L. Hou, J. He and K. Li, Food Chem., 2019, 287, 55–60 CrossRef CAS PubMed.
- Y. Li, Y. Li, S. Xu, K. Li and Y. Lu, Spectrosc. Spectr. Anal., 2011, 31, 1069–1073 CAS.
- L. Zhu, C. Guan, B. Zhou, Z. Zhang, R. Yang, Y. Tang and J. Yang, Polym. Polym. Compos., 2017, 25, 627–634 CAS.
- B. Lasio, L. Malfatti and P. Innocenzi, J. Photochem. Photobiol., A, 2013, 271, 93–98 CrossRef CAS.
- B. Bag and A. Pal, Org. Biomol. Chem., 2011, 9, 4467 RSC.
- H. Ozay and O. Ozay, Chem. Eng. J., 2013, 232, 364–371 CrossRef CAS.
- E. Araya-Hermosilla, D. Muñoz, S. Orellana, A. Yáñez, A. F. Olea, F. Oyarzun-Ampuero and I. Moreno-Villoslada, React. Funct. Polym., 2014, 81, 14–21 CrossRef CAS.
- K. Y. Lee and D. J. Mooney, Prog. Polym. Sci., 2012, 37, 106–126 CrossRef CAS PubMed.
- S. K. Papageorgiou, F. K. Katsaros, E. P. Kouvelos, J. W. Nolan, H. Le Deit and N. K. Kanellopoulos, J. Hazard. Mater., 2006, 137, 1765–1772 CrossRef CAS PubMed.
- Y. Cheng, X. Luo, J. Betz, G. F. Payne, W. E. Bentley and G. W. Rubloff, Soft Matter, 2011, 7, 5677 RSC.
- R. Hernández, J. Sacristán and C. Mijangos, Macromol. Chem. Phys., 2010, 211, 1254–1260 CrossRef.
- M. P. Filippov and R. Kohn, Chem. Zvesti, 1974, 817–819, 6 Search PubMed.
- K. Sakugawa, A. Ikeda, A. Takemura and H. Ono, J. Appl. Polym. Sci., 2004, 93, 1372–1377 CrossRef CAS.
- W. Mackie, Carbohydr. Res., 1971, 20, 413–415 CrossRef CAS PubMed.
- T. Salomonsen, H. M. Jensen, D. Stenbæk and S. B. Engelsen, Carbohydr. Polym., 2008, 72, 730–739 CrossRef CAS.
- A. Pielesz and M. K. K. Bąk, Int. J. Biol. Macromol., 2008, 43, 438–443 CrossRef CAS PubMed.
- M. Nakauma, T. Funami, Y. Fang, K. Nishinari, K. I. Draget and G. O. Phillips, Food Hydrocolloids, 2017, 69, 318–328 CrossRef CAS.
- I. Donati, S. Holtan, Y. A. Mørch, M. Borgogna and M. Dentini, Biomacromolecules, 2005, 6, 1031–1040 CrossRef CAS PubMed.
- L. Li, Y. Fang, R. Vreeker, I. Appelqvist and E. Mendes, Biomacromolecules, 2007, 8, 464–468 CrossRef CAS PubMed.
- W. Chen, N. Li, Y. Ma, M. L. Minus, K. Benson, X. Lu, X. Wang, X. Ling and H. Zhu, Biomacromolecules, 2019, 20, 4476–4484 CrossRef CAS PubMed.
- G. Sen, R. P. Singh and S. Pal, J. Appl. Polym. Sci., 2010, 115, 63–71 CrossRef CAS.
- F. Sabbagh and I. I. Muhamad, J. Taiwan Inst. Chem. Eng., 2017, 72, 182–193 CrossRef CAS.
- C. Sartori, D. S. Finch, B. Ralph and K. Gilding, Polymer, 1997, 38, 43–51 CrossRef CAS.
- H. S. Samanta and S. K. Ray, Carbohydr. Polym., 2014, 99, 666–678 CrossRef CAS PubMed.
- S. El Atouani, F. Bentiss, A. Reani, R. Zrid, Z. Belattmania, L. Pereira, A. Mortadi, O. Cherkaoui and B. Sabour, Phycol. Res., 2016, 64, 185–193 CrossRef CAS.
- A. Bradshaw, M. Salt, A. Bell, M. Zeitler, N. Litra and A. M. Smith, J. Exp. Biol., 2011, 214, 1699–1706 CrossRef CAS PubMed.
- A. R. Fajardo, M. B. Silva, L. C. Lopes, J. F. Piai, A. F. Rubira and E. C. Muniz, RSC Adv., 2012, 2, 11095 RSC.
- F. Ding, X. Shi, Z. Jiang, L. Liu, J. Cai, Z. Li, S. Chen and Y. Du, J. Mater. Chem. B, 2013, 1, 1729 RSC.
- S. E. Bakarich, M. In H. Panhuis, S. Beirne, G. G. Wallace and G. M. Spinks, J. Mater. Chem. B, 2013, 1, 4939 RSC.
- Y. Ma and Q. Feng, J. Solid State Chem., 2011, 184, 1008–1015 CrossRef CAS.
- D. E. Apostolides and C. S. Patrickios, Polym. Int., 2018, 67, 627–649 CrossRef CAS.
- M. Zhu, H. Jin, T. Shao, Y. Li, J. Liu, L. Gan and M. Long, Mater. Des., 2020, 192, 108723 CrossRef CAS.
- R. Pereira, A. Tojeira, D. C. Vaz, A. Mendes and P. Bártolo, Int. J. Polym. Anal. Charact., 2011, 16, 449–464 CrossRef CAS.
- S. K. Papageorgiou, E. P. Kouvelos, E. P. Favvas, A. A. Sapalidis, G. E. Romanos and F. K. Katsaros, Carbohydr. Res., 2010, 345, 469–473 CrossRef CAS PubMed.
- A. S. Montaser, M. Rehan and M. E. El-Naggar, Int. J. Biol. Macromol., 2019, 124, 1016–1024 CrossRef CAS PubMed.
- F. Martínez-Gómez, M. V. Encinas, B. Matsuhiro and J. Pavez, J. Appl. Polym. Sci., 2015, 132(32) DOI:10.1002/app.42398.
- A. Beratto, C. Agurto, J. Freer, C. Peña-Farfal, N. Troncoso, A. Agurto and R. del P. Castillo, Appl. Spectrosc., 2017, 71, 2263–2277 CrossRef CAS PubMed.
- S. H. Bjørnøy, D. C. Bassett, S. Ucar, B. L. Strand, J.-P. Andreassen and P. Sikorski, Acta Biomater., 2016, 44, 254–266 CrossRef PubMed.
- M. Heinemann, H. Meinberg, J. Büchs, H.-J. Koß and M. B. Ansorge-Schumacher, Appl. Spectrosc., 2005, 59, 280–285 CrossRef CAS PubMed.
- A. A. Naddaf and H. J. Bart, Defect Diffus. Forum, 2011, 312–315, 193–198 Search PubMed.
- H. Mondal, M. Karmakar, P. K. Chattopadhyay and N. R. Singha, Carbohydr. Polym., 2019, 213, 428–440 CrossRef CAS PubMed.
- Z. Modrzejewska, K. Nawrotek, W. Maniukiewicz and T. Douglas, J. Mol. Struct., 2014, 1074, 629–635 CrossRef CAS.
- R. Galli, S. Tamosaityte, M. Koch, K. H. Sitoci-Ficici, R. Later, O. Uckermann, R. Beiermeister, M. Gelinsky, G. Schackert, M. Kirsch, E. Koch and G. Steiner, in Advanced Microscopy Techniques IV; and Neurophotonics II, ed. E. Beaurepaire, P. T. C. So, F. Pavone and E. M. Hillman, OSA, Washington, D.C., 2015, p. 95360Y Search PubMed.
- P. Ray, D. Gidley, J. V. Badding and A. D. Lueking, Microporous Mesoporous Mater., 2019, 277, 29–35 CrossRef CAS.
- A. M. M. A. da Costa and A. M. Amado, Polymer, 2000, 41, 5361–5365 CrossRef CAS.
- A. Amorim da Costa and A. Amado, Solid State Ionics, 2001, 145, 79–84 CrossRef CAS.
- D. C. Furuya, S. A. da Costa, R. C. de Oliveira, H. G. Ferraz, A. Pessoa Junior and S. M. da Costa, Mater. Res., 2017, 20, 377–386 CrossRef CAS.
- R. P. Dumitriu, G. R. Mitchell and C. Vasile, Polym. Int., 2011, 60, 222–233 CrossRef CAS.
- Z. Kroneková, M. Pelach, P. Mazancová, L. Uhelská, D. Treľová, F. Rázga, V. Némethová, S. Szalai, D. Chorvát, J. J. McGarrigle, M. Omami, D. Isa, S. Ghani, E. Majková, J. Oberholzer, V. Raus, P. Šiffalovič and I. Lacík, Sci. Rep., 2018, 8, 1637 CrossRef PubMed.
- X.-W. Shi, C.-Y. Tsao, X. Yang, Y. Liu, P. Dykstra, G. W. Rubloff, R. Ghodssi, W. E. Bentley and G. F. Payne, Adv. Funct. Mater., 2009, 19, 2074–2080 CrossRef CAS.
- C. Baldock, L. Rintoul, S. F. Keevil, J. M. Pope and G. A. George, Phys. Med. Biol., 1998, 43, 3617–3627 CrossRef CAS PubMed.
- O. Guselnikova, P. Postnikov, A. Trelin, V. Švorčík and O. Lyutakov, ACS Sens., 2019, 4, 1032–1039 CrossRef CAS PubMed.
- A. Georgiev, D. Yordanov, D. Dimov, J. Assa, E. Spassova and G. Danev, Spectrochim. Acta, Part A, 2015, 140, 444–450 CrossRef CAS PubMed.
- J. Lu, Y. Li, D. Hu, X. Chen, Y. Liu, L. Wang, M. A. Ashraf and Y. Zhao, Saudi J. Biol. Sci., 2016, 23, S22–S31 CrossRef CAS PubMed.
- M. G. Tosato, D. E. Orallo, S. M. Ali, M. S. Churio, A. A. Martin and L. Dicelio, J. Photochem. Photobiol., B, 2015, 153, 51–58 CrossRef CAS PubMed.
- H. Yu, Y. Guo, C. Yao, D. F. Perepichka and H. Meng, J. Mater. Chem. C, 2016, 4, 11055–11058 RSC.
- O. Zavorotynska, S. Deledda, J. Vitillo, I. Saldan, M. Guzik, M. Baricco, J. Walmsley, J. Muller and B. Hauback, Energies, 2015, 8, 9173–9190 CrossRef CAS.
- N. Buzgar and A. I. Apopei, ANALELE ŞTIINŢIFICE ALE UNIVERSITĂŢII “AL. I. CUZA” IAŞI, 2009, http://geology.uaic.ro/auig/articole/2009%20no2/1_L10-Buzgar%20-%20pag%2097-112.pdf Search PubMed.
- M. Todica, R. Stefan, C. V. Pop and L. Olar, Acta Phys. Pol., A, 2015, 128, 128–135 CrossRef CAS.
- S. Hur and T. C. Bruice, J. Am. Chem. Soc., 2002, 124, 7303–7313 CrossRef CAS PubMed.
- C. Chen, T. Wang, Y. Fu and M. Liu, Chem. Commun., 2016, 52, 1381–1384 RSC.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- M. Kim, W. G. Chen, J. W. Kang, M. J. Glassman, K. Ribbeck and B. D. Olsen, Adv. Mater., 2015, 27, 4207–4212 CrossRef CAS PubMed.
- H. Nithya, S. Selvasekarapandian, P. C. Selvin, D. A. Kumar, M. Hema and J. Kawamura, J. Solid State Electrochem., 2012, 16, 1791–1797 CrossRef CAS.
- G. Mariotto, M. Montagna, G. Viliani, R. Campostrini and G. Carturan, J. Non-Cryst. Solids, 1988, 106, 384–387 CrossRef CAS.
- O. F. Nielsen, T. Lindström, P. A. Lund, Q. Shen, J. Weidlein, V. P. Spiridonov and T. G. Strand, Acta Chem. Scand., 1982, 36, 623–625 CrossRef.
- X. Chen, C. Dong, K. Wei, Y. Yao, Q. Feng, K. Zhang, F. Han, A. F.-T. Mak, B. Li and L. Bian, NPG Asia Mater., 2018, 10, 788–799 CrossRef CAS.
- X. Zhao, N. Huebsch, D. J. Mooney and Z. Suo, J. Appl. Phys., 2010, 107, 063509 CrossRef PubMed.
- J. Wang, J. Wei, S. Su and J. Qiu, J. Nanomater., 2019, 2019, 1–15 Search PubMed.
- K. M. Gattás-Asfura, C. A. Fraker and C. L. Stabler, J. Biomed. Mater. Res., Part A, 2011, 99, 47–57 CrossRef PubMed.
- H. Zhang, H. Peng, Y. Li, Y. Xu and W. Weng, Polymer, 2015, 80, 130–137 CrossRef CAS.
- B. Zhang, I. M. Jayalath, J. Ke, J. L. Sparks, C. S. Hartley and D. Konkolewicz, Chem. Commun., 2019, 55, 2086–2089 RSC.
- J. P. Gong, Soft Matter, 2010, 6, 2583 RSC.
- L. Zhu, J. Qiu and E. Sakai, RSC Adv., 2017, 7, 43755–43763 RSC.
- M. X. Wang, C. H. Yang, Z. Q. Liu, J. Zhou, F. Xu, Z. Suo, J. H. Yang and Y. M. Chen, Macromol. Rapid Commun., 2015, 36, 465–471 CrossRef CAS PubMed.
- Y. Deng and S. W. Cranford, J. Appl. Mech., 2018, 85(11), 111001 CrossRef.
- Q. Chen, D. Wei, H. Chen, L. Zhu, C. Jiao, G. Liu, L. Huang, J. Yang, L. Wang and J. Zheng, Macromolecules, 2015, 48, 8003–8010 CrossRef CAS.
- J. Wei, J. Wang, S. Su, S. Wang and J. Qiu, J. Mater. Chem. B, 2015, 3, 5284–5290 RSC.
- Y. Cui, M. Tan, A. Zhu and M. Guo, RSC Adv., 2014, 4, 56791–56797 RSC.
- C. Shao, M. Wang, H. Chang, F. Xu and J. Yang, ACS Sustainable Chem. Eng., 2017, 5, 6167–6174 CrossRef CAS.
- N. T. Nguyen, A. H. Milani, J. Jennings, D. J. Adlam, A. J. Freemont, J. A. Hoyland and B. R. Saunders, Nanoscale, 2019, 11, 7921–7930 RSC.
- Y. Noda, Y. Hayashi and K. Ito, J. Appl. Polym. Sci., 2014, 131(15) DOI:10.1002/app.40509.
- A. Bin Imran, K. Esaki, H. Gotoh, T. Seki, K. Ito, Y. Sakai and Y. Takeoka, Nat. Commun., 2014, 5, 5124 CrossRef CAS PubMed.
- K. Kato, T. Yasuda and K. Ito, Polymer, 2014, 55, 2614–2619 CrossRef CAS.
- S. A. Ferreira, P. J. G. Coutinho and F. M. Gama, Langmuir, 2010, 26, 11413–11420 CrossRef CAS PubMed.
- D. Kaneko, N. Q. Thi le, T. Shimoda and T. Kaneko, Polym. J., 2010, 42, 829–833 CrossRef CAS.
- Z. Xiong, S. Li and Y. Xia, New J. Chem., 2016, 40, 9951–9957 RSC.
- Z.-X. Zhang, S. S. Liow, K. Xue, X. Zhang, Z. Li and X. J. Loh, ACS Appl. Polym. Mater., 2019, 1, 1769–1777 CrossRef CAS.
- J. Sun and H. Tan, Materials, 2013, 6, 1285–1309 CrossRef CAS PubMed.
- R. Niu, Z. Qin, F. Ji, M. Xu, X. Tian, J. Li and F. Yao, Soft Matter, 2017, 13, 9237–9245 RSC.
- N. P. Levenhagen and M. D. Dadmun, Polymer, 2017, 122, 232–241 CrossRef CAS.
- G. A. Appuhamillage, J. C. Reagan, S. Khorsandi, J. R. Davidson, W. Voit and R. A. Smaldone, Polym. Chem., 2017, 8, 2087–2092 RSC.
- B. Zhang, Z. Gao, G. Gao, W. Zhao, J. Li and X. Ren, Macromol. Mater. Eng., 2018, 303, 1800072 CrossRef.
- M. C. Darnell, J.-Y. Sun, M. Mehta, C. Johnson, P. R. Arany, Z. Suo and D. J. Mooney, Biomaterials, 2013, 34, 8042–8048 CrossRef CAS PubMed.
- W. Zhao, Z. Han, L. Ma, S. Sun and C. Zhao, J. Mater. Chem. B, 2016, 4, 8016–8024 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09210j |
| ‡ These authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2021 |