
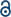 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective removal of cationic micro-pollutants using disulfide-linked network structures†
Mehmet Sahin Atasa,
Sami Dursuna,
Hasan Akyildiza,
Murat Citir b,
Cafer T. Yavuz*c and
Mustafa Selman Yavuz*a
b,
Cafer T. Yavuz*c and
Mustafa Selman Yavuz*a
aMetallurgy and Materials Engineering Department, Selcuk University, Konya, Turkey. E-mail: selmanyavuz@gmail.com
bMaterials Science and Nanotechnology Engineering, Abdullah Gül University, Melikgazi 38039, Kayseri, Turkey
cGraduate School of EEWS, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, 34141 Republic of Korea. E-mail: yavuz@kaist.ac.kr
First published on 15th May 2017
Abstract
Micropollutants are found in all water sources, even after thorough treatments that include membrane filtration. New ones emerge as complex molecules are continuously produced and discarded after used. Treatment methods and sorbent designs are mainly based on non-specific interactions and, therefore, have been elusive. Here, we developed swellable covalent organic polymers (COP) with great affinity towards micropollutants, such as textile industry dyes. Surprisingly, only cationic dyes in aqueous solution were selectively and completely removed. Studies of the COPs surfaces led to a gating capture, where negatively charged layer attracts cationic dyes and moves them inside the swollen gel through diffusive and hydrophobic interaction of the hydrocarbon fragments. Despite its larger molecular size, crystal violet has been taken the most, 13.4 mg g−1, surpassing all competing sorbents. The maximum adsorption capacity increased from 12.4 to 14.6 mg and from 8.9 to 11.4 mg when the temperature of dye solution was increased from 20 to 70 °C. The results indicated that disulfide-linked COPs are attractive candidates for selectively eliminating cationic dyes from industrial wastewater due to exceptional swelling behaviour, low-cost and easy synthesis.
Introduction
Micropollutants are highly water-soluble, often charged, organic molecules that are increasingly found in ground and surface water.1,2 These organic molecules mainly come from chemical and pharmaceutical industries. Although safety protocols are in place for newly discovered molecules and their environmental fate, there are still many unknowns, especially if they undergo chemical transformations like oxidation. Conventional sorbents like activated carbon suffer from low kinetics and specificity when it comes to extremely hydrophilic micropollutants.2Among the leading sources for micropollutants (Table S1 in ESI†), industrial use of synthetic dyes has increased considerably over the last few decades, particularly in textiles, plastics and papers.3–7 Textile industry is the most prominent where effluents containing large amounts of dye residue have to be treated before being discharged into wastewater streams. If not done properly, water pollution with toxic dyes, metal ions and organic contaminants pose serious environmental hazards, especially for aquatic biosystems, where symbiotic processes are affected by the contaminant induced inhibition of the photosynthetic activity.8
In recent years, a number of techniques were employed for the removal of organic pollutants from effluents, including precipitation, membrane filtration, coagulation, ion exchange, adsorption and chemical oxidation.9–15 Among these methods, adsorption is one of the promising techniques,16–19 mainly due to the effective procedure, operational simplicity, low cost and low energy requirements.1 The most widely used adsorbents include mesoporous silica,20 organoclays,21 powdered activated sludge,22 activated carbon,23 magnetic nanoparticles,24,25 zeolite,26 molecular sieves,27,28 activated aluminium oxide29 and porous polymers.30–34 Recently, we have shown that a fluorinated porous polymer can separate water-soluble organic molecules based on their sizes and charges.2
Network polymers have been increasingly studied for their tunable functionalities, controllable pore geometries and wide range of applications.35–42 Although conventional polymers and their derivatives, such as poly(acrylic acid),43 cellulose,44 carbonaceous materials,45,46 poly(orthocarbonate),47 microporous polymers,48,49 poly(lactic acid),50 polyaniline,51–53 were used as superabsorbents to remove oils, metal ions, toxic dyes and organic solvents from water, some of these materials were synthesized by using transition metal catalysts and sometimes at high temperatures.54 Therefore, new, low cost and effective adsorbents are always needed. One particular challenge is that high selectivity requires targeted chemistry, and such control on chemical architecture brings additional costs in material production.41
Covalent Organic Polymers (COPs) are highly stable and inexpensive solids41,55 that are often made from commercially available monomers with simple chemical transformations. In this study, we used two inexpensive disulfide-linked covalent organic polymers, COP-65 and COP-66, for selective dye uptake. Surprisingly, only cationic dyes were captured and the capacities did not follow simple surface monolayer coverage. These COPs were generated through disulfide linkages (–S–S–) by easy oxidative coupling under catalyst-free mild conditions. Adsorption and kinetics of textile dye removal from aqueous solutions were analysed with respect to the initial dye concentration, temperature, pH and sorbent dosage. Our findings point to significant potential for swellable polymers in effective water treatment needs.
Experimental
Materials
Trimethylolpropane tris(3-mercaptopropionate) (98%, 3SH) and pentaerythritol tetrakis(3-mercaptopropionate) (98%, 4SH) were obtained from Sigma-Aldrich. Dimethyl sulfoxide (99.5%, DMSO), tetrahydrofuran (98%, THF) and methanol (98%, MeOH) were purchased from Merck Chemical Company. Cationic dye Rhodamine B (RB) was obtained from Alfa Aesar, Methylene Blue (MB) and Crystal Violet (CV) were purchased from Merck Chemical Company and used as received. Anionic dyes Alizarin Yellow R (AY), Methyl Orange (MO) and Orange G Sodium Salt (OG) were purchased from Alfa Aesar, Sigma-Aldrich and Amresco, respectively. All chemicals were used as received without any further purification.Synthesis of covalent organic polymers (COPs)
Disulfide-linked covalent organic polymers (COPs) were reproduced through catalyst-free oxidative coupling polymerization according to our earlier method (Fig. 1).56 In a typical synthesis, trimethylolpropane tris(3-mercaptopropionate) (3SH) or pentaerythritol tetrakis(3-mercaptopropionate) (4SH) was added into DMSO, and the solution was heated at 80 °C for 24 h under vigorous stirring. The reaction mixture was cooled down to room temperature. The precipitates were centrifuged and soaked in THF for 12 h. Washing procedure was repeated four times. Finally, a gel-like material was dried at 50 °C for 2 h and 100 °C for 5 h under vacuum to obtain COP-65 (by using 3SH) or COP-66 (by using 4SH). FTIR spectroscopy (VERTEX-70 Bruker, Germany) was used for the structural confirmations of each synthesized COPs at room temperature (Fig. S1 in the ESI†). Solid-state cross polarization magic angle spinning (CP/MAS) NMR spectra were recorded on a Bruker Avance III 400 WB NMR spectrometer (Fig. S2 in the ESI†).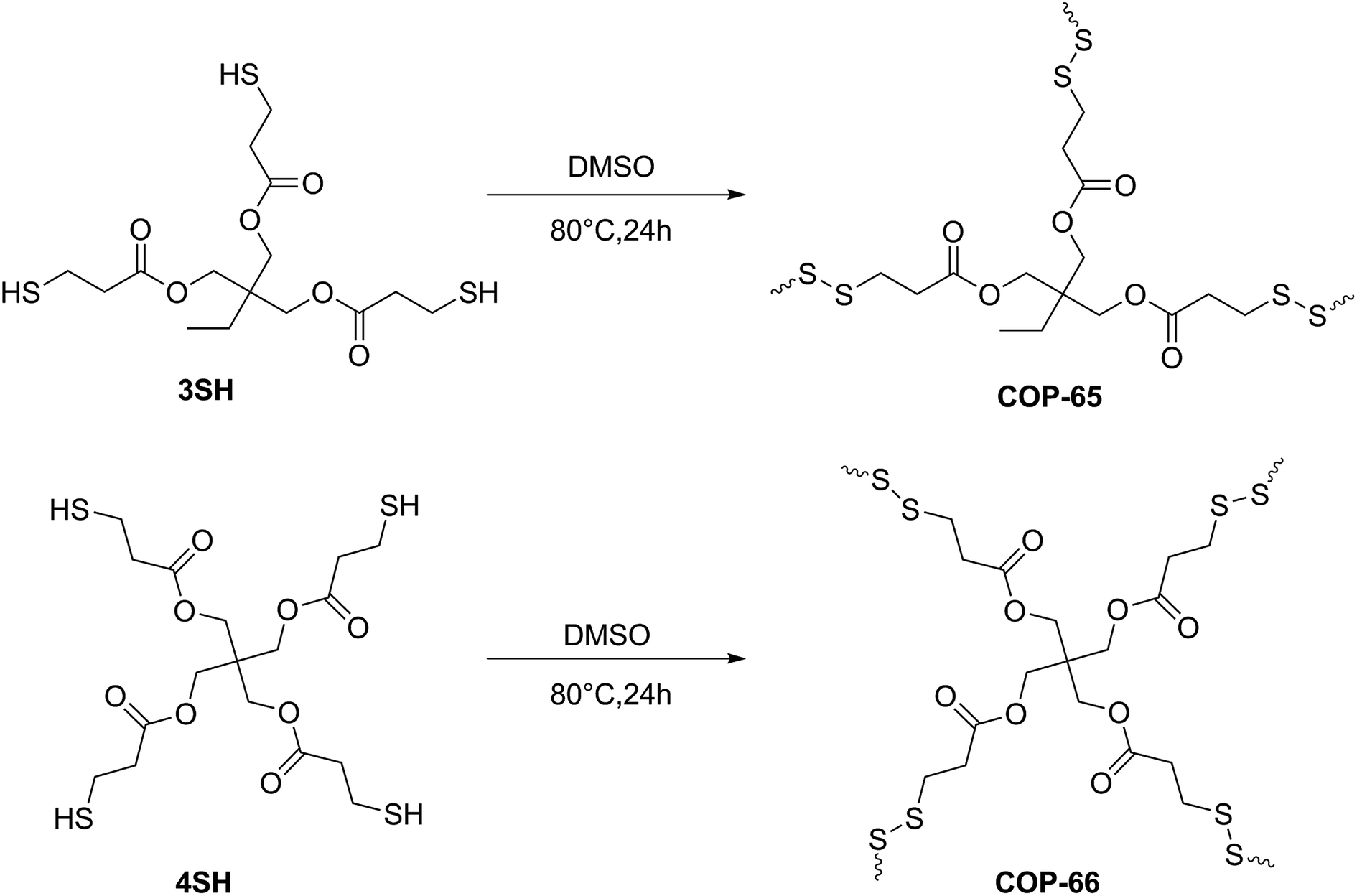 | ||
| Fig. 1 Schematic illustration for one pot, open air and easy synthesis of COPs by oxidative coupling of trimethylolpropane tris(3-mercaptopropionate) and pentaerythritol tetrakis(3-mercaptopropionate) in DMSO. | ||
Adsorption studies
All the adsorption experiments were carried out in 20 mL glass vials. Before adsorption experiments, calibration curves for each dye, CV, MB, RB, AY, MO and OG solutions (500 mg L−1) were prepared by using Biochrom Libra S22 model UV-Vis spectrophotometer. The dye uptake study was performed using an incubator shaker. Samples were placed in an incubator shaker (ZHWY-200B, Labwit, Shanghai, China), which was set at a speed of 150 rpm and temperature of 30 °C.In a typical adsorption study, 100 mg of COPs was added to a 3 mL of an aqueous micropollutant solution (500 mg L−1) at room temperature. After mixing by the shaker at 200 rpm for 24 h, the COPs were removed from the solution and supernatant was analysed by UV-Vis. For example, the adsorbed amount of CV dye was calculated from the difference between the concentrations of stock CV dye solution and the supernatant CV solution by UV-Vis spectrophotometry at the wavelength of 594 nm. Other cationic dyes MB (668 nm) and RB (554 nm), and anionic dyes as AY (374 nm), MO (463 nm) and OG (481 nm) were analysed similarly.
pH variation
The effect of the initial pH on the percentage removal of CV, MB and RB through adsorption onto COPs from aqueous dye solution was studied. In that order, the experiments were conducted at various pH values, ranging from 2.0 to 10.0 with a 500 mg L−1 initial dye concentration and 100 mg of COPs (adsorbent) at room temperature for 24 h. The pH was adjusted with 0.1 M hydrochloric acid (HCl) or 0.1 M sodium hydroxide (NaOH) by using a pH meter.Contact time effect
A mixture of aqueous initial CV dye concentrations of 500 mg L−1 and 100 mg of COPs (adsorbent) was shaken with an incubator shaker at 200 rpm for 5 h equilibrium time at room temperature. Every 30 min, a sample was taken for UV-Vis analysis.Temperature
The effect of the temperature for the removal of CV by using COPs was studied at different temperatures, namely, 25, 40, 55, 70 and 90 °C. Again, the experiments were performed with a solution containing an aqueous dye solution (concentration of 500 mg L−1) and 100 mg of COPs (adsorbent) using a incubator shaker at 200 rpm for 24 h equilibrium time.Dye concentration
The effect of the contact time for the removal of CV by using COPs was studied at different dye concentration such as, 100, 200, 300, 400 and 500 mg L−1 for 5 h equilibrium time under the same conditions as the temperature variation study.Desorption and recyclability
To evaluate the possibility of regeneration of the dye-saturated adsorbent, desorption experiments were carried out using tetrahydrofuran/water (50/50) solution in an ultrasonic generator. 100 mg COPs (adsorbents) were added to 5 mL of dye solutions and the mixture was shaken on a rotary shaker at 200 rpm for 24 h. The initial dye concentration was 500 mg L−1. At the end of the adsorption period, the dye-adsorbed COPs adsorbents were soaked into 3 mL tetrahydrofuran for desorption process. The suspensions were shaken on a rotary shaker at 200 rpm for 24 h. Then the supernatant solutions were analysed by using UV-Vis spectrometry.Characterisation
In order to evaluate the porosity of COPs, N2 adsorption isotherms were obtained with a Micromeritics Tristar II 3020 accelerated surface area and porosimetry analyser at 77 K, after the samples were degassed at 150 °C for 5 h under vacuum prior to the measurements. The adsorption desorption isotherms were obtained to give BET (P/Po = 0.01–0.25) and Langmuir (P/Po = 0.1–0.35) surface areas. The UV-Vis spectra from 190–900 nm were recorded using a Biochrom Libra S22 spectrometer using quartz cuvettes. Measurements of pH were obtained using an Orion Research model SA250 digital pH meter. Zeta potential determinations of COP-65 and COP-66 were performed in aqueous 10 mM NaCl solutions by using Nano ZS, Malvern Instruments.Results and discussion
The hydrophobic, aprotic behaviour of COP-65 and COP-66 led us to investigate adsorbent characteristics in removal of contaminants with complex organic molecule nature.56 Due to the easy detection and the industrial relevance, we chose common textile dyes to be screened for removal activity. We, therefore, classified the target dyes in two groups: (1) anionic, and (2) cationic. Three dyes for each group were selected: Methyl Orange (MO), Alizarin Yellow R (AY) and Orange G Sodium Salt (OG) as anionic dyes and Crystal Violet (CV), Methylene Blue (MB) and Rhodamine B (RB) as cationic (Fig. 2). For all adsorption experiments, 100 mg of sorbent was treated with 3 mL aqueous solution (500 mg L−1) of the selected dye. | ||
| Fig. 2 Chemical structures of the micro-pollutants; (a) anionic dyes: Alizarin Yellow (AY), Methyl Orange (MO) and Orange G (OG); (b) cationic dyes: Methylene Blue (MB), Crystal Violet (CV) and Rhodamine B (RB). | ||
The micropollutant dye molecules have varying sizes as well. We have recently shown that a rigid microporous polymer (COP-99) can do both size and charge dependent separation.2 Here, however, the network is made up of purely aliphatic monomers and prone to swelling with no initial porosity. We anticipate a different mechanism, where guest molecules need to penetrate by expanding the network branches.
Interestingly, both COPs showed a selective uptake towards cationic dyes (Fig. 3). CV was the most captured dye molecule with capacities reaching up to 13.4 mg g−1 (with COP-65). RB uptake reached 10.9 mg g−1 and MB has a maximum of 9.5 mg g−1 loading. However, the uptake of anionic dyes (AY, MO and OG) by both COPs was negligible. The adsorption capacities of anionic dyes were 0.425 mg g−1 (AY), 0.575 mg g−1 (MO) and 0.325 mg g−1 (OG) by COP-65; and 0.337 mg g−1 (AY), 0.445 mg g−1 (MO) and 0.267 mg g−1 (OG) by COP-66.
 | ||
| Fig. 3 (a) Adsorption capacities of COP-65 and COP-66 for six common anionic and cationic dyes, (b) digital pictures of anionic and cationic dye uptake experiment (before and after 24 h) by COP-65. | ||
The high capture capacities for Crystal Violet (CV) allowed us to compare our results with that of others, surpassing all of the other low cost literature examples (Table 1).57–62 These values suggest that the inexpensive COPs have great potential in selective cationic dye removal from aqueous solution. In that, COPs behave uniquely and can serve as extractors from aqueous medium. This, in contrast to well known all-purpose sorbents like activated carbons, is quite different and only recently been explored.2
In order to understand why cationic dyes were taken up selectively, we measured surface charges of both COP structures. By using zeta potential readings, the surface charges of COP-65 and COP-66 were measured as −14.6 mV and −8.53 mV, respectively. Negative values generally correspond to the anionic species and electronegative atoms with lone pairs. We suspect that the network building by a single monomer with thiol ends provides high thiol coverage of the surface, hence lone pairs. Deprotonation of the thiols also generate anionic sulphides and perhaps sulfonates from the oxidative coupling reaction. Ester groups on the branches of the network polymers also contribute to the electron donating nature of the surface.
The surface charges explain the favourable interaction, but our findings indicate that a swelling behaviour is mainly responsible for the selective uptake. Despite the larger hydrodynamic molecular size of the CV, it is adsorbed the most. This leads to a conviction that uptake is not only due to surface coverage of the dye molecules. Taking all these possibilities, we suggest a “gating approach” where surface charges form an electrostatic barrier for rejection of similar charges. Then the hydrophobic interior is swollen by the interaction of hydrocarbon backbone of the dye molecules. A similar mechanism was observed when linear or branched hydrocarbons were selectively separated on a metal–organic framework (MOF), Fe2(BDP)3.63 Most recently, using analogous concepts, Eddaoudi and co-workers showed a very difficult separation of propylene from propane by tailor-made MOF structure, KAUST-7.64
Surface negative charge and the possibility of the deprotonation prompted us to study in detail how the surface behaves. We, therefore, carried out pH effect on the uptake capacities by both network polymers (Fig. 4). It is imperative that at lower pH values, a less negative surface is present, leading to a lower uptake of cations. The experiments were conducted at various pH values (ranging from 2.0 to 10.0) with a 500 mg L−1 initial dye concentration and 100 mg of COP-65 and COP-66 (adsorbents) at room temperature for a day. The pH was adjusted by adding either HCl or NaOH. The results indicate when the pH was changed from 2.0 to higher values up to 8.0, the amount of cationic dyes adsorbed by COP-65 and COP-66 as shown in Fig. 4a and b, respectively. At lower pH values, the hydrogen ion (H+) ion concentration in the aqueous system is higher, which makes the surface of COPs less negatively charged due to protonation.22 The pH of the aqueous solution is above 7, more negatively charged surfaces of both COPs are formed due to the excess hydroxide ions (OH−). As the surfaces of COP-65 and COP-66 become negatively charged at pH 8, there is significant electrostatic attraction between the negatively charged surface of COPs and the cationic dye molecules, which leads to a maximum dye adsorption. However, above pH 8, the hydroxide ions are in competition with the sorbent to combine with cationic species, which lead to lower dye adsorptions by COPs due to the lack of substrate availability.22
 | ||
| Fig. 4 Adsorption performance of cationic dyes by (a) COP-65 (b) COP-66 in varying pH conditions. | ||
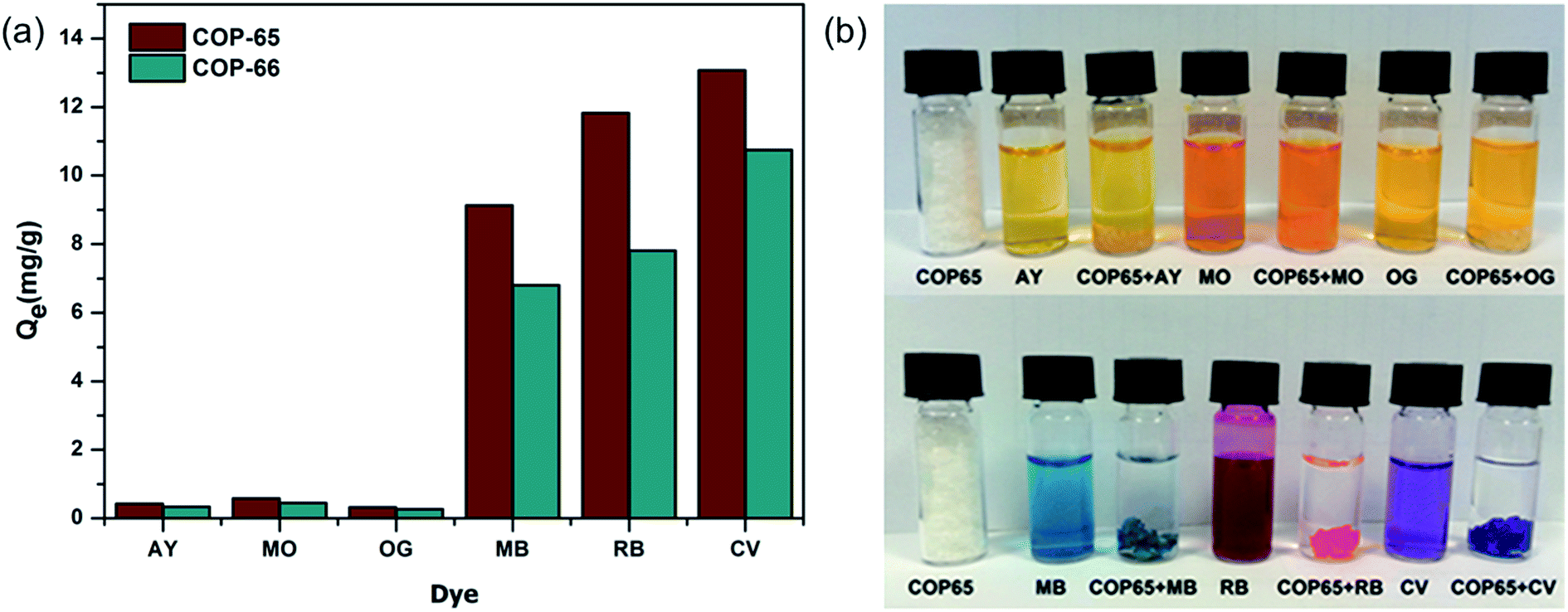
In order to study the kinetics, we analysed the adsorption of Crystal Violet (CV) with respect to contact time and temperature (Fig. 5). The rate of CV sorption was determined in varying time intervals by mixing 500 mg L−1 of aqueous CV solution with 100 mg of COP-65 or COP-66. Just after the addition of COP-65 or COP-66, the dye adsorption started immediately. The adsorption was increased sharply within 30 min and attained equilibrium after 2 h (Fig. 5a). There is no significant increase in removal of CV dye after 2 h contact time.
 | ||
| Fig. 5 Effects of (a) contact time (inset shows the first hour) and (b) temperature on adsorption capacity of Crystal Violet (CV) on COP-65 and COP-66. | ||
Effect of temperature on the adsorption of CV by COP-65 and COP-66 at equilibrium was investigated at five different temperatures: 25, 40, 55, 70 and 90 °C (Fig. 5b). We observed that the amount of adsorbed CV by COP-65 and COP-66 increased from 12.4 to 14.6 mg g−1 and from 8.9 to 11.4 mg g−1, respectively, as the temperature was increased from 25 to 70 °C. The enhanced adsorption at higher temperatures could be a result of an increase in the mobility of the dye molecules (higher diffusion rate) and an easier swelling within COPs at higher temperatures, which could enable the dye molecules to penetrate further inside.65 On the other hand, we found no significant change in the CV dye adsorption efficiency after 70 °C, as it nears to the boiling point of water.
We also studied the dose of the dye molecules in adsorption capacities. Fig. 6a shows the adsorption capacity of COP-65 against the contact time for varying initial concentrations (100, 200, 300, 400 and 500 mg L−1) of the Crystal Violet (CV). A fast adsorption was observed for all concentrations during the first 15 min due to the available surface area of the COPs for the adsorption of CV. Higher CV dye concentration led to an increase in the adsorption capacity of COP-65 due to the driving force of the concentration gradient. The amounts of dye (mg g−1) adsorbed on the COPs (Qe) increased from 4.516 to 11.238 mg g−1 when the initial dye concentration changes from 100 to 500 mg L−1. Equilibrium of dye adsorption capacity was attained for all dye concentrations after 4 h.
 | ||
| Fig. 6 (a) Effect of contact time on adsorption capacity of COP-65 with different concentrations of Crystal Violet (CV), (b) recycling experiment of COP-65 for the adsorption and desorption of CV (500 mg L−1). | ||
Recyclability of the sorbents is ultimately important for any commercial feasibility. The COP-65 was tested in terms of its reusability for three consecutive cycles. 100 mg of COP-65 was mixed into the 500 mg L−1 of CV solution. The desorption of almost all adsorbed CV was achieved by treating with THF (a widely available industrial solvent) for 6 h (Fig. 6b). COP-65 showed 94.4% adsorption of CV in the second cycle.56 Thereafter, the adsorbent showed slight decline in its performance, both in adsorption as well as desorption. The use of the same COP-65 in the third cycle showed 93.1% dye adsorption. We suspect that the remaining adsorbed dye molecules lead to decrease in both uptake and recovery of CV. Since the adsorption is purely physisorptive, mechanical or sonochemical approaches could extract all trapped molecules. Our studies are underway in that direction.
The equilibrium adsorption isotherm plays an important role in predictive modelling for design and optimization of adsorption systems.66 In order to describe the relationship between the amount of dye adsorbed and its equilibrium concentration in solution, we used two of the most commonly used models, Langmuir and Freundlich isotherms. Langmuir isotherm theory is based upon the assumption of monolayer adsorption on a homogeneous adsorbent.67,68 The linear form of the Langmuir isotherm model can be written by using the equation below:
 | (1) |
 | (2) |
Linear plots of Ce/qe versus Ce and ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) qe versus lnCe are shown in Fig. 7. The values of qm and KL in the Langmuir isotherm model and the values of KF and n in the Freundlich isotherm model are presented in Table 2. These results indicate that the CV dye in the aqueous solution is favourably adsorbed by the COP-65. The R2 value with respect to sorption of CV for Langmuir isotherm model was noted to be 0.99657, which was 0.85131 for Freundlich isotherm model. It could be concluded from these observations that the adsorption of CV by using COP-65 was better defined by the Langmuir than Freundlich isotherm model.
qe versus lnCe are shown in Fig. 7. The values of qm and KL in the Langmuir isotherm model and the values of KF and n in the Freundlich isotherm model are presented in Table 2. These results indicate that the CV dye in the aqueous solution is favourably adsorbed by the COP-65. The R2 value with respect to sorption of CV for Langmuir isotherm model was noted to be 0.99657, which was 0.85131 for Freundlich isotherm model. It could be concluded from these observations that the adsorption of CV by using COP-65 was better defined by the Langmuir than Freundlich isotherm model.
 | ||
| Fig. 7 The linear fittings for (a) Langmuir and (b) Freundlich adsorption isotherms for the sorption of Crystal Violet (CV) by COP-65. | ||
| Langmuir | Freundlich | ||||
|---|---|---|---|---|---|
| qm (mg g−1) | KL (L mg−1) | R2 | KF (L mg−1) | n | R2 |
| 13.245 | 0.063 | 0.99657 | 2.287 | 2.886 | 0.85131 |
Conclusions
We have demonstrated selective cationic micropollutant (in this case textile dyes) removal from aqueous solutions by using covalent organic polymers (COPs) that were synthesized by catalyst-free oxidative coupling polymerization of tetravalent or trivalent aliphatic thiols. COP-65 and COP-66 were obtained from commercially available and inexpensive monomers, and the resultant swellable insoluble networks exhibited unique uptake behaviour for three different cationic dyes. Interestingly, only cationic dyes were taken in favourably by these sorbents. Crystal Violet (CV) was captured the highest, despite its larger hydrodynamic structure. As temperature, initial dye concentrations or contact time increases, higher adsorption of CV was observed. The experimental data correlated well by the Langmuir adsorption isotherm compared to Freundlich ones. The present study concludes that the disulfide COPs could be employed effectively as affordable adsorbents for the removal of cationic micropollutants from aqueous media. In the future, new and improved polymer networks will be evaluated for a variety of potential applications and investigated with respect to different parameters that include a multivariate study of experimental design and chemometrics.Acknowledgements
This study was carried out as a Master thesis by Mehmet Sahin Atas at the Graduate School of Natural and Applied Science at Selcuk University, Konya, Turkey. Support for this work is provided by the Scientific Research Foundation (BAP) of Selçuk University, Scientific and Technological Research Council of Turkey (TUBITAK MAG) (Project Number 214M232) and Academic Staff Training Program (ÖYP) (Project Number 2015-ÖYP-128), which authors gratefully acknowledge. C. T. Y. thanks for the support from National Research Foundation (NRF) of Korea, No: NRF-2016R1A2B4011027.References
- A. Alsbaiee, B. J. Smith, L. Xiao, Y. Ling, D. E. Helbling and W. R. Dichtel, Nature, 2016, 529, 190–194 CrossRef CAS PubMed.
- J. Byun, H. A. Patel, D. Thirion and C. T. Yavuz, Nat. Commun., 2016, 7, 13377 CrossRef CAS PubMed.
- B. Miccoli, V. Cauda, A. Bonanno, A. Sanginario, K. Bejtka, F. Bella, M. Fontana and D. Demarchi, Sci. Rep., 2016, 6, 29763 CrossRef CAS PubMed.
- M. Ibáñez, V. Borova, C. Boix, R. Aalizadeh, R. Bade, N. S. Thomaidis and F. Hernández, J. Hazard. Mater., 2017, 323, 26–35 CrossRef PubMed.
- G. Crini, Bioresour. Technol., 2006, 97, 1061–1085 CrossRef CAS PubMed.
- R. Gong, M. Li, C. Yang, Y. Sun and J. Chen, J. Hazard. Mater., 2005, 121, 247–250 CrossRef CAS PubMed.
- F.-C. Wu and R.-L. Tseng, J. Hazard. Mater., 2008, 152, 1256–1267 CrossRef CAS PubMed.
- C. J. Madadrang, H. Y. Kim, G. Gao, N. Wang, J. Zhu, H. Feng, M. Gorring, M. L. Kasner and S. Hou, ACS Appl. Mater. Interfaces, 2012, 4, 1186–1193 CAS.
- M.-S. Chiou and G.-S. Chuang, Chemosphere, 2006, 62, 731–740 CrossRef CAS PubMed.
- J.-L. Gong, B. Wang, G.-M. Zeng, C.-P. Yang, C.-G. Niu, Q.-Y. Niu, W.-J. Zhou and Y. Liang, J. Hazard. Mater., 2009, 164, 1517–1522 CrossRef CAS PubMed.
- T.-H. Kim, C. Park and S. Kim, J. Cleaner Prod., 2005, 13, 779–786 CrossRef.
- R. J. Stephenson and S. J. Duff, Water Res., 1996, 30, 781–792 CrossRef CAS.
- D. P. Mesquita, A. L. Amaral and E. C. Ferreira, Chem. Eng. J., 2016, 285, 349–357 CrossRef CAS.
- F. Bella, D. Pugliese, J. R. Nair, A. Sacco, S. Bianco, C. Gerbaldi, C. Barolo and R. Bongiovanni, Phys. Chem. Chem. Phys., 2013, 15, 3706–3711 RSC.
- F. Bella, M. Imperiyka and A. Ahmad, J. Photochem. Photobiol., A, 2014, 289, 73–80 CrossRef CAS.
- V. Gupta, J. Environ. Manage., 2009, 90, 2313–2342 CrossRef CAS PubMed.
- A. Gürses, Ç. Doğar, M. Yalçın, M. Açıkyıldız, R. Bayrak and S. Karaca, J. Hazard. Mater., 2006, 131, 217–228 CrossRef PubMed.
- J. Y. Song, B. N. Bhadra and S. H. Jhung, Microporous Mesoporous Mater., 2017, 243, 221–228 CrossRef CAS.
- R. K. Motkuri, H. V. R. Annapureddy, M. Vijaykumar, H. T. Schaef, P. F. Martin, B. P. McGrail, L. X. Dang, R. Krishna and P. K. Thallapally, Nat. Commun., 2014, 5, 4368 CAS.
- K. Y. Ho, G. McKay and K. L. Yeung, Langmuir, 2003, 19, 3019–3024 CrossRef CAS.
- H. A. Patel, H. C. Bajaj and R. V. Jasra, Ind. Eng. Chem. Res., 2008, 48, 1051–1058 CrossRef.
- M. Doğan, M. Alkan, A. Türkyilmaz and Y. Özdemir, J. Hazard. Mater., 2004, 109, 141–148 CrossRef PubMed.
- Y. Sharma, U. S. Upadhyay and F. Gode, J. Appl. Sci. Environ. Sanit., 2009, 4, 21–24 Search PubMed.
- C. Junyong, H. Yongmei, L. Yan and G. Jiajia, RSC Adv., 2013, 3, 7254–7258 RSC.
- D. Das, J. Plazas-Tuttle, I. V. Sabaraya, S. S. Jain, T. Sabo-Attwood and N. B. Saleh, Environ. Sci.: Nano, 2017, 4, 60–68 RSC.
- M. R. Delgado and C. O. Arean, Energy, 2011, 36, 5286–5291 CrossRef CAS.
- H. Huang, C. Yang, H. Zhang and M. Liu, Microporous Mesoporous Mater., 2008, 111, 254–259 CrossRef CAS.
- D. Lozano-Castello, J. Alcaniz-Monge, D. Cazorla-Amoros, A. Linares-Solano, W. Zhu, F. Kapteijn and J. Moulijn, Carbon, 2005, 43, 1643–1651 CrossRef CAS.
- A. Genz, A. Kornmüller and M. Jekel, Water Res., 2004, 38, 3523–3530 CrossRef CAS PubMed.
- S. Debnath, A. Shome, S. Dutta and P. K. Das, Chem.–Eur. J., 2008, 14, 6870–6881 CrossRef CAS PubMed.
- B. Pan, B. Pan, W. Zhang, L. Lv, Q. Zhang and S. Zheng, Chem. Eng. J., 2009, 151, 19–29 CrossRef CAS.
- Y. Zhang, S. Wei, Y. He, F. Nawaz, S. Liu, H. Zhang and F.-S. Xiao, J. Mater. Chem., 2010, 20, 4609–4614 RSC.
- B. Ashourirad, P. Arab, T. Islamoglu, K. A. Cychosz, M. Thommes and H. M. El-Kaderi, J. Mater. Chem. A, 2016, 4, 14693–14702 CAS.
- X. Zhu, S. An, Y. Liu, J. Hu, H. Liu, C. Tian, S. Dai, X. Yang, H. Wang, C. W. Abney and S. Dai, AIChE J., 2017 DOI:10.1002/aic.15699.
- S. H. Je, O. Buyukcakir, D. Kim and A. Coskun, Chem, 2016, 1 482–493 Search PubMed.
- P. Li, J. A. Modica, A. J. Howarth, E. L. Vargas, P. Z. Moghadam, R. Q. Snurr, M. Mrksich, J. T. Hupp and O. K. Farha, Chem, 2016, 1, 154–169 Search PubMed.
- Q.-L. Zhu and Q. Xu, Chem, 2016, 1, 220–245 Search PubMed.
- Q. Sun, B. Aguila, G. Verma, X. Liu, Z. Dai, F. Deng, X. Meng, F.-S. Xiao and S. Ma, Chem, 2016, 1, 628–639 Search PubMed.
- D. Banerjee, S. K. Elsaidi, B. Aguila, B. Li, D. Kim, M. J. Schweiger, A. A. Kruger, C. J. Doonan, S. Ma and P. K. Thallapally, Chem.–Eur. J., 2016, 22, 17581–17584 CrossRef CAS PubMed.
- S. Kuecken, J. Schmidt, L. Zhi and A. Thomas, J. Mater. Chem. A, 2015, 3, 24422–24427 CAS.
- H. A. Patel, F. Karadas, A. Canlier, J. Park, E. Deniz, Y. Jung, M. Atilhan and C. T. Yavuz, J. Mater. Chem., 2012, 22, 8431–8437 RSC.
- H. A. Patel and C. T. Yavuz, Chem. Commun., 2012, 48, 9989–9991 RSC.
- T. Ono, T. Sugimoto, S. Shinkai and K. Sada, Adv. Funct. Mater., 2008, 18, 3936–3940 CrossRef CAS.
- G. Marcì, G. Mele, L. Palmisano, P. Pulito and A. Sannino, Green Chem., 2006, 8, 439–444 RSC.
- Z. L. Fan, X. J. Qin, H. X. Sun, Z. Q. Zhu, C. j. Pei, W. D. Liang, X. M. Bao, J. An, P. Q. La and A. Li, ChemPlusChem, 2013, 78, 1282–1287 CrossRef CAS.
- X. Gui, J. Wei, K. Wang, A. Cao, H. Zhu, Y. Jia, Q. Shu and D. Wu, Adv. Mater., 2010, 22, 617–621 CrossRef CAS PubMed.
- H. B. Sonmez and F. Wudl, Macromolecules, 2005, 38, 1623–1626 CrossRef CAS.
- A. Li, H.-X. Sun, D.-Z. Tan, W.-J. Fan, S.-H. Wen, X.-J. Qing, G.-X. Li, S.-Y. Li and W.-Q. Deng, Energy Environ. Sci., 2011, 4, 2062–2065 CAS.
- P. D. Mines, J. Byun, Y. Hwang, H. A. Patel, H. R. Andersen and C. T. Yavuz, J. Mater. Chem. A, 2016, 4, 632–639 CAS.
- Z. Xue, Z. Sun, Y. Cao, Y. Chen, L. Tao, K. Li, L. Feng, Q. Fu and Y. Wei, RSC Adv., 2013, 3, 23432–23437 RSC.
- M. M. Ayad and A. A. El-Nasr, J. Phys. Chem. C, 2010, 114, 14377–14383 CAS.
- T. K. Mahto, A. R. Chowdhuri and S. K. Sahu, J. Appl. Polym. Sci., 2014, 131, 40840 CrossRef.
- L. Wang, X. L. Wu, W. H. Xu, X. J. Huang, J. H. Liu and A. W. Xu, ACS Appl. Mater. Interfaces, 2012, 4, 2686–2692 CAS.
- L. Zhang, Y. Liu, S. Wang, B. Liu and J. Peng, RSC Adv., 2015, 5, 99618–99626 RSC.
- H. A. Patel, F. Karadas, J. Byun, J. Park, E. Deniz, A. Canlier, Y. Jung, M. Atilhan and C. T. Yavuz, Adv. Funct. Mater., 2013, 23, 2270–2276 CrossRef CAS.
- H. A. Patel, M. S. Yavuz and C. T. Yavuz, RSC Adv., 2014, 4, 24320–24323 RSC.
- H. Ali and S. K. Muhammad, J. Environ. Sci. Technol., 2008, 1, 143–150 CrossRef CAS.
- R. Dhodapkar, N. Rao, S. Pande, T. Nandy and S. Devotta, React. Funct. Polym., 2007, 67, 540–548 CrossRef CAS.
- S. Khattri and M. Singh, Water, Air, Soil Pollut., 2000, 120, 283–294 CrossRef CAS.
- S. Khattri and M. Singh, Adsorpt. Sci. Technol., 1999, 17, 269–282 CrossRef CAS.
- C. Namasivayam, M. D. Kumar, K. Selvi, R. A. Begum, T. Vanathi and R. Yamuna, Biomass Bioenergy, 2001, 21, 477–483 CrossRef CAS.
- H. Parab, M. Sudersanan, N. Shenoy, T. Pathare and B. Vaze, Clean: Soil, Air, Water, 2009, 37, 963–969 CrossRef CAS.
- Z. R. Herm, B. M. Wiers, J. A. Mason, J. M. van Baten, M. R. Hudson, P. Zajdel, C. M. Brown, N. Masciocchi, R. Krishna and J. R. Long, Science, 2013, 340, 960 CrossRef CAS PubMed.
- A. Cadiau, K. Adil, P. M. Bhatt, Y. Belmabkhout and M. Eddaoudi, Science, 2016, 353, 137 CrossRef CAS PubMed.
- H. Asfour, O. Fadali, M. Nassar and M. El-Geundi, J. Chem. Technol. Biotechnol., Chem. Technol., 1985, 35, 21–27 CrossRef.
- M. S. Sajab, C. H. Chia, S. Zakaria, S. M. Jani, M. K. Ayob, K. L. Chee, P. S. Khiew and W. S. Chiu, Bioresour. Technol., 2011, 102, 7237–7243 CrossRef CAS PubMed.
- P. Monash and G. Pugazhenthi, Adsorption, 2009, 15, 390–405 CrossRef CAS.
- K. Periasamy and C. Namasivayam, Ind. Eng. Chem. Res., 1994, 33, 317–320 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04775d |
| This journal is © The Royal Society of Chemistry 2017 |