
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective Baeyer–Villiger oxidation of lignin model compounds with tin beta zeolite catalyst and hydrogen peroxide†
John A. Jenningsab,
Sean Parkina,
Eric Munsonc,
Sean P. Delaneyc,
Julie L. Calahanc,
Mark Isaacs d,
Kunlun Honge and
Mark Crocker*ab
d,
Kunlun Honge and
Mark Crocker*ab
aDepartment of Chemistry, University of Kentucky, Lexington, KY, USA. E-mail: mark.crocker@uky.edu; Tel: +859-257-0295
bUniversity of Kentucky Center for Applied Energy Research, Lexington, KY, USA
cDepartment of Pharmaceutical Sciences, University of Kentucky, Lexington, KY, USA
dEuropean Bioenergy Research Institute, Aston University, Birmingham, UK
eCenter for Nanophase Materials Sciences and Chemical Science Division, Oak Ridge National Laboratory, Oak Ridge, TN, USA
First published on 16th May 2017
Abstract
Lignin depolymerization represents a promising approach to the sustainable production of aromatic molecules. One potential approach to the stepwise depolymerization of lignin involves oxidation of the benzylic alcohol group in β-O-4 and β-1 linkages, followed by Baeyer–Villiger oxidation (BVO) of the resulting ketones and subsequent ester hydrolysis. Towards this goal, BVO reactions were performed on 2-adamantanone, a series of acetophenone derivatives, and lignin model compounds using a tin beta zeolite/hydrogen peroxide biphasic system. XRD, 119Sn MAS NMR spectroscopy, DRUVS and XPS were used to determine tin speciation in the catalyst, the presence of both framework Sn and extra framework SnO2 being inferred. Conversion of ketones to BVO products was affected by electron donation as well as steric hindrance, 4′-methoxyacetophenone affording the highest yield of ester (81%). As the size and complexity of the ketone increased, excess hydrogen peroxide was typically needed for successful BVO. Yields of ester products derived from β-O-4 and β-1 lignin models were modest due to the formation of polymeric material stemming from direct ring hydroxylation.
1. Introduction
Lignin is biosynthesized to defend plant-life against chemical and biological attack.1 As the largest natural source of aromatics, products of lignin depolymerization have potential as both renewable fuel (e.g., benzene, toluene, and xylenes (BTX)) and fine chemicals (e.g., vanillin). However, most lignin produced by the pulp and paper industries is currently burned on-site as a low-grade fuel. Due to lignin's characteristic irregularity and poor solubility, lignin is challenging to utilize on an industrial scale.In lieu of using lignin, which is difficult to analyze, models of various linkage motifs are commonly used to investigate depolymerization strategies. The most abundant of these linkages is the β-O-4 structure, which represents up to 60% of the linkages found in lignin.2 A benzylic alcohol moiety and a bridging aryl ether bond are characteristic of the β-O-4 linkage. Many lignin depolymerization strategies focus on cleaving the bridging ether groups using thermal and/or reductive techniques, requiring high temperatures and pressures. As an alternative, Stahl and coworkers3 performed an extensive study of stoichiometric oxidants, metal-catalyzed aerobic oxidations, and metal-free catalytic aerobic oxidations. The authors found the most selective and efficient benzylic oxidation system to be 4-acetamido-2,2,6,6-tetramethylpiperidine-N-oxyl/oxygen, yielding almost quantitative benzylic oxidation for “dimeric” model compounds (i.e., compounds containing two aromatic groups linked in β-O-4 fashion).3 The authors also reported that in some less selective oxidation systems, after γ-oxidation, models were converted to substituted aldehydes via retro aldol reactions. According to Stahl and coworkers,3 the utility of this retro aldol reaction is limited due to formation of unidentified products, presumably phenolic radical coupling products. Retro aldol reactions have been reported for both α and γ-ketones.3–5
After benzylic oxidation, Stahl and coworkers cleaved the β-O-4 linkage via Dakin oxidation, affording an 88% yield of 3,4-dimethoxybenzoic acid and 42% yield of guaiacol (Scheme 1). The low yield of guaiacol is derived from phenolic radical coupling initiated by hydrogen peroxide, which is capable of both one and two-electron oxidations. Indeed, poor isolated yields of phenolic compounds is a common problem in the oxidation of aromatic molecules.6
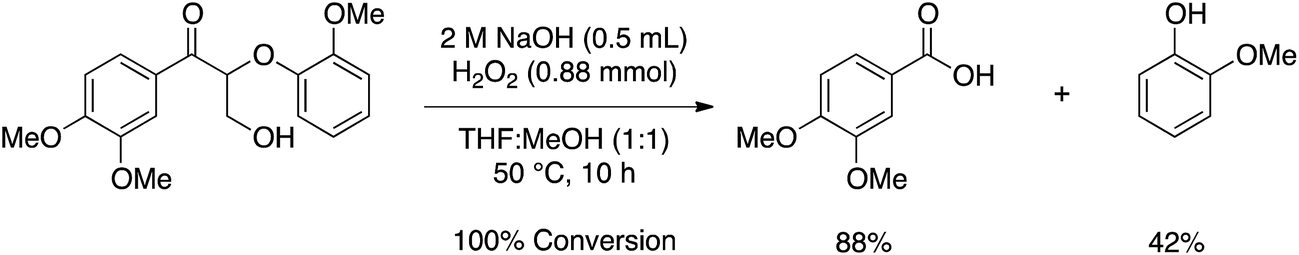 | ||
| Scheme 1 Dakin oxidation of a lignin dimer model compound performed by Stahl and coworkers.3 | ||
Westwood and coworkers7 also reported almost quantitative benzylic oxidation of lignin model compounds using a 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)/oxygen system. After zinc-based reduction/cleavage the authors obtained up to 92% isolated yields of substituted propylarenes, although the yields of phenolic moieties were not reported.
Wang and coworkers5 also observed almost quantitative benzylic oxidation of lignin model compounds using vanadyl sulfate/TEMPO/oxygen (Scheme 2). In addition to benzylic ketones, the reaction yielded an unexpected α-retro aldol product (20% yield). Benzylic (Cα) retro aldol reactions simplify models by elimination of formaldehyde, increasing the reactivity in oxidizing systems.4,5 Indeed, Wang et al.5 found that models with no γ-carbon were more active to further oxidation.
 | ||
| Scheme 2 Benzylic oxidation performed by Wang et al.5 | ||
Following benzylic oxidation, Wang et al.5 depolymerized the ketone with a copper/phenanthroline/superoxide system to form a benzoic acid and a phenol, the authors noting a significant decrease in phenol yields in models with electron-donating methoxy groups.
In one of the few reports resulting in high yields of phenols, Stahl and coworkers8 used a redox-neutral system (excess formic acid and sodium formate) to effectively depolymerize oxidized lignin and lignin model compounds to discrete products. Depolymerization products of models included phenols, which were obtained in excellent yields (Scheme 3). The absence of a one-electron reactant supports the hypothesis that phenols produced in a peroxide-free environment can be isolated in excellent yields. However, this very successful example of β-O-4 cleavage suffers from a large excess of formic acid and is homogeneous in nature.
 | ||
| Scheme 3 Depolymerization of a lignin model compound performed by Stahl and coworkers.8 | ||
Although selective benzylic oxidation is a rather facile first step, there are few methods for which the phenolic portions of models produced by Cβ–O4 bond cleavage are isolated in high yield. To avoid the production of phenolate radicals, which readily repolymerize, benzylic ketones can in principle be converted directly to esters via Baeyer–Villiger Oxidation (BVO), and subsequently hydrolyzed in a one-electron free medium. In another publication concerning benzylic oxidation of lignin model compounds, Patil et al.9 reported the first well-characterized homogeneous BVO of an aromatic ketone using an oxidation system generating performic acid in situ (Scheme 4).
 | ||
| Scheme 4 Lignin model depolymerization strategy reported by Patil et al.9 | ||
As a consequence of the acidic nature of the reaction, a high yield of 3,4-dimethoxybenzoic acid from hydrolysis of the β-O-4 model was reported (Scheme 4, entry (a)). However, the complete absence of the phenol co-product was attributed to polymerization. In the same report a BVO product (not hydrolyzed in situ) was reported in 10% yield (>99% selectivity) (Scheme 4, entry (b)).9
Recently, several heterogeneous systems for the oxidation of lignin model compounds have been identified.10–13 However, these catalysts do not yield ester products resulting from oxygen insertion. Rather, reports of heterogeneous oxidation result in small molecules resulting directly from Cα–Cβ or Cβ–O4 bond cleavage. In the present study an oxidation system consisting of hydrogen peroxide (a weaker oxidant than performic acid) and a heterogeneous tin beta zeolite catalyst was explored in an effort to avoid phenolic polymerization. The nature of this reaction system was anticipated to facilitate ready isolation of the desired products.
Most reports of heterogeneously catalyzed BVO concern cyclic ketones,14 with few reports of acyclic ketones, and even fewer of benzylic ketones. BVO of aromatic aldehydes to formate esters using hydrothermally synthesized tin beta zeolite has been reported,15 but employed highly active peroxyimidic acids. Panchgalle et al.16 reported BVO of acetophenone derivatives using an ionic liquid as co-oxidant, but provided no details of the catalyst synthesis, characterization, or role of the ionic liquid. Hydrothermally synthesized tin beta zeolite has also been reported as being active in several other Lewis acid catalyzed reactions such as Meerwein–Ponndorf–Verley (MPV) reductions.17 However, synthetic challenges such as long crystallization times (e.g., 20 days), and hazardous synthetic reagents (hydrofluoric acid) make large-scale hydrothermal synthesis of tin beta zeolite problematic.18
As an alternative to hydrothermal synthesis, tin beta zeolite has recently been synthesized using post-synthetic (PS) modification strategies.18–22 Indeed, PS incorporation of tin into beta zeolite has presented numerous advantages over hydrothermal synthesis, e.g., higher tin loading, introduction of multiple active sites, and smaller beta particle size.18 PS tin beta zeolite has been used for many of the same heterogeneously catalyzed reactions as its hydrothermally synthesized analogue, including BVO,14,18,20–22 MPV reductions,14 Oppenauer oxidations (OPO),14 hydration of epoxides20 and glucose isomerization.19 In this work, we applied a PS tin beta zeolite/hydrogen peroxide oxidation system to acetophenone derivatives and dimeric lignin model compounds.
2. Results and discussion
2.1. Tin beta synthesis and characterization
Beta zeolite (Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al, 25:1) was first treated with 13 M nitric acid to afford dealuminated beta zeolite (De-Al beta) with a Si/Al ratio of >1800 (Table 1). The De-Al beta was treated with butyltin trichloride in toluene, followed by triethylamine to promote formation of Sn–O–Si bonds via hydrochloric acid elimination (procedure modified from Corma and coworkers).23 The catalyst was then calcined at 500 °C for 3 h to remove remaining ligands, yielding tin beta zeolite with a Si/Sn ratio of 12 (Sn-beta, 3.7 wt% Sn).
:Al, 25:1) was first treated with 13 M nitric acid to afford dealuminated beta zeolite (De-Al beta) with a Si/Al ratio of >1800 (Table 1). The De-Al beta was treated with butyltin trichloride in toluene, followed by triethylamine to promote formation of Sn–O–Si bonds via hydrochloric acid elimination (procedure modified from Corma and coworkers).23 The catalyst was then calcined at 500 °C for 3 h to remove remaining ligands, yielding tin beta zeolite with a Si/Sn ratio of 12 (Sn-beta, 3.7 wt% Sn).
| Zeolite | Si/Al ratio | Surface aread (m2 g−1) | Pore volumee (cm3 g−1) | NH3 ads. (μmol g−1) | ||
|---|---|---|---|---|---|---|
| Micro | Meso | Macro | ||||
| a Micropore (2 nm), mesopore (2 nm–50 nm) macropore (>50 nm).b Determined by ICP analysis.c Determined by PIXE.d Determined by BET method.e Micropore volume calculated from t-plot. | ||||||
| H-beta | 25 | 480 | 0.142 | 0.253 | 0.161 | — |
| De-Al beta | >1800b | 472 | 0.135 | 0.227 | — | ∼1 |
| Sn-beta | >1800b,c | 479 | 0.142 | 0.158 | — | 28.4 |
The surface area and micropore volumes measured at each step of tin beta zeolite synthesis (Table 1) showed little variation, indicating that removal of framework aluminum did not lead to collapse of the zeolite lattice. Ammonia temperature-programmed desorption (NH3-TPD) was performed to measure the relative acidity of De-Al beta zeolite and tin beta zeolite. De-Al beta zeolite adsorbed a minimal amount of ammonia, consistent with near complete removal of Al from the framework, while tin beta zeolite adsorbed 28.4 μmol of ammonia per gram of catalyst. Most of the ammonia desorbed below 525 K during TPD, indicating that tin incorporation resulted in weakly acidic sites. Pore-size distribution measurements on the tin beta zeolite showed that 55% of the total pore volume corresponded to the mesopore range. This is noteworthy given that the presence of mesopores is critical to the reaction of bulky lignin model compounds, and ultimately lignin itself.
The preservation of beta zeolite structure was verified via XRD (Fig. 1). Additionally, subtle changes in the d302 interlayer spacing confirmed that first dealumination had occurred, with subsequent inclusion of tin. Indeed, the interlayer spacing of H-beta zeolite was reduced from 3.864 Å (2θ = 22.54°) to 3.817 Å (2θ = 22.82°) upon dealumination but increased to 3.856 Å (2θ = 22.59°) after incorporation of tin, consistent with the findings of Tang et al.20 In addition to incorporation of tin into the beta zeolite framework, the presence of crystalline extra framework (EFW) tin was also observed (2θ = 26.7° and 34.0°).19 Application of the Scherrer equation indicated the average tin dioxide particle size to be 48 nm. EFW species are incorporated as a consequence of tin hydrolysis by adventitious water during grafting, or from calcination due to the high tin loading (3.7 wt%). The presence of water before calcination hydrolyzes Sn–Cl bonds, forming low-coordinate Sn–OH species; upon calcination, Sn–OH bonds in close spatial proximity dehydrate forming catalytically inactive SnO2 crystals as opposed to framework Sn–O–Si bonds. Dijkmans et al.21 found the maximum tin loading for the PS tin beta zeolite used in their report without formation of EFW species to be approximately 2%.
 | ||
| Fig. 1 X-ray diffractograms of tin dioxide, H-beta, De-Al-beta, and Sn-beta. | ||
Fig. 2 shows SEM images of De-Al beta and Sn-beta. SEM images of the dealuminated beta zeolite show highly textured particles characteristic of beta zeolite. Images of beta zeolite following tin incorporation show textured particles as well as a smaller (∼40 nm) secondary phase that is structurally different and which can be assigned to EFW tin dioxide.
 | ||
| Fig. 2 SEM images of De-Al beta zeolite (left) and Sn-beta (right). | ||
In an effort to evaluate tin speciation, 119Sn magic-angle-spinning (MAS) nuclear magnetic resonance (NMR) spectroscopy was employed. 119Sn MAS NMR spectroscopy has been reported to effectively distinguish between EFW and framework tin species based on coordination number. Removal of water aids in the elucidation of open tetrahedral, closed tetrahedral and octahedral tin species present in the catalyst. To remove adsorbed water, tin beta zeolite was heated at 200 °C overnight in vacuo prior to data collection. Spectra of both the hydrated and dehydrated samples were collected, effectively monitoring the dehydration process.
As shown in Fig. 3, 119Sn MAS NMR spectra for hydrated and dehydrated tin beta zeolite contain resonances for both octahedral and tetrahedral tin. The observed resonances in the spectrum of hydrated tin beta zeolite at −630 ppm to −720 ppm are assigned to the presence of hexa-coordinate hydrated tetrahedral tin species. Davis and coworkers19 observed similar resonances centered at −688 ppm and −700 ppm for hydrated species. Penta-coordinate hydrous tin species present in the spectrum of hydrated tin beta zeolite resonate at −579 ppm, and are absent after dehydration. In a recent report, Yakimov et al.24 observed penta-hydrated tin species at −581 ppm. Crystalline tin dioxide resonating at −606 ppm was also observed.
 | ||
| Fig. 3 Comparison of 119Sn MAS NMR spectra of tin dioxide, dehydrated tin beta zeolite, and hydrated tin beta zeolite. | ||
In the spectrum of the dehydrated tin beta zeolite (Fig. 3), a broad resonance at −437 ppm indicates the presence of both open and closed tetrahedral tin species, which were previously hydrated.20,25 Tetrahedrally coordinated tin species reported by Davis and coworkers19 were centered at −424 ppm and −443 ppm after dehydration, the authors assigning the downfield resonance to open tin sites (strongly acidic, SnOH(OSi)3) and the upfield resonance to closed tin sites (weakly acidic, Sn(OSi)4). Following dehydration, crystalline tin dioxide was still present as indicated by a resonance centered at −604 ppm corresponding to EFW tin (Fig. 3).
Diffuse reflectance UV-Vis spectroscopy is a sensitive technique that can distinguish between octahedral and tetrahedral Sn4+ to O2− ligand to metal charge transfer.20 A local intensity maximum at 200 nm suggests incorporation of tin into tetrahedral sites (ESI Fig. S1†), consistent with the finding of Davis et al.19 who reported an intensity maximum at 203 nm. However, a broad intensity maximum at 281 nm arises from the presence of hexa-coordinate polymeric tin (EFW tin), which largely dominates the spectrum. Consequently, the spectrum of tin beta zeolite is similar to that of bulk tin dioxide (maximum at 270 nm).
Tin speciation was also investigated by X-ray photoelectron spectroscopy (XPS). The spectrum of tin beta zeolite contained broad signals centered around 486.0 eV and 494.6 eV corresponding to tin 3d3/2 and 3d5/2 photoelectrons (Fig. 4). In comparison, the 3d3/2 and 3d5/2 photoelectrons of crystalline tin dioxide produced signal maxima at 487.0 eV and 495.5 eV, the signals being much narrower (FWHM = 1.6 eV) than the corresponding signals for dehydrated tin beta (FHWM = 3.7 eV). We propose that line broadening in the spectrum of dehydrated tin beta is due to the presence of overlapping signals from a mixture of octahedral and tetrahedral Sn4+ species, consistent with the data presented above.
 | ||
| Fig. 4 XPS spectra of tin beta zeolite and tin dioxide in the Sn 3d region. | ||
In summary, characterization of the synthesized tin beta zeolite confirmed the presence of tetrahedral Sn4+ species. Given that Lewis acid sites are believed to be responsible for Baeyer–Villiger catalysis,14 such low coordinate Sn species should function as active sites. These correspond to the “open” and “closed” tetrahedral tin sites observed by 119Sn MAS NMR spectroscopy, although other coordinatively unsaturated amorphous tin species on the catalyst surface could also contribute to catalysis.
2.2. BVO of simple cyclic and acyclic ketones
In nearly all cases, literature reports of heterogeneous BVO concern cyclic ketones. Thus, in order to compare the catalyst in this work with those reported in the literature, 2-adamantanone was oxidized using the tin beta zeolite/H2O2 system (Scheme 5). 2-Adamantanone is less susceptible to the hydroxylation side-reactions discussed below, which makes 2-adamantanone a valuable probe molecule to aid in discerning reactivity trends. After 24 h at 80 °C the corresponding lactone was obtained in 83% yield (>99% selectivity), comparable to previous reports for tin beta zeolite/H2O2.18,22 | ||
| Scheme 5 BVO of 2-adamantanone. | ||
The tin beta zeolite/hydrogen peroxide oxidation system was then used to investigate BVO of acetophenone 1 (Table 2). Low yields of phenyl acetate (1a, 33%) were observed as a result of poor electron donation to the carbonyl oxygen, slowing nucleophilic addition of hydrogen peroxide.26,27 Alkyl migration from the Criegee intermediate is considered to be the rate-limiting step, however, when using weak oxidants such as hydrogen peroxide, the rate-limiting step can become nucleophilic addition.28 While this result is disappointing, angiosperm and gymnosperm lignins are the result of coniferyl (G) and sinapyl (S) alcohol polymerization, these monolignols containing one and two methoxy group(s), respectively. Therefore, addition of an electron-donating group (EDG) was investigated to determine if it positively affects catalyst activity.26,27 Indeed, when EDGs were present (i.e., methyl and methoxy groups, corresponding to 2 and 3, respectively), a higher yield of the corresponding phenyl acetate (2a, 3a) was observed. Due to an EDG influenced increase in the basicity of the carbonyl oxygen,27 3 was converted in 81% yield to 3a at 45 °C after 24 h. When the temperature was increased to 80 °C selectivity decreased from 90% to 83%, and a notable darkening of the reaction mixture was observed, implying formation of phenolic resins. To demonstrate facile cleavage of the ester, after BVO 4′-methoxyphenyl acetate (3a) was hydrolyzed to 4-methoxyphenol (9b, 98% yield) using potassium carbonate and methanol at room temperature for 30 min.29
| Model number | Model | Conv. (%) | Selectivity (%) | Yield (%) | Product number | Product |
|---|---|---|---|---|---|---|
| a All reactions were performed in 1,2-dichloroethane at 80 °C for 24 h unless otherwise noted.b Reaction was conducted at 45 °C. Control reactions were run with crystalline tin oxide (2% conversion) and dealuminated beta zeolite (1% conversion), yielding only trace amounts of 4′-methoxyphenyl acetate, 3a, at 80 °C. Mequinol, 9b, was observed in 9% yield in the reaction of model 3. Phenol was observed in 23% yield in the reaction of model 1. | ||||||
| 1 | 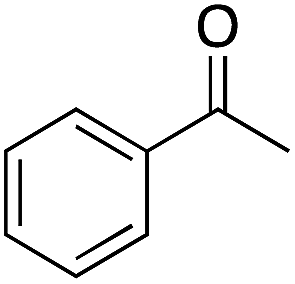 |
54 | 41 | 33 | 1a |  |
| 2 | 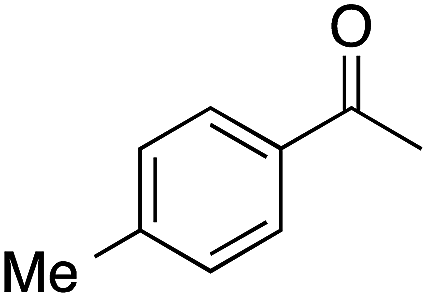 |
99 | 60 | 59 | 2a |  |
| 3b |  |
90 | 90 | 81 | 3a |  |
| 4 |  |
58 | 47 | 27 | 4a |  |
| 5 | 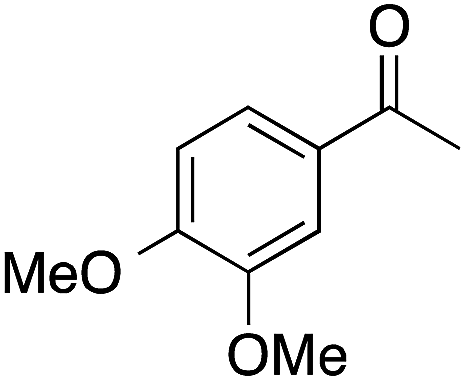 |
91 | 64 | 58 | 5a | 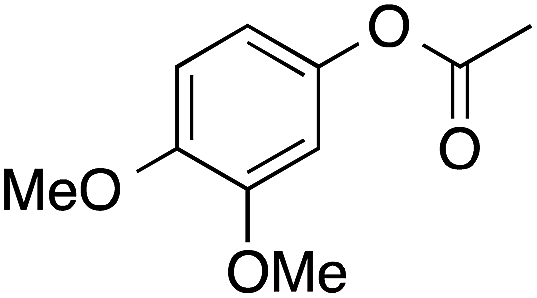 |
Considering that 4 contains an EDG in the ortho-position, surprisingly low yields of 4a were observed (Table 2). This could be due to hindrance of the ortho-substituent to both coordination of 4 to the catalyst surface, and alkyl migration. A similar effect was observed for 5, which contains both meta and para-methoxy groups, and which gave 5a in 58% yield. In reactions with poor selectivity, such as the BVO of 1 and 4, water-soluble resins were also obtained. Likely the product of direct hydroxylation (discussed below) or phenolic radical coupling following ester hydrolysis, resin formation was much more prevalent at 80 °C than 45 °C (as in the case of model 3), presumably due to the increased rate of hydrogen peroxide homolysis. Additionally, mequinol was detected in 9% yield from hydrolysis of model 3a at 45 °C (Table 2). Hydrolysis could have occurred during the reaction, during reaction work up, or in the gas chromatograph inlet.
Given the high yield of 3a, compound 3 was selected for a catalyst reusability study (Fig. 5). After each sequential cycle the catalyst was thermally regenerated at 500 °C in air for 3 h. Minimal loss in activity (3% yield) was observed after the 3rd catalytic cycle.
 | ||
| Fig. 5 Results of catalyst reusability study. Cycles one and two were run in duplicate and yields are reported as averages of the two runs. Cycle three is reported as a single run due to limited catalyst availability. | ||
Using 3 as a probe molecule, solvent effects in the tin beta zeolite/H2O2 oxidation system were explored at 45 °C (Table 3). Initially, water-miscible solvents were evaluated (e.g., ethanol), however they generally afforded modest yields. Acetonitrile was also tested, since in the presence of H2O2 it forms peroxyimidic acid, a more powerful oxidant/nucleophile than H2O2. However, results were disappointing, a 24% yield of 4-methoxyphenol being obtained from ester hydrolysis. Other solvents (e.g., toluene) were also determined to be compatible with this oxidation system. They were not considered further in this study due to the low solubility of lignin in non-polar solvents, but provide potential for other heterogeneous BVO applications.
| Solvent | Conversion (%) | Selectivity (%) | Yield 3a (%) |
|---|---|---|---|
| a Selectivity and yields are expressed in terms of 4′-methoxyphenyl acetate, 3a. Reactions were conducted at 45 °C unless otherwise noted.b In the case of BVO with acetonitrile, ester hydrolysis resulted in 24% yield of 4-methoxyphenol, 9b.c Reaction conducted at 80 °C.d 9b was observed in 31% yield. | |||
| 1,4-Dioxane | 48 | 95 | 45 |
| Acetonitrileb | 40 | 3 | 1 |
| Toluene | 72 | 95 | 68 |
| Ethanol | 55 | >99 | 55 |
| Ethanolc,d | 99 | 49 | 49 |
| 1,2-Dichloroethane | 90 | 90 | 81 |
| 1,2-Dichloroethanec | 99 | 83 | 83 |
Of the solvents evaluated, 1,2-dichloroethane (which is water immiscible) gave the highest yield of phenyl acetate. The resistance to oxidation and immiscibility of chlorinated solvents has previously been reported to yield high conversions in BVO reactions as demonstrated with chlorobenzene.18 The use of a biphasic system limits the solubility of hydrogen peroxide in the organic solvent, decreasing the prevalence of side reactions such as direct ring hydroxylation and ester hydrolysis, which occur readily at temperatures above 45 °C. In BVO, hydrogen peroxide acts as a two-electron oxidant, performing nucleophilic addition to form the Criegee intermediate. However, as the temperature is increased hydrogen peroxide decomposition is accelerated. Homolysis of the peroxide bond forms hydroxyl radicals that are very active one-electron oxidants. Hydroxyl radicals can perform direct ring hydroxylation as well as many other side-reactions.26 In competition with BVO, direct ring-hydroxylation reactions can involve both starting material and BVO products. Without the presence of an organic layer (i.e., using a single aqueous phase at 80 °C), reactions resulted in the production of a dark insoluble phenolic resin or tar, as has been reported for similar reactions (i.e., hydrogen peroxide mediated oxidation of benzene to phenol).6 Interestingly, at low temperatures the highest selectivity was obtained when ethanol was the solvent. However when the reaction temperature was increased, lower selectivity and an increased yield of unidentifiable products was observed. In the case of ethanol at 80 °C, mequinol (9b) was obtained in 31% yield, whereas 9b was not observed when the solvent was DCE, consistent with decreased ester hydrolysis in the biphasic system.
2.3. BVO of lignin model compounds
Using the tin beta zeolite/hydrogen peroxide oxidation system, selected lignin dimer model compounds, similar to the retro aldol products of α-position oxidation observed by Wang et al.,5 were oxidized in good to moderate yields. In the reaction of these lignin model compounds, increased amounts of hydrogen peroxide were used due to their lower reactivity as compared to acetophenone derivatives, which can be attributed to their increased steric bulk. Ketone compounds representing the product of benzylic alcohol oxidation in β-O-4 and β-1 linkages were converted to their respective BVO products as shown in Table 4.| Model Number | Model | Conv. (%) | Selectivity (%) | Yield (%) | Product number | Product |
|---|---|---|---|---|---|---|
| a Product yields were determined by 1H-NMR spectroscopy using an internal standard. Mequinol, 9b was observed in 3% and 2% yields in the reaction of models 6 and 7, respectively.b Isolated product yield. | ||||||
| 6 |  |
86 | 37 | 32 | 6a |  |
| 7 | 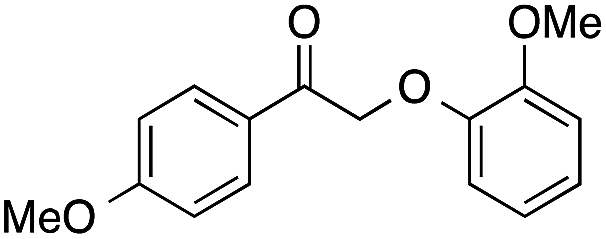 |
81 | 26 | 21 | 7a |  |
| 8 |  |
41 | 22 | 9 | 8a |  |
| 9 |  |
94 | 23 | 22b, 15 | 9a, 9b |  |
Small amounts of the corresponding phenol were detected in the reactions of 6 and 7, likely resulting from hydrolysis of the ester during aqueous workup or thermal cleavage upon injection during gas chromatography. In the case of compounds 6–8, no 4-methoxybenzoic acid resulting from alkyl migration was present.
The crystal structure of the BVO product of lignin dimer 6 is shown below (Fig. 6a). The crystal structure confirms that the product, 6a, is the result of aryl migration. 6a is over twice the length (15.3 Å) of the pore openings in zeolite beta (7 Å), inhibiting its diffusion through the micropore system. Although in some confirmations (via rotation about the C8–C9 bond) 6a may be able to enter the pore system, conformational requirements of the reaction intermediates (specifically the Criegee intermediate) make the occurrence of BVO in micropores unlikely. Consequently, catalysis likely takes place at tin centers located in mesopores. The crystal structure of BVO product 7a was also determined (Fig. 6b; see also Tables S1–S9 in the ESI† for additional details of the structures of 6a and 7a).
 | ||
| Fig. 6 (a) Crystal structure of the Baeyer–Villiger oxidation product 6a of 2-(4-methoxyphenoxy)-1-(4-methoxyphenyl)-ethanone (6). (b) Crystal structure of the Baeyer–Villiger oxidation product 7a of 2-(4-methoxyphenoxy)-1-(4-methoxyphenyl)-ethanone (7). Crystals of 7a were twinned by non-merohedry, and there were two molecules in the asymmetric unit. For the sake of clarity, only one of the independent molecules is shown. | ||
The occurrence of selective aryl migration is contrary to the findings of Patil and coworkers9 who found that alkyl migration was preferred over aryl migration for all lignin model compounds in their homogeneous oxidation system. In our work the p-methoxybenzene migrating group was presumably better able to stabilize positive charge accumulation in the Criegee intermediate, consistent with DFT studies of acetophenones.27
Regioselective aryl migration was observed for all cases excluding BVO of anisoin 9, where mixed selectivity was observed. Anisoin, 9, was converted almost quantitatively with 7 eq. of hydrogen peroxide at 80 °C to products consistent with BVO, the reaction resulting primarily in alkyl migration. The observed change in selectivity is a result of polarization of the Cβ-OH group that stabilizes the partial positive charge during Cβ alkyl migration in the Criegee intermediate. Hemiacetal decomposition followed by aldehyde oxidation after BVO resulted in 4′-methoxybenzoic acid, 9a (22%). Mequinol, 9b, was also detected (15%), resulting from aryl migration.
Patil et al.9 first reported homogenous BVO of lignin model dimer compounds under a formic acid/hydrogen peroxide environment, achieving a maximum yield of 10% of the BVO product of alkyl migration (Scheme 4). In the same report, using a more complex model they reported a 78% yield of benzoic acid from cleavage of the ester but observed polymerization of the phenolic co-product. In this work we obtained ester yields of respectively 32% and 21% in the BVO of β-O-4 model compounds 6 and 7 using tin beta zeolite/H2O2, which can be cleaved in a second step to recover phenolic moieties. Furthermore, β-1 model compounds were also successfully oxidized. Moreover, the heterogeneous nature of this catalytic system makes this reaction more industrially applicable due to generation of water as a co-product and separation of the catalyst by filtration.
On the other hand, the effects of direct hydroxylation of the aromatic groups are reflected in the moderate yields. Ester yields were found to be affected by reaction time, temperature and hydrogen peroxide concentration. Low yields can be caused by hydrolysis of BVO products to form phenols that are susceptible to multiple hydroxylations, as well as H2O2 homolysis, which limits the conversion of the ketone reactant. In an effort to improve product yields, staging of the H2O2 addition was investigated. However, the addition of aliquots of H2O2 over a three-day period (14 eq. initially and 7 eq. after 24 h) led to mass balances as low as 14%, due to formation of insoluble phenolic resins.
3. Experimental methods
3.1. Catalyst characterization
X-ray diffractograms were collected on a Phillips PW 3040 X-ray diffractometer using Cu Kα radiation (λ = 1.54184 Å) and a step size of 0.02°. X-ray diffractograms were referenced to the International Centre for Diffractogram Data (ICDD) database. Elemental concentrations were determined by Proton-Induced X-ray Emission (PIXE) (Elemental Analysis Inc., Lexington, KY).Room temperature solid state NMR spectra were acquired using a Tecmag Redstone spectrometer (Tecmag, Inc., Houston, TX) operating at 111.917 MHz for 119Sn (7.05 T static magnetic field). Samples were packed into 7.5 mm zirconia rotors and sealed with Teflon or Kel-F end caps (Revolution NMR, LLC, Fort Collins, CO). Experiments were performed using a 7.5 mm double resonance MAS probe (Varian, Palo Alto, CA). All 119Sn spectra were acquired under MAS at 5 kHz at ambient conditions. 119Sn chemical shifts are reported relative to samarium stannate at −102.6 ppm with an accuracy of ±0.4 ppm. 119Sn spectra were collected with a single pulse on 119Sn followed by acquisition. All spectra were acquired with a 50 second recycle delay and a 2.56 ms acquisition time. Dehydrated samples were heated to 473 K in vacuo overnight, prior to data collection.
Scanning electron microscopy (SEM) was performed on a Hitachi S-2700 microscope equipped with a LaB6 gun and a PGT EDS analyzer with thin window detector. Samples were gold-coated prior to imaging. Brunauer–Emmett–Teller (BET) surface area and micropore volume measurements were determined by nitrogen adsorption at 77 K using a Micromeritics Gemini VII analyzer. Prior to measurement, samples were degassed at 423 K under nitrogen. X-ray Photoelectron Spectroscopy (XPS) was performed on a Kratos Axis HSi X-ray photoelectron spectrometer equipped with a charge neutralizer and magnetic focusing lens, using a monochromated Mg Kα X-ray source (hν = 1253.6 eV). Spectra were referenced to the adventitious C 1s peak at 284.6 eV. Prior to analysis, XPS samples were dried under high vacuum at 303 K and then transferred to an in situ stage where they were dried at 673 K in vacuo for 1 h. UV-Vis Diffuse Reflectance spectra were collected on a Varian Cary 5000 spectrometer using barium sulfate as a reference. NH3-TPD experiments were performed on a Micromeritics AutoChem II analyzer using 0.500 g of sample. In each case the sample was first dehydrated at 673 K under argon for 1 h, cooled to 363 K and saturated with NH3 (1% in helium, 50 sccm) for 1 h. Next, the sample was purged with He (120 sccm) for 1 h. The sample was then heated to 1023 K at 10 K min−1. Effluent gas was analyzed using a mass spectrometer (Pfeiffer Thermostar GSD301), the signal at m/z = 15 being used to monitor NH3.
Single crystal X-ray diffraction data were collected at 90.0(2) K on a Bruker-Nonius X8 Proteum diffractometer with graded-multilayer focused Cu Kα X-rays. Raw data were integrated, scaled, merged and corrected for Lorentz-polarization effects using the APEX2 package.30 Corrections for absorption were applied using SADABS.31 The structure was solved by direct methods (SHELXT)32 and refined against F2 by weighted full-matrix least-squares (SHELXL-2014).33 Hydrogen atoms were found in difference maps but subsequently placed at calculated positions and refined using a riding model. Non-hydrogen atoms were refined with anisotropic displacement parameters. The final structure model was checked using an R-tensor34 and by Platon/checkCIF.35 Atomic scattering factors were taken from the International Tables for Crystallography.36 Crystal structure data are deposited at the Cambridge Crystallographic Data Center (CCDC deposition numbers 1541264 and 1541265).†
3.2. Synthesis of grafted tin beta zeolite (Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :Al ratio = 25:1)
:Al ratio = 25:1)
Commercial H-beta-zeolite (25 g), obtained from Clariant® (Si:Al ratio = 25:1), was treated with 13 M nitric acid and heated at 353 K overnight (solid/liquid ratio = 1 g/20 mL). The mixture was filtered and washed with deionized water until neutral. This dealumination procedure was repeated a second time. Two batches were mixed and dried in vacuo overnight at 343 K, yielding De-Al beta (42.8 g). De-Al beta was added to a solution of butyltin trichloride (7.6 mL, 45.6 mmol) in anhydrous toluene (150 mL) under nitrogen and stirred at room temperature for 1 h. The solution was neutralized by addition of triethylamine (17.6 mL, 126.2 mmol) and stirred for 1 h. The suspension was then filtered, washed with toluene (500 mL), and dried overnight in a vacuum oven. The catalyst was heated in air to 623 K for 30 min followed by calcination at 773 K for 3 h, yielding 37.3 g of Sn-beta zeolite.
3.3. Synthesis of lignin model compounds
Model compounds 6 (ref. 37) and 7 (ref. 38) were synthesized according to literature procedures which are included in the ESI.†3.4. Procedure for Baeyer–Villiger oxidations
All reagents were purchased from Fisher Scientific or Sigma Aldrich and were used without further purification, unless stated otherwise. In a typical reaction, tin beta zeolite (0.150 g), 4-methoxyacetophenone, 3 (0.451 g, 3 mmol), 1,2-dichloroethane (3 g) and 30% hydrogen peroxide (2 mL, 20 mmol) were added to a 50 mL round-bottomed flask equipped with a jacketed water-cooled condenser, and heated at the desired temperature with stirring for 24 h. After completion of the reaction, the suspension was filtered through a Nylon® membrane (0.45 μm pore size). The catalyst was rinsed with dichloroethane (5 mL × 2) and water (5 mL × 2). The biphasic mixture was separated and the aqueous layer was extracted with dichloroethane (2 mL × 3) and dried over magnesium sulfate. In the case of reusability experiments, the catalyst was calcined at 500 °C for 3 h and allowed to cool to room temperature prior to being reused. For the hydrolysis of 3a, potassium carbonate (0.455 g, 3.3 mmol) in methanol (3 g) was added to the reaction mixture. The mixture was stirred for 30 min at room temperature and was then neutralized with hydrochloric acid to afford 4-methoxyphenol 9b (0.307 g, 98% yield).3.5. Determination of phenyl acetate yields using gas chromatography mass spectrometry (GCMS)
GCMS analyses were performed using an Agilent 7890 GC with a tandem Agilent 5975C MS detector. The column used was a DB-1701 (60 m × 0.25 mm × 0.25 μm) and the temperature program was as follows: 45 °C for 3 min, ramp to 280 °C at 4 °C min−1, and hold for 10 min. The flow rate was set to 1 mL min−1 using helium as the carrier gas. The inlet was maintained at 260 °C, and the MS source was set at 70 eV. An external calibration curve of anisole or dodecane was used to calibrate BVO products in the reaction mixture. Analysis of lignin model dimer compounds proved difficult (due to the low response factors observed for oxidized dimer model compounds). In lieu of calibrated GCMS yields, 1H-NMR spectroscopy with a ferrocene internal standard was used to determine product yields.4. Conclusions
The tin beta zeolite/H2O2 oxidation system was applied to 2-adamantanone, several acetophenone derivatives and oxidized lignin β-O-4 and β-1 linkage models. Selective aryl migration was observed in all cases excluding anisoin, where both aryl and alkyl migration were observed. The oxidation system presented in this work yields esters that can be cleaved in a simple hydrolysis reaction, yielding phenolic moieties that are typically difficult to isolate from β-O-4 oxidation reactions. Yields of ester products derived from β-O-4 and β-1 lignin models were generally modest due to the formation of polymeric material stemming from direct ring hydroxyl. While preventing the formation of byproducts is challenging, if the reaction were run at low conversion (shorter residence time and/or lower temperature) then the selectivity should increase due to decreased ring hydroxylation and resin formation. Naturally, this would require a means for separating the products and starting material, so that the latter could be recycled. To our knowledge, this is the first report of heterogeneous BVO of lignin model dimer compounds.Acknowledgements
This material is based on work supported by the National Science Foundation under Cooperative Agreement No. 1355438. Funding from the British Council under the Global Innovation Initiative for the GB3-Net project is also gratefully acknowledged. The single-crystal diffractometer was funded by the NSF (MRI CHE-0319176). Diffuse reflectance UV-Vis spectra were measured at the Center for Nanophase Materials Sciences, which is a DOE Office of Science User Facility. The authors would also like to thank Shelley Hopps, Dr Mark Meier, Dr Justin Mobley, Yang Song, Gerald Thomas, and Soledad Yao for their help in the preparation of this manuscript.References
- A. Barakat, A. Bagniewska-Zadworna, C. J. Frost and J. E. Carlson, BMC Plant Biol., 2010, 10, 1–11 CrossRef PubMed.
- J. Zakzeski, A. L. Jongerius, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemSusChem, 2012, 5, 1602–1609 CrossRef CAS PubMed.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS PubMed.
- A. Wu, B. O. Patrick, E. Chungand and B. R. James, Dalton Trans., 2012, 41, 11093–11106 RSC.
- M. Wang, J. Lu, X. Zhang, L. Li, H. Li, N. Luo and F. Wang, ACS Catal., 2016, 6, 6086–6090 CrossRef CAS.
- L. Balducci, D. Bianchi, R. Bortolo, R. D'Aloisio, M. Ricci and R. Tassinari, Angew. Chem., 2003, 115, 5087–5090 CrossRef.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., 2015, 127, 260–264 CrossRef.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- N. D. Patil, S. G. Yao, M. S. Meier, J. K. Mobley and M. Crocker, Org. Biomol. Chem., 2015, 13, 3243–3254 CAS.
- J. Mottweiler, M. Puche, C. Räuber, T. Schmidt, P. Concepción, A. Corma and C. Bolm, ChemSusChem, 2015, 8, 2106 CrossRef CAS PubMed.
- Y. Gao, J. Zhang, X. Chen, D. Ma and N. Yan, ChemPlusChem, 2014, 79, 825 CrossRef CAS.
- M. R. Sturgeon, M. H. O'Brien, P. N. Ciesielski, R. Katahira, J. S. Kruger, S. C. Chmely, J. Hamlin, K. Lawrence, G. B. Hunsinger, T. D. Foust, R. M. Baldwin, M. J. Biddy and G. T. Beckham, Green Chem., 2014, 16, 824 RSC.
- J. S. Kruger, N. S. Cleveland, S. Zhang, R. Katahira, B. A. Black, G. M. Chupka, T. Lammens, P. G. Hamilton, M. J. Biddy and G. T. Beckham, ACS Catal., 2016, 6, 1316 CrossRef CAS.
- J. Dijkmans, W. Schutyser, M. Dusselier and B. Sels, Chem. Commun., 2016, 52, 6712–6715 RSC.
- A. Corma, V. Fornés, S. Iborra, M. A. Mifsud and M. Renz, J. Catal., 2004, 221, 67–76 CrossRef CAS.
- S. P. Panchgalle, U. R. Kalkote, P. S. Niphadkar, P. N. Joshi, S. P. Chavan and G. M. Chaphekar, Green Chem., 2004, 6, 308–309 RSC.
- A. Corma, M. E. Domine, L. Nemeth and S. Valencia, J. Am. Chem. Soc., 2002, 124, 3194–3195 CrossRef CAS PubMed.
- P. Li, G. Liu, H. Wu, Y. Liu, J.-G. Jiang and P. Wu, J. Phys. Chem. C, 2011, 115, 3663–3670 CAS.
- R. Bermejo-Deval, R. Gounder and M. E. Davis, ACS Catal., 2012, 2, 2705–2713 CrossRef CAS.
- B. Tang, W. Dai, G. Wu, N. Guan, L. Li and M. Hunger, ACS Catal., 2014, 4, 2801–2810 CrossRef CAS.
- J. Dijkmans, J. Demol, K. Houthoofd, S. Huang, Y. Pontikes and B. Sels, J. Catal., 2015, 330, 545–557 CrossRef CAS.
- J. Jin, X. Ye, Y. Li, Y. Wang, L. Li, J. Gu, W. Zhao and J. Shi, Dalton Trans., 2014, 43, 8196–8204 RSC.
- A. Corma, M. T. Navarro and M. Renz, J. Catal., 2003, 219, 242–246 CrossRef CAS.
- A. V. Yakimov, Y. G. Kolyagin, S. Tolborg, P. N. Vennestrøm and I. I. Ivanova, J. Phys. Chem. C, 2016, 120, 28083–28092 CAS.
- R. Bermejo-Deval, R. S. Assary, E. Nikolla, M. Moliner, Y. Román-Leshkov, S.-J. Hwang, A. Palsdottir, D. Silverman, R. F. Lobo and L. A. Curtiss, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9727–9732 CrossRef CAS PubMed.
- C. Gambarotti and J. R. Bjørsvik, J. Phys. Org. Chem., 2015, 28, 619–628 CrossRef CAS.
- L. Reyes, J. R. Alvarez-Idaboy and N. Mora-Diez, J. Phys. Org. Chem., 2009, 22, 643–649 CrossRef CAS.
- M. Renz and B. Meunier, Eur. J. Org. Chem., 1999, 737–750 CrossRef CAS.
- G. Buechi and S. M. Weinreb, J. Am. Chem. Soc., 1971, 93, 746–752 CrossRef CAS.
- Bruker, APEX2, Bruker-AXS, Madison WI, USA, 1992 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- S. Parkin, Acta Crystallogr., Sect. A: Found. Crystallogr., 2000, 56, 157–162 CrossRef.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- A. J. C. Wilson, International Tables for Crystallography: Mathematical, physical, and chemical tables, International Union of Crystallography, 1992, vol. 3, pp. 476–511 Search PubMed.
- J. H. Lee, M. Kim and I. Kim, J. Org. Chem., 2014, 79, 6153–6163 CrossRef CAS PubMed.
- B. Landers, C. Berini, C. Wang and O. Navarro, J. Org. Chem., 2011, 76, 1390–1397 CrossRef CAS PubMed.
- http://www.sigmaaldrich.com/spectra/fnmr/FNMR010821.PDF.
- http://www.sigmaaldrich.com/spectra/fnmr/FNMR000488.PDF.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1541264 and 1541265. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra03830e |
| This journal is © The Royal Society of Chemistry 2017 |