
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and in vivo proof of concept of a BODIPY-based fluorescent probe as a tracer for biodistribution studies of a new anti-Chagas agent
Gonzalo Rodríguezab,
Javier Nargolia,
Andrés Lópezc,
Guillermo Moynac,
Guzmán Álvareza,
Marcelo Fernándezd,
Carlos A. Osorio-Martínez†
ab,
Mercedes González*a and
Hugo Cerecetto*ab
aGrupo de Química Medicinal, Laboratorio de Química Orgánica, Instituto de Química Biológica, Facultad de Ciencias, Universidad de la República, 11400 Montevideo, Uruguay. E-mail: megonzal@fq.edu.uy; hcerecetto@cin.edu.uy; Tel: +598 25250800
bÁrea de Radiofarmacia, Centro de Investigaciones Nucleares, Facultad de Ciencias, Universidad de la República, Mataojo 2055, 11400 Montevideo, Uruguay
cDepartamento de Química del Litoral, Universidad de la República, CENUR Litoral Norte, Ruta 3 km 363, Paysandú 60000, Uruguay
dLaboratorio de Experimentación Animal, Centro de Investigaciones Nucleares, Facultad de Ciencias, Universidad de la República, Uruguay
First published on 24th January 2017
Abstract
The potential use of amide-containing thiazoles, especially (2E,2Z)-3-allyl-4-[((E)-4-cinnamylpiperazin-1-yl)carbonyl]-2-[2-((E)-3-(furan-2-yl)propenylidene)hydrazono]-2,3-dihydrothiazole (1), as drugs for the treatment of Chagas disease has been recently described. The therapeutic application of 1 requires further pre-clinical studies, including in vivo biodistribution. In this sense, a BODIPY-fluorophore based probe for this drug (1-BODIPY) was developed and investigated for its potential as an in vivo tracer. The fluorescent tracer was synthesized, physicochemically and in vitro biologically characterised, and its in vivo biodistribution evaluated. The in vitro studies demonstrated that the fluorescent probe could simulate the in vivo behaviour of compound 1. Furthermore, the in vivo proof of concept showed that the 1-BODIPY biodistribution involves organs that are associated with the parasitic disease. These findings allow us to establish future administration routes and regimens in the treatment of Chagas disease with 1.
1. Introduction
Chagas disease or American trypanosomiasis, caused by the parasite Trypanosoma cruzi, is a widespread illness that affects nearly 8 million people worldwide.1 The parasite reaches the secondary lymphoid organs, the heart, skeletal muscles, neurons in the intestine, esophagus, and other tissues.2 The disease is characterized by mega syndromes, which may affect the esophagus, the colon and the heart, in about 30% of infected people. The current chemotherapy is restricted to nifurtimox and benznidazole, two drugs that were developed more than 40 years ago, which have limited efficacy and a high occurrence of adverse effects.3 Therefore, there is a considerable need for new compounds able to become drugs for Chagas disease. In this sense, we have made multiple efforts identifying new lead compounds with potential activity against T. cruzi,4 finding one of the most successful anti-Chagas chemotypes, the 2,3-dihydrothiazole system.5 Especially, the thiazole (2E,2Z)-3-allyl-4-[((E)-4-cinnamylpiperazin-1-yl)carbonyl]-2-[2-((E)-3-(furan-2-yl)propenylidene)hydrazono]-2,3-dihydrothiazole (1, Fig. 1) is one of the most effective in vivo anti-T. cruzi agent belonging to this chemotype. This compound showed excellent in vivo anti-T. cruzi behaviour with concomitant safety to be used as drug, i.e. absence of mutagenic and clastogenic effects,5b suggesting that it is surpassing the lead optimization stage in drug development and leading to a candidate for preclinical studies. | ||
| Fig. 1 Anti-Chagas agent 1 and the proposed structural modification to prepare in vivo fluorescent-imaging probe. | ||
Being Chagas disease an organ-specific illness, the biodistribution profile of new drug candidates is a relevant issue in preclinical studies. As part of our ongoing studies in the preclinical behaviour of thiazole 1, we decided to study its in vivo biodistribution using a fluorescent probe analogue.6 Considering that compound 1 was the result of a series of structural optimization efforts,5a,b where the chemotype furanylpropenylidenehydrazonothiazole (“west” of the structure, Fig. 1) was identified as the potential anti-T. cruzi pharmacophore, we proposed probes modified with a fluorophore on the “eastern” region of the structure (Fig. 1).
The fluorophore incorporated into the analogue should minimally alter the stereoelectronic properties in order to retain the bio- and physicochemical behaviour of thiazole 1. In this sense, we selected the 4,4-difluoro-4-bora-3a,4a-diaza-s-indacenyl framework (known as BODIPY) among the different described fluorophores due to its excellent photochemical characteristics and known chemistry.7,8 Additionally, compared to other fluorophores (for example those in the ALEXA series), BODIPY has a hydrophilic/lipophilic balance9 and a structural size that when it substitutes the phenylpropenyl group (Fig. 1) the generated probe could simulate physicochemical and biological properties of 1.
Here we report on the synthesis of a furanylpropenylidenehydrazonothiazole bearing a BODIPY moiety with the intent of developing a probe for the biodistribution studies of thiazole 1. Physicochemical, photophysical, and biological in vitro and in vivo properties of the labelled-compound were studied.
2. Results and discussion
2.1 Design of the probes
In order to minimize the structural perturbations on the “eastern” region of compound 1, we proposed two different linkers. On the one hand, an amide connection between the piperidinyl and 8-phenyl-BODIPY moieties was planned (Scheme 1). On the other hand, a methylene linkage between the piperidinyl and 1,3,5,7-tetramethyl-BODIPY moieties was suggested (Scheme 2).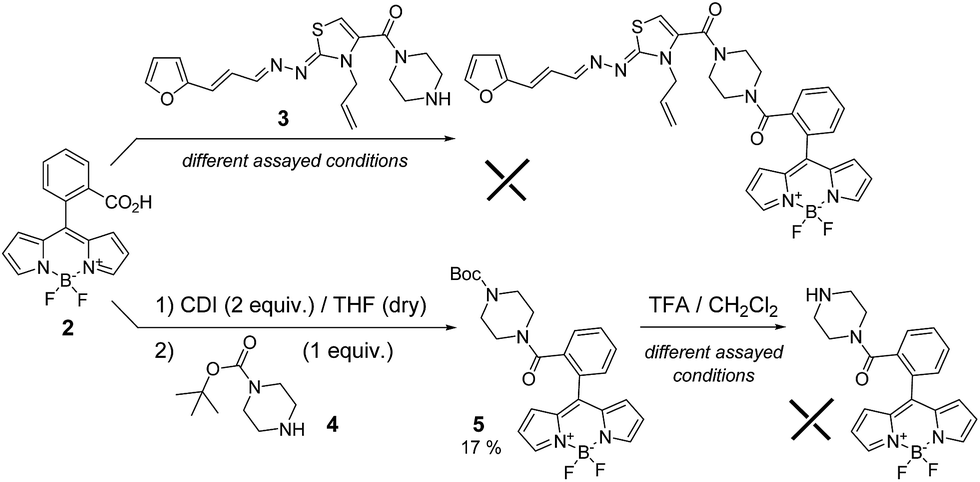 | ||
| Scheme 1 Attempts to obtain the probe with amide linker. | ||
 | ||
| Scheme 2 Attempt and synthesis of probe 1-BODIPY. | ||
2.2 Synthesis of the 1-BODIPY probe
The maximum of emission of the unsubstituted BODIPY fluorophore is near 500 nm, but modifying different BODIPY positions one could modulate this maximum. For this reason, we used the meso-substituted 8-phenyl-BODIPY derivative functionalised with an acid group (2)10 to perform the amidation process with furanylpropenylidenehydrazonothiazole derivative 3 (Scheme 1). Different coupling conditions were assayed unsuccessfully, most likely as result of the steric hindrance. For that reason, we tried the same reaction with the less bulky amine 4 (Scheme 1). In this case, derivative 5 was generated in low yield (17%). However, subsequent deprotection in presence of different concentrations of trifluoroacetic acid (TFA) and different time periods led in all cases to the complete decomposition of the starting material (Scheme 1).In view of these negative results, we selected an alternative synthetic route involving an SN2 reaction on the 8-chloromethyl-BODIPY derivative 6 (Scheme 2),11 which shifts the emission maximum as result of the tetramethyl-substitution. Firstly, we assayed the SN2 reaction with amine 4 (Scheme 2) as a model, generating the desired product 7 with adequate yield (34%). However, as was the case of amide 5, deprotection with TFA led to complete decomposition of the starting material. The low stability of the BF2 system present in BODIPY under acidic conditions was recently described.12 To avoid this, we performed the SN2 directly with furan intermediate 8 (Scheme 2).5b Under these conditions we were able to obtain the desired probe 1-BODIPY (Scheme 2) in moderate yield (24%), and its structure was unambiguously confirmed by multinuclear NMR spectroscopy and mass spectrometry.
2.3 Physicochemical and biological in vitro properties of 1-BODIPY probe
We studied the photophysical properties of 1-BODIPY in different solvents covering a range of polarities. Although the fluorescence was dependent on solvent polarity (Fig. 2), the analogue had a maximum in the fluorescence emission at 530 nm and displayed negligible changes in the emission properties in the different solvents studied. Additionally, the quantum yield in DMSO compares favourably with that of other BODIPY probes (φ = 0.32 with respect to 1,3,5,7,8-pentamethyl-BODIPY). | ||
| Fig. 2 (a) Absorbance spectra of 1 and 1-BODIPY in EtOH; (b) normalized fluorescence excitation and emission spectra of 1-BODIPY in different solvents. λexcitation 460 nm; λemission 550 nm; [1-BODIPY]= 2.0 × 10−5 M; T = 298 K. Inset shows the appearance of solutions under ambient light (left) and under a hand-held 365 nm UV lamp (right); (c) fluorescence excitation and emission spectra (non-normalized) of 1-BODIPY in different solvents. | ||
In order to validate the in vivo behaviour of 1-BODIPY, we performed in vitro studies related to its bioactivity. In this sense, we determined the lipophilicity parameter log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P at physiological pH 7.4, also known as the distribution coefficient (logD7.4). Both molecules were hydrophobic with similar positive logD7.4 (Table 1). Consequently, we expected that the biological behaviour of 1 and 1-BODIPY would be similar. To confirm this hypothesis, we evaluated the in vitro bioactivity of the probe 1-BODIPY against epimastigote forms of T. cruzi. 1-BODIPY displayed an IC50 at the micromolar level comparable to that of the parent compound 1 (Table 1).
P at physiological pH 7.4, also known as the distribution coefficient (logD7.4). Both molecules were hydrophobic with similar positive logD7.4 (Table 1). Consequently, we expected that the biological behaviour of 1 and 1-BODIPY would be similar. To confirm this hypothesis, we evaluated the in vitro bioactivity of the probe 1-BODIPY against epimastigote forms of T. cruzi. 1-BODIPY displayed an IC50 at the micromolar level comparable to that of the parent compound 1 (Table 1).
| Compound | logD7.4 |
IC50 (μM) |
|---|---|---|
| 1 | 1.92 ± 0.18 | 3.1 ± 0.3 (ref. 5b) |
| 1-BODIPY | 1.26 ± 0.08 | 9.1 ± 0.5 |
These results verified the biological similarity between 1 and its fluorescent analogue 1-BODIPY.
Moreover, with the in vivo oral administration of 1-BODIPY in mind, we studied the in vitro stability of the probe in simulated gastric conditions.13 In an aqueous hydrochloric acid solution resembling the fasted state (pH 1.2, 37 °C), 1-BODIPY showed no evidence of decomposition for at least 2 hours. This indicates that the probe would be stable for the time periods involved in gastric emptying in fasting conditions.
2.4 In vivo biodistribution studies of 1-BODIPY probe
In order to determine the biodistribution behaviour of 1-BODIPY, we analyzed its organ accumulation over the time in mice using two drug vehicles and two different administration routes, i.e. intravenously (i.v.) and orally (p.o.).At the first time point evaluated (15 min after administration), clear differential organ accumulations were evident (see example in Fig. 3), and until 24 h post-administration it was possible to detect the presence of the probe in the organism of the treated mice. On the other hand, the same microemulsion used to dissolve 1-BODIPY in the biological in vitro studies (see Experimental section, Table 1) quenched fluorescence in vivo (Fig. 3b). For this reason we opted for DMSO/H2O as vehicle for the probe in all the studies.
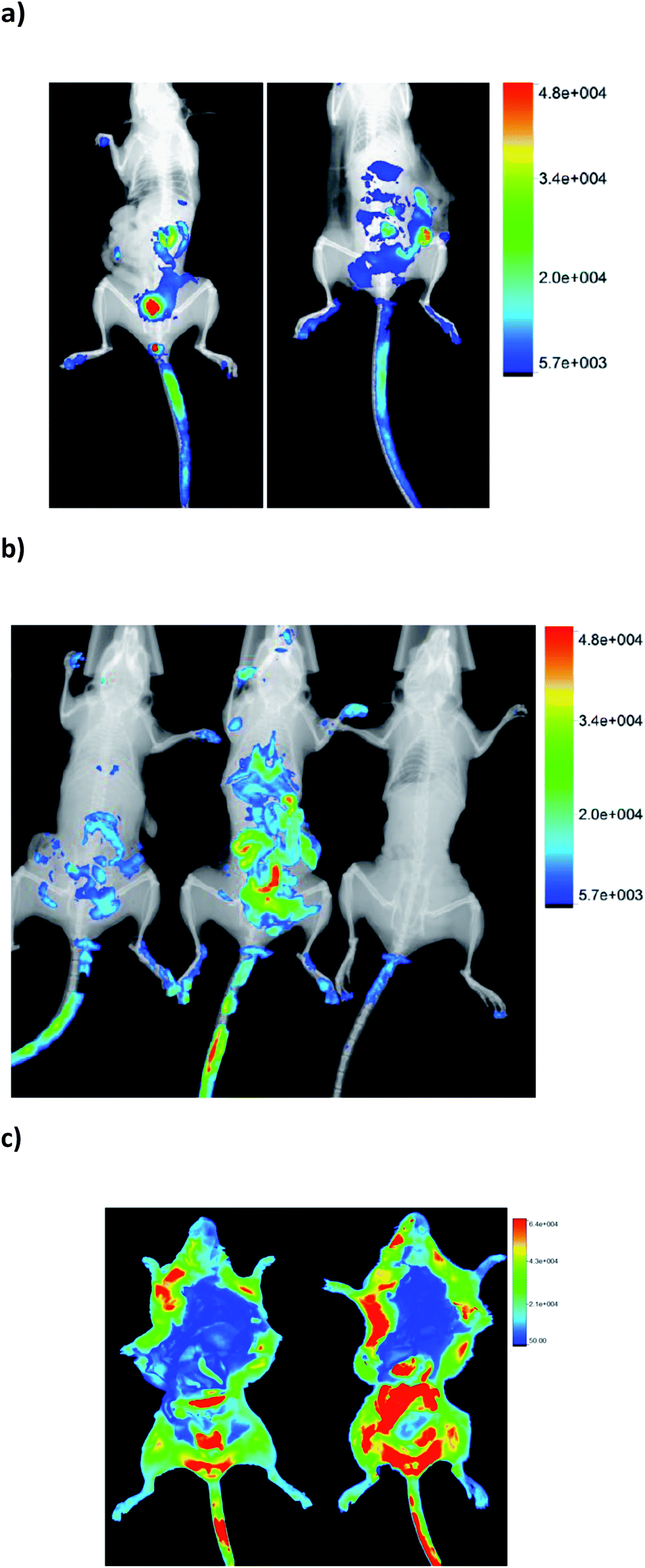 | ||
| Fig. 3 In vivo real-time imaging of black mice acquiring whole body images before and after i.v. injection of 1-BODIPY, via the mouse tail vein: (a) images captured at 30 min (left) and 120 min (right) post-injection of 1-BODIPY dissolved in DMSO/H2O; (b) images captured at 120 min in mouse treated with 1-BODIPY dissolved in the microemulsion as vehicle (see Experimental section) (left) or with 1-BODIPY dissolved in DMSO/H2O (center) and in mouse treated with PBS (control) (right). (c) In vivo imaging of white mice acquiring whole body images after 60 min of i.v. injection, via the mouse tail vein, (left) and p.o. (right) of 1-BODIPY. | ||
Additionally, we performed ex vivo studies where each relevant organ, including spleen, kidney, lung, liver, stomach, bladder, and specially the Chagas involved organs, i.e. heart and intestines, was extracted and its fluorescence measured (Fig. 4). In order to consider the different volume of the organs, the values were expressed as the ratio between fluorescence intensity and organ weight. Furthermore, in order to normalize different experiments, time, and animals, the fluorescence of the heart, as Chagas disease target organ, was used as reference. Thus, the fluorescence emission was always expressed as the ratio between the fluorescence of the organ under study and that measured for the heart.
 | ||
| Fig. 4 (a) Example of ex vivo disposition of organs and fluorescence emissions; (b) organ fluorescence per organ weight and normalized with respect to heart fluorescence at each time and mouse after i.v. injection of 1-BODIPY (dissolved in DMSO/H2O); (c) organ fluorescence per organ weight and normalized respect to heart fluorescence at each time and mouse after p.o. administration of 1-BODIPY (dissolved in DMSO/H2O). | ||
The biodistribution clearly depends on the administration route (Fig. 3c and 4). Regardless of the route, the probe was eliminated through the urinal and gastrointestinal tracts, i.e. presence of fluorescence in kidney, especially in p.o., bladder, stomach, and intestines. On the other hand, when the probe was administered p.o. it was detected significantly in liver, at all time points evaluated. Consequently it could be susceptible of being metabolized by hepatic detoxifying systems. Indeed, in the i.v. via, the fluorescence was detected in liver after 24 hours post-administration.
In order to investigate the behavior of 1-BODIPY in heart, we analyzed the fluorescence of this organ against time and administration route (Fig. 5). For i.v. administration, a rapid and significant accumulation was evidenced after 15 min, and even after 24 h it was possible to detect fluorescence (Fig. 5a). Similarly, for the p.o. administration the presence of 1-BODIPY in heart tissue was significant from 30 min to 24 h post-administration (Fig. 5b).
 | ||
| Fig. 5 Heart ex vivo fluorescence per organ weight: after i.v. administration (a) and p.o. administration (b). (*) p = 0.06; (**) p = 0.03; (***) p = 0.02. | ||
3. Experimental section
All commercial reagents were used without further purification. Solvents were dried and distilled previous their use. Compounds 6 and 8 were prepared according to literature.5b,11 Thin-layer chromatography (TLC) was performed on Merck silica gel F-254 plastic plates, and visualization was carried out under UV light (254 nm). Flash chromatography was conducted using Merck silica gel (40–63 mm mesh size). Melting points were determined on an ELECTROTHERMAL IA-9100 melting points apparatus and are uncorrected. NMR experiments were carried out on Bruker DPX-400 and AVANCE III 400 spectrometers operating at 1H, 13C, 11B, and 19F frequencies of 400.13, 100.62, 128.39, and 376.50 MHz, respectively. TMS was used as internal standard for 1H and 13C measurements, while 11B and 19F spectra were referenced indirectly to BF3·OEt2 and CFCl3, respectively. Chemical shifts (δ) are reported in ppm, and coupling constants (J) in Hz. Mass spectra were obtained on a Shimadzu GCMS-QP 2010 Ultra single quadrupole mass spectrometer using electron impact (EI) ionization. To determine the purity of the compound, elemental microanalysis obtained on a Carlo Erba Model EA1108 elemental analyzer from vacuum-dried sample was used. The analytical results for C, H and N, were within ±0.4 of the theoretical values.3.1 Synthesis of 3-allyl-4-{[4-(4,4-difluoro-1,3,5,6-tetramethyl-4-bora-3a,4a-diaza-s-indacen-8-yl)methyl]piperazin-1-yl}carbonyl-2(Z)-[(1E,2E)-3-(furan-2-yl)propenylidene]hydrazono-2,3-dihydrothiazole (1-BODIPY)
Under nitrogen atmosphere, a mixture of BODIPY 6 (1 equiv.), compound 8 (1 equiv.), K2CO3 (1 equiv.), KI (1 equiv.), and dry THF (5 mL per each 10 mg of 6) was heated at reflux until disappearance of compound 8. After that, the solvent was evaporated and the residue was dissolved in CH2Cl2. The organic layer was washed with brine (three times), dried with anhydrous Na2SO4, and the solvent evaporated to dryness. The residue was purified by column chromatography using a gradient of ethyl acetate in hexane as mobile phase. Compound 1-BODIPY was obtained as a violet solid (24%). Mp 190–191 °C; 1H NMR (CDCl3) δ: 2.50 (6H, s, CH3), 2.55 (6H, s, CH3), 2.62 (4H, bs, piperazinyl–CH2), 3.61 (4H, bs, piperazinyl–CH2), 3.84 (2H, s, N–CH2–C), 4.68 (2H, ddd, J = 5.7, J = 1.5, and J = 1.5, CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.16 (2H, m, CH2–CHCH2), 5.91 (1H, ddt, J = 17.5, J = 10.2, and J = 5.7, CH2–CHCH2), 6.10 (2H, s, indacenyl–H), 6.12 (1H, s, thiazolyl–H), 6.45 (2H, m, furyl–H), 6.64 (1H, d, J = 15.9, CHCH–CHN), 6.97 (1H, dd, J = 15.9 and J = 10.0, CHCH–CHN), 7.44 (1H, bs, furyl–H), 8.06 (1H, d, J = 10.0, CHCH–CHN); 13C NMR (CDCl3) δ: 14.6, 17.5, 42.4, 47.2, 51.7, 52.0, 104.0, 110.3, 111.9, 112.1, 117.7, 122.5, 124.8, 132.3, 132.8, 132.9, 138.8, 141.8, 143.1, 152.8, 154.0, 155.3, 160.2, 167.6; 19F NMR (CDCl3) δ: −146.38 (q, 1JBF = 32.8); 11B NMR (CDCl3) δ: 0.56 (t, 1JBF = 32.8); EI-MS (m/z, abundance) 631 (M+, 47), 371 (31), 260 (100).
CH2), 5.16 (2H, m, CH2–CHCH2), 5.91 (1H, ddt, J = 17.5, J = 10.2, and J = 5.7, CH2–CHCH2), 6.10 (2H, s, indacenyl–H), 6.12 (1H, s, thiazolyl–H), 6.45 (2H, m, furyl–H), 6.64 (1H, d, J = 15.9, CHCH–CHN), 6.97 (1H, dd, J = 15.9 and J = 10.0, CHCH–CHN), 7.44 (1H, bs, furyl–H), 8.06 (1H, d, J = 10.0, CHCH–CHN); 13C NMR (CDCl3) δ: 14.6, 17.5, 42.4, 47.2, 51.7, 52.0, 104.0, 110.3, 111.9, 112.1, 117.7, 122.5, 124.8, 132.3, 132.8, 132.9, 138.8, 141.8, 143.1, 152.8, 154.0, 155.3, 160.2, 167.6; 19F NMR (CDCl3) δ: −146.38 (q, 1JBF = 32.8); 11B NMR (CDCl3) δ: 0.56 (t, 1JBF = 32.8); EI-MS (m/z, abundance) 631 (M+, 47), 371 (31), 260 (100).
3.2 Spectroscopy
2.5 mM stock solutions of 1 and 1-BODIPY were prepared in the appropriate solvent. The fluorescence spectra of solutions of the desired concentration were recorded at 298 K using an excitation wavelength of 460 nm with excitation and emission slit widths of 5 nm. UV-vis absorption and emission spectra were recorded with a Varioskan™ Flash spectral scanner multimode reader.The quantum yield of 1-BODIPY was determined at 298 K according to the literature.14 1,3,5,7,8-Pentamethyl BODIPY was used as a standard (φ = 1). Excitation was carried out at 490 nm, and emission was measured at 530 nm. The quantum yield was calculated following the expression φsample = φstandard × [emissionsample/emissionstandard].
3.3 log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) D7.4 determination15
D7.4 determination15
A pH 7.4 phosphate buffer solution was saturated with n-octanol and n-octanol was saturated with a pH 7.4 aqueous buffer solution. Both were shaken vigorously and then allowed to sit for at least 24 h to ensure complete separation of the two phases. Once this occurred, compounds 1 and 1-BODIPY were dissolved in the n-octanol saturated with aqueous buffer, and then the same volume of the aqueous buffer saturated with n-octanol was added. Partitions were shaken for 24 hours at room temperature. After that, both the octanolic standard and the aqueous phase, after equilibration of each partition, were analysed by UV-spectrophotometry. The logD7.4 values were determined as log10absorbancebuffer/absorbancen-octanol. The determinations were done in triplicate.
3.4 In vitro anti-T. cruzi activity16
T. cruzi epimastigotes (Tulahuen 2 strain) were grown at 28 °C in BHI–tryptose milieu supplemented with 5% fetal bovine serum. Cells from a 10 day-old culture (stationary phase) were inoculated into 50 mL of fresh milieu to give an initial concentration of 1 × 106 cells per mL. Cell growth was followed by measuring the absorbance of the culture at 600 nm every day. Before inoculation, the milieu was supplemented with different doses of 1-BODIPY dissolved in a microemulsion. The microemulsion was prepared as follow: 460 mg polyoxyl-40 hydrogenated castor oil, 180 mg of soya phosphatidylcholine and 1.0 g of cholesterol were pulverized and mixed in a porcelain mortar. This mixture was dissolved in chloroform and the solution was evaporated in vacuo to dryness. In parallel, 360 mg of sodium oleate was dissolved in phosphate buffer and shaken for 12 h at room temperature in an orbital shaker. This solution was then added to the mix containing the compounds and the mixture was homogenized and immersed in an ultrasonic bath at full power for 30–60 min until reaching the desired homogeneity and consistency. Cultures with non-treated epimastigotes and 0.4% microemulsion were included as negative controls, while cultures with 8 μM of nifurtimox were used as positive controls. The percentage of growth inhibition (PGI) was calculated as follows: PGI (%) = {1 − [(Ap − A0p)/(Ac − A0c)]} × 100, where Ap = A600 of the culture containing the drug at day 5; A0p = A600 of the culture containing the drug just after adding the inoculum (day 0); Ac = A600 of the culture in the absence of drug (negative control) at day 5; A0c = A600 in the absence of the drug at day 0. In order to determine the 50% inhibitory concentration (IC50) values, parasite growth was followed in the absence (negative control) and presence of increasing concentrations of the corresponding drug. At day 5, the absorbance of the culture was measured and related to the control. The IC50 value was taken as the concentration of drug needed to reduce the absorbance ratio to 50%. All IC50 values in this work were obtained by analysis with the OriginLab8.5® software package, using sigmoidal regression (PGI vs. logarithm of the compounds concentration) and samples for triplicate. The positive control PGI was always around 50%.3.5 Stability in simulated gastric pH
The simulated gastric pH was obtained using a pH 1.2 aqueous hydrochloric acid solution. 1-BODIPY was dissolved in DMSO and added to the hydrochloric acid solution to reach a concentration of 100 μM, and placed in a shaking water bath at 37 °C. At time points 15, 30, 60, 90 and 120 min sample tubes, in triplicate, were removed and extracted with EtOAc. The organic phases were analyzed by TLC.3.6 In vivo fluorescence imaging
:50, v/v) or in the microemulsion described below. Three subjects per group were imaged at each time points (15 min, 30 min, 60 min, 120 min, and 24 h post-injection). PBS treated mice were used as negative controls. Spleen, kidney, lung, liver, stomach, bladder, heart, and intestines were removed for ex vivo fluorescent organ imaging from one mouse per group at each time point. All the animals' procedures were performed in compliance with national laws and guidelines of the University and were approved by the University ethic committee (Comisión Honoraria de Experimentación Animal, Universidad de la República, Uruguay).Two-dimensional regions of interest (ROIs) were selected using the equipment software in order to measure organs intensities. To calculate the total organ fluorescence intensity in each scan, fluorescence intensities were normalized respect to organ weight and with respect to the fluorescence intensity of heart.
4. Conclusions
We studied the design, preparation, spectral behaviour, and in vivo ability of a BODIPY-fluorescent probe that mimic an anti-Chagas agent.The differential organ distribution of the probe was evidenced resulting good targeting in critic involved-Chagas disease organs. The intravenous administration produced a rapid and constant distribution of the probe during the time.
Acknowledgements
The authors would like to thanks Comisión Sectorial de Investigación Científica (CSIC)-UdelaR (CSIC-No. 661), PEDECIBA-QUÍMICA, and Agencia Nacional de Investigación e Innovación (ANII) of Uruguay for financial support. GR and CAO-M thank ANII for their scholarships.Notes and references
- A. J. Rassi, A. Rassi and J. Marcondes de Rezende, Infect. Dis. Clin., 2012, 26, 275 CrossRef PubMed.
- (a) H. B. Tanowitz, F. S. Machado, L. A. Jelicks, J. Shirani, A. C. de Carvalho, D. C. Spray, S. M. Factor, L. V. Kirchhoff and L. M. Weiss, Prog. Cardiovasc. Dis., 2009, 51, 524 CrossRef PubMed; (b) C. E. Gullo, C. F. Estofolete, C. D. Gil, A. B. Christiano and J. G. Netinho, Revista do Colégio Brasileiro de Cirurgiões, 2012, 39, 146 CrossRef PubMed.
- (a) H. Cerecetto and M. González, Pharmaceuticals, 2010, 3, 810 CrossRef CAS PubMed; (b) M. González and H. Cerecetto, Expert Opin. Ther. Pat., 2011, 21, 699 CrossRef PubMed.
- (a) H. Cerecetto and M. González, Mini-Rev. Med. Chem., 2008, 8, 1355 CrossRef CAS PubMed; (b) J. Varela, E. Serna, S. Torres, G. Yaluff, N. I. Vera de Bilbao, P. Miño, X. Chiriboga, H. Cerecetto and M. González, Molecules, 2014, 19, 8488 CrossRef PubMed.
- (a) G. Álvarez, J. Varela, P. Márquez, M. Gabay, C. E. Arias Rivas, K. Cuchilla, G. A. Echeverría, O. E. Piro, M. Chorilli, S. M. Leal, P. Escobar, E. Serna, S. Torres, G. Yaluff, N. I. Vera de Bilbao, M. González and H. Cerecetto, J. Med. Chem., 2014, 57, 3984 CrossRef PubMed; (b) G. Álvarez, J. Varela, E. Cruces, M. Fernández, M. Gabay, S. M. Leal, P. Escobar, L. Sanabria, E. Serna, S. Torres, S. J. Figueredo Thiel, G. Yaluff, N. I. Vera de Bilbao, H. Cerecetto and M. González, Antimicrob. Agents Chemother., 2015, 59, 1398 CrossRef PubMed; (c) G. Álvarez, J. Martínez, J. Varela, E. Birriel, E. Cruces, M. Gabay, S. M. Leal, P. Escobar, B. Aguirre-López, N. Cabrera, M. Tuena de Gómez-Puyou, A. Gómez Puyou, R. Pérez-Montfort, G. Yaluff, S. Torres, E. Serna, N. Vera de Bilbao, M. González and H. Cerecetto, Eur. J. Med. Chem., 2015, 100, 246 CrossRef PubMed.
- (a) S. Biffi, S. Dal Monego, C. Dullin, C. Garrovo, B. Bosnjak, K. Licha, P. Welker, M. M. Epstein and F. Alves, PLoS One, 2013, 8, e57150 CAS; (b) I. E. Serdiuk, M. Reszka, H. Myszka, K. Krzymiński, B. Liberek and A. D. Roshal, RSC Adv., 2016, 6, 42532 RSC.
- (a) Z. Dost, S. Atilgan and E. U. Akkaya, Tetrahedron, 2006, 62, 8484 CrossRef CAS; (b) H. Sun, X. Dong, S. Liu, Q. Zhao, X. Mou, H. Y. Yang and W. Huang, J. Phys. Chem. C, 2011, 115, 19947 CrossRef CAS; (c) Y. Ni and J. S. Wu, Org. Biomol. Chem., 2014, 12, 3774 RSC; (d) H. Lu, J. Mack, Y. C. Yang and Z. Shen, Chem. Soc. Rev., 2014, 43, 4778 RSC.
- (a) C. A. Osorio-Martínez, A. Urías-Benavides, C. F. A. Gómez-Durán, J. Bañuelos, I. Esnal, I. López Arbeloa and E. Peña-Cabrera, J. Org. Chem., 2012, 77, 5434 CrossRef PubMed; (b) J. O. Flores-Rizo, I. Esnal, C. A. Osorio-Martínez, C. F. A. Gómez-Durán, J. Bañuelos, I. López Arbeloa, K. H. Pannell, A. J. Metta-Magaña and E. Peña-Cabrera, J. Org. Chem., 2013, 78, 5867 CrossRef CAS PubMed; (c) L. Betancourt-Mendiola, I. Valois-Escamilla, T. Arbeloa, J. Bañuelos, I. López Arbeloa, J. O. Flores-Rizo, R. Hu, E. Lager, C. F. A. Gómez-Durán, J. L. Belmonte-Vázquez, M. R. Martínez-González, I. J. Arroyo, C. A. Osorio-Martínez, E. Alvarado-Martínez, A. Urías-Benavides, B. D. Gutiérrez-Ramos, B. Zhong Tang and E. Peña-Cabrera, J. Org. Chem., 2015, 80, 5771 CrossRef CAS PubMed.
- (a) L. C. Zanetti-Domingues, C. J. Tynan, D. J. Rolfe, D. T. Clarke and M. Martin-Fernandez, PLoS One, 2013, 8, e74200 CAS; (b) L. D. Hughes, R. J. Rawle and S. G. Boxer, PLoS One, 2014, 9, e87649 Search PubMed.
- D. Wang, J. Fan, X. Gao, B. Wang, S. Sun and X. Peng, J. Org. Chem., 2009, 74, 7675 CrossRef CAS PubMed.
- J. Rosenthal and S. J. Lippard, J. Am. Chem. Soc., 2010, 132, 5536 CrossRef CAS PubMed.
- (a) L. Yang, R. Simionescu, A. Lough and H. Yan, Dyes Pigm., 2011, 91, 264 CrossRef CAS; (b) F. Heisig, S. Gollos, S. J. Freudenthal, A. El-Tayeb, J. Iqbal and C. E. Müller, J. Fluoresc., 2014, 24, 213 CrossRef CAS PubMed; (c) M. Yu, J. K.-H. Wong, C. Tang, P. Turner, M. H. Todd and P. J. Rutledge, Beilstein J. Org. Chem., 2015, 11, 37 CrossRef PubMed.
- (a) L. Di and E. H. Kerns, Drug-like Properties: Concepts, Structure Design and Methods: from ADME to Toxicity, Elsevier Science, USA, 2010 Search PubMed; (b) E. B. Asafu-Adjaye, P. J. Faustino, M. A. Tawakkul, L. W. Anderson, L. X. Yu, H. Kwon and D. A. Volpe, J. Pharm. Biomed. Anal., 2007, 43, 1854 CrossRef CAS PubMed.
- Y. Wu, X. Peng, B. Guo, J. Fan, Z. Zhang, J. Wang, A. Cui and Y. Gao, Org. Biomol. Chem., 2005, 3, 1387 CAS.
- A. Andrés, M. Rosés, C. Ràfols, E. Bosch, S. Espinosa, V. Segarra and J. M. Huerta, Eur. J. Pharm. Sci., 2015, 76, 181 CrossRef PubMed.
- E. Aguilera, J. Varela, E. Birriel, E. Serna, S. Torres, G. Yaluff, N. Vera de Bilbao, B. Aguirre-López, N. Cabrera, S. Díaz Mazariegos, M. Tuena de Gómez-Puyou, A. Gómez-Puyou, R. Pérez-Montfort, L. Minini, A. Merlino, H. Cerecetto, M. González and G. Alvarez, ChemMedChem, 2016, 11, 1328 CrossRef CAS PubMed.
Footnote |
| † Current address: Grupo de Investigación e Innovación en Ciencias Básicas, GICBAS, Universidad de la Costa, Colombia. |
| This journal is © The Royal Society of Chemistry 2017 |